

Key Points
IL21-mediated induction of CD25 expression on naïve human B cells requires STAT3.
A lack of response to IL-2 may amplify humoral immunodeficiency in patients with STAT3, IL2RG, or IL21R mutations due to unresponsiveness to IL21.
Abstract
B-cell responses are guided by the integration of signals through the B-cell receptor (BCR), CD40, and cytokine receptors. The common γ chain (γc)-binding cytokine interleukin (IL)-21 drives humoral immune responses via STAT3-dependent induction of transcription factors required for plasma cell generation. We investigated additional mechanisms by which IL-21/STAT3 signaling modulates human B-cell responses by studying patients with STAT3 mutations. IL-21 strongly induced CD25 (IL-2Rα) in normal, but not STAT3-deficient, CD40L-stimulated naïve B cells. Chromatin immunoprecipitation confirmed IL2RA as a direct target of STAT3. IL-21–induced CD25 expression was also impaired on B cells from patients with IL2RG or IL21R mutations, confirming a requirement for intact IL-21R signaling in this process. IL-2 increased plasmablast generation and immunoglobulin secretion from normal, but not CD25-deficient, naïve B cells stimulated with CD40L/IL-21. IL-2 and IL-21 were produced by T follicular helper cells, and neutralizing both cytokines abolished the B-cell helper capacity of these cells. Our results demonstrate that IL-21, via STAT3, sensitizes B cells to the stimulatory effects of IL-2. Thus, IL-2 may play an adjunctive role in IL-21–induced B-cell differentiation. Lack of this secondary effect of IL-21 may amplify the humoral immunodeficiency in patients with mutations in STAT3, IL2RG, or IL21R due to impaired responsiveness to IL-21.
Introduction
The primary function of B cells is to produce antigen (Ag)-specific antibodies that neutralize and clear pathogens. Antibody (Ab) production is mediated by 2 populations of effector B cells: memory cells, which circulate throughout the body and rapidly respond to reencounter with the initiating Ag, and long-lived plasma cells, which constitutively secrete large quantities of high-affinity, isotype-switched Ab. Both populations are generated from naïve B cells during germinal center (GC) reactions occurring within secondary lymphoid tissues.1-3 GCs are established when B cells encounter specific Ag and receive instructive signals from T follicular helper (Tfh) cells, which provide signals for their growth, survival, selection, and differentiation.4,5
B-cell differentiation is influenced by many cytokines, including interleukin (IL)-2, IL-4, IL-6, IL-10, IL-12, IL-13, IL-15, transforming growth factor-β6-10 and IL-21.11-13 IL-4 and IL-13 induce class switching, leading to expression and secretion of immunoglobulin (Ig)G and IgE by naïve B cells,6,9,14 whereas IL-10 and IL-21 induce naïve and memory cells to differentiate into plasmablasts producing IgM, IgG, and IgA.6,12,13,15 Some cytokines induce secretion of particular Ig subclasses by human naïve B cells, with IL-4 and IL-13 inducing IgG46,9 and IL-10 and IL-21 inducing IgG1 and IgG3.11,12,16,17 There is also significant interplay between different cytokines: IL-4 enhances IL-21–induced switching to IgG,16 and these cytokines synergize to induce IgE.18 Similarly, transforming growth factor-β and IL-10 cooperate to induce IgA production by naïve B cells,7 and IL-2 enhances the effects of IL-10 on memory B-cell differentiation.19,20 On the other hand, IL-4 inhibits IL-21–induced isotype switching to, and secretion of, IgA.13,16
IL-21 has emerged as the most potent cytokine influencing human B cells. It induces secretion of IgM, IgG, and IgA from all subsets of mature B cells.13,21 The IL-21 receptor comprises a specific IL-21R chain and the common γ chain (γc), an integral component of the receptors for IL-2, IL-4, IL-7, IL-9, and IL-15.22 Binding of IL-21 to its receptor activates JAK1 and JAK3, resulting in phosphorylation and activation of STAT1, STAT3, and STAT5, thereby initiating gene transcription and effector function in responding cells.22 The predominant mechanism underlying IL-21–induced B-cell differentiation is STAT3-mediated induction of BLIMP-1,12,13,23-25 a transcriptional repressor critical for the generation of plasma cells and normal Ab responses in vivo.1,26
Loss-of-function mutations in STAT3 cause Autosomal Dominant Hyper-IgE Syndrome (AD-HIES).27,28 A feature of this condition is impaired humoral immunity following infection and vaccination.29-31 We have previously established that naïve B cells from these individuals fail to differentiate into Ag-specific memory cells in vivo and Ab-secreting cells in response to IL-21 in vitro.23 We have now investigated additional mechanisms by which IL-21/STAT3 signaling modulates human B-cell responses and how defects in this pathway contribute to poor serological immunity in patients with immunodeficiencies.
Methods
Human blood and tissue samples
Buffy coats from healthy donors and spleens from cadaveric organ donors were provided by the Australian Red Cross Blood Service and tonsillar tissue from patients undergoing tonsillectomy. Peripheral blood was collected from patients with mutations in STAT3, STAT1, IL21R, IL2RG, and IL2RA.21,23,32,33 Epstein-Barr virus-transformed lymphoblastoid B-cell lines (LCLs) were established as described.23 Human experiments were approved by the relevant institutional ethics committees at St. Vincent's Hospital, Royal Prince Alfred Hospital, Sydney Children's Hospital Network, Westmead Hospital, The Canberra Hospital (in Australia), as well as at National Institute for Allergy and Infectious Diseases/National Institutes of Health and Rockefeller University. The study was conducted in accordance with the Declaration of Helsinki.
mAbs
The following mAbs were used: FITC-anti-CD20, APC-anti-CD10, APC-anti-IgG, PE-anti-CD27, APC-anti-CD25, PE-anti-IL-2Rβ, PE-anti-IL-2Rγ, APC-anti-CD38, PE-anti-CD4, Alexa Fluor 647-anti-CXCR5, PE-anti-pSTAT3 (pY705), Alexa Fluor 647-anti-pSTAT1 (pY701), Alexa Fluor 488-anti-pSTAT5 (pY694), APC-anti-IL-2 (BD Biosciences); FITC-anti-CD45RA, PE-anti-IL-21, neutralizing anti-IL-2 (eBioscience); and Alexa Fluor 647-anti-CD226 (DNAM1) (Biolegend).
B-cell phenotyping and isolation
Naïve B cells were isolated from peripheral blood, tonsil, or splenic mononuclear cells using negative isolation (Invitrogen) followed by sorting naïve B cells (CD20+CD27−CD10−IgG−) after labeling with specific mAbs (FACSAria; BD). The purity of the recovered population was typically >98%.
In vitro activation and analysis of cultured human lymphocytes
Sorted naïve B cells were cultured with CD40L18,23 alone or together with IL-10 (100 U/mL), IL-21 (0-50 ng/mL; PeproTech), and/or IL-2 (50 ng/mL; Millipore). For phenotypic analysis, cells were labeled with 5 (and 6)-carboxyfluorescein diacetate succinimidyl ester, cultured in 48-well plates (∼2 × 105 cells per 400 μL/well) for 5 days, then harvested and incubated with specific mAbs for fluorescence-activated cell sorter analysis. The frequency of plasmablasts (CD27hiCD38hi)34 and expression of CD25, IL-2Rβ, and IL-2Rγ were determined using FlowJo software (Tree Star, Inc.). Ig secretion was determined by enzyme-linked immunosorbent assay13 on supernatants of naïve B cells (∼5 × 103 cells per 200 μL/well) cultured with CD40L alone or together with IL-2 and/or IL-21 for 10 to 12 days. CD4+ T-cell subsets were isolated from human tonsils and stimulated with bead-bound antibodies against CD2, CD3, and CD28 (T cell activation and expansion [TAE] beads; Miltenyi); intracellular cytokine expression was determined as described.35 For T- and B-cell co-cultures, sorted naïve B cells were cultured in 96-well plates with sorted, mitomycin C-treated CD4+ T-cell subsets (4.5 × 104 T and B cells per 200 μL/well)35 with or without TAE beads and/or neutralizing anti-IL-2 Ab or IL-21R-Fc (R&D) for 7 days.
Microarrays
Naïve B cells were purified from normal human donors or STAT3-deficient patients and cultured in 48-well plates (∼2 × 105 cells per 400 μL/well) for 4 to 5 days with CD40L alone or together with IL-21. Cultured cells were harvested and RNA extracted (RNeasy Mini Kit; Qiagen). Microarrays were performed using GeneChip Human Gene 1.0 ST Arrays (Affymetrix). Microarray data were analyzed using GenePattern software (version 3.2.3, Broad Institute, Cambridge, MA). The GEO accession number for the microarray data is GSE51587.
Quantitative polymerase chain reaction
Sorted naïve B cells were cultured for 5 days with CD40L alone or together with IL-21. Expression of IL2RA (forward, 5′-GAAATGCAAAGTCCAATGCAG-3′; reverse, 5′-AATTCTCTCTGTGGCTTCATTTTC-3′) was determined using the Roche LightCycler 480 Probe Master Mix and System and standardized to GAPDH (forward, 5′-CTCTGCTCCTCCTGTTCGAC-3′; reverse, 5′-ACGACCAAATCCGTTGACTC-3′).
Chromatin immunoprecipitation assay
LCLs were fixed with formaldehyde, washed with cold phosphate-buffered saline containing Protease Inhibitor Cocktail (Roche), resuspended in Nuclei Buffer, and homogenized. Lysates were sonicated, depleted of insoluble material, and immunoprecipitated with anti-STAT3 or mouse IgG. Immunoprecipitated DNA was used as a template for quantitative polymerase chain reaction using SensiMix Probe Master Mix (Bioline) and primers for GAPDH (forward, 5′-TTGCAACCGGGAAGGAAA-3′; reverse, 5′-TAGCCTCGCTCCACCTGACTT-3′) and the promoter regions of PRDM1 (forward, 5′-TGCAGGAAGGTGGTAGGAAACGG-3′; reverse, 5′-TCGCTGGTGCGGAAACTGCTT-3′) and IL2RA (forward, 5′-TGTCATCCCCAAAACTCCCG-3′; reverse, 5′-ACGTCACCAAGTAAAGGGCA-3′).
Expression of phosphorylated STATs
LCLs were cultured in the absence or presence of IL-2, IL-21 or both cytokines. Expression of phospho-STAT1, STAT3, or STAT5 was determined as previously described.18
Results
IL-21 induces IL-2RA in normal, but not STAT3-deficient, naïve B cells
We have previously demonstrated that naïve B cells from STAT3-deficient individuals have impaired PRDM1/BLIMP-1 up-regulation in response to IL-21.23 To identify other STAT3-dependent genes modulated by IL-21, we analyzed gene expression in normal and STAT3-deficient naïve B cells stimulated with CD40L alone or together with IL-21. PRDM1 was the gene most strongly induced in CD40L/IL-21-stimulated normal naïve B cells compared with those stimulated with CD40L alone (Figure 1A). As expected, PRDM1 was not induced in IL-21–stimulated STAT3-deficient naïve B cells (Figure 1A). Similarly, other genes involved in plasma cell formation, PERP1,36 XBP1,26 IGJ, IGKC, and IGLC, were also up-regulated in normal but not STAT3-deficient naïve B cells in response to IL-21 (Figure 1A). Interestingly, the second most-highly expressed gene in CD40L/IL-21–stimulated normal naïve B cells was IL2RA (Figure 1A). IL2RA encodes CD25 (IL-2Rα), which, when complexed with IL-2Rβ and IL-2Rγ/γc, forms the high-affinity IL-2 receptor.22 Induction of IL2RA by IL-21 required functional STAT3, as its level of expression in CD40L/IL-21–stimulated STAT3-deficient naïve B cells did not differ from CD40L stimulation alone (Figure 1A).
Figure 1.
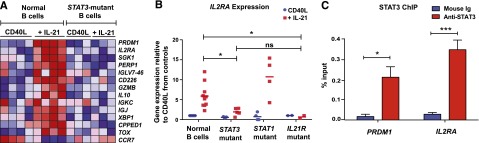
IL-21 directly induces IL2RA in normal, but not STAT3MUT, naïve B cells. (A) Naïve B cells were purified from the peripheral blood of normal donors (n = 4) and STAT3-deficient AD-HIES patients (n = 3) and then cultured with CD40L alone (“CD40L”) or together with IL-21 (“+IL-21”). RNA was extracted after 4 days and microarrays performed using Affymetrix Human Gene 1.0 ST Arrays. Genes with marked differences in expression between normal and STAT3-mutant (STAT3MUT) cells are shown. (B) Naïve B cells from normal donors (n = 10) or patients with loss-of-function mutations in STAT3 (n = 6), STAT1 (n = 4), or IL21R (n = 2) were cultured with CD40L alone (blue) or together with IL-21 (red). RNA was extracted after 5 days and used to determine expression of IL2RA by quantitative polymerase chain reaction. Results show expression levels relative to B cells cultured with CD40L alone. Each symbol represents an individual experiment using cells from a different donor or patient; the horizontal line represents the mean, *P < .05. (C) Chromatin immunoprecipitation (ChIP) was performed on normal LCLs using mouse Ig or anti-STAT3 Ab. Immunoprecipitated chromatin was assessed for the presence of PRDM1 and IL2RA Results are expressed relative to gene expression in the input DNA and represent the mean ± SEM from 3 separate experiments using different LCLs. *P < .05, ***P < .005.
To confirm the microarray data, we examined IL2RA mRNA in normal and STAT3-mutant (STAT3MUT) naïve B cells cultured with CD40L with or without IL-21 for 5 days. As IL-21 can activate STAT1,23 we also examined IL2RA induction in naïve B cells from individuals with loss-of-function STAT1 mutations.37 Although IL-21 increased IL2RA expression in CD40L-stimulated normal or STAT1-deficient naïve B cells by 6- to 10-fold (Figure 1B), it had only a modest effect on IL2RA in STAT3MUT naïve B cells (Figure 1B). Not surprisingly, IL-21 failed to increase IL2RA expression in CD40L-stimulated IL-21R–deficient naïve B cells (Figure 1B).
Impaired induction of IL2RA by IL-21 in STAT3MUT naïve B cells suggested IL2RA may be a direct transcriptional target of STAT3. To confirm this, we performed chromatin immunoprecipitation assays using normal LCLs. As a control, we assessed STAT3 binding to the PRDM1 promoter. Immunoprecipitation with anti-STAT3 Ab significantly enriched for PRDM1, confirming PRDM1 is a direct STAT3 target. A similar degree of enrichment was observed for IL2RA, demonstrating that IL2RA is also directly targeted by STAT3 (Figure 1C).
IL-21–induced expression of CD25 is impaired on STAT3-deficient naïve B cells
Assessment of the defect in IL2RA up-regulation in STAT3MUT naïve B cells was extended by assessing the expression of CD25, as well as IL-2Rβ and IL-2Rγ/γc, on naïve B cells from controls and STAT3MUT patients following a 5-day culture with CD40L with or without IL-21. Whereas IL-21 strongly increased CD25 on normal B cells, there was a significant reduction in IL-21–induced CD25 expression on STAT3MUT naive B cells (Figure 2A,C). In contrast to CD25, IL-21 had minimal effect on IL-2Rβ and γc on CD40L-stimulated normal and STAT3MUT naïve B cells (Figure 2A).
Figure 2.

IL-21–induced expression of CD25 is impaired on STAT3MUT naïve B cells. (A-C) Naive B cells from normal donors or patients with loss-of-function mutations in STAT3, STAT1, IL2RG, or IL21R were cultured with CD40L alone (blue) or in combination with IL-21 (red). (A) Expression of IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132, γc) on normal and STAT3MUT B cells or of (B-C) IL-2Rα (CD25) on normal and STAT1MUT, STAT3MUT, IL-21RMUT, or IL2RγMUT B cells was determined after 5 days (or on day 0 for B). The histogram plots in A and B are representative of experiments performed on naïve B cells isolated from 6 normal donors, 6 STAT3-deficient patients, 1 STAT1-deficient patient, 2 IL-2Rγ–deficient patients, and 3 IL-21R–deficient patients. The summary graph in C depicts the mean ± SEM of CD25 expression on cultured B cells from the indicated numbers of patients. (D-E) Naïve B cells from normal donors (blue, n = 7), STAT3-deficient AD-HIES patients (red, n = 6), IL-2Rγ–deficient X-SCID patients (green, n = 2), or IL-21R–deficient patients (black, n = 3) were cultured with CD40L alone (D) or in the presence of IL-21 (E). The mean fluorescence intensity (MFI) of CD25 expression was determined at the indicated times. Results represent mean ± SEM for the indicated number of controls and patients. *P < .005, **P < .001, ***P < .0001.
To further explore the requirements for IL-21–induced CD25 expression, we examined individuals with defects in IL-21 signaling, ie, loss-of-function mutations in IL21R, IL2RG (encoding IL-2Rγ), or STAT1. Consistent with quantitative polymerase chain reaction data, CD25 induction on CD40L/IL-21–stimulated STAT1-deficient naïve B cells was comparable to normal B cells (Figure 2B). However, this effect of IL-21 was abolished by mutations in IL21R and IL2RG (Figure 2B-C). The inability of IL-21 to increase CD25 expression on IL-21R– or γc-deficient naïve B cells confirms that both of these are essential, nonredundant components of IL-21 signaling in B cells. Furthermore, STAT3 is the predominant effector of IL-21R/γc signaling to induce CD25 on IL-21–stimulated naive B cells.
Because induction of CD25 on activated lymphocytes is transient,22 it was possible that lower CD25 levels on IL-21–stimulated STAT3MUT naïve B cells reflected alterations in kinetics of expression on these cells. When normal naïve B cells were cultured with CD40L alone, CD25 expression increased after 2 days (Figure 2D). This was greatly enhanced by exogenous IL-21, being maximal after 3-4 days, exceeding that on CD40L-stimulated cells by 3.5-fold (Figure 2D-E). Induction of CD25 on STAT3MUT naïve B cells followed kinetics similar to normal B cells, with detectable increases in the presence of IL-21 over CD40L alone after 2 to 4 days (compare Figure 2D-E). However, CD25 on IL-21–stimulated STAT3MUT naïve B cells was significantly reduced compared with normal B cells (Figure 2D-E). The residual STAT3 function in STAT3MUT naïve B cells likely explains the modest increase in CD25 expression in response to IL-21. Consistent with the requirement for IL-21R and γc as components of the IL-21R complex, induction of CD25 was completely abrogated at all times on naïve B cells from patients with IL2RG and IL21R mutations (Figure 2D-E). The identical response of γc and IL21R-deficient B cells suggests that IL-21 acts directly through the IL-21R/γc complex to induce CD25 rather than indirectly by inducing IL-2, which then promotes expression of its own receptor through CD25.
IL-21–induced autocrine IL-10 secretion does not up-regulate CD25 on naïve B cells
Another CD40L/IL-21–induced STAT3-dependent gene expressed by naïve B cells was IL10 (Figure 1A), which can also activate STAT3 and promote human B-cell differentiation.6,22 Thus, impaired production of IL-10 by IL-21–stimulated STAT3MUT naïve B cells may underlie their inability to up-regulate CD25. We therefore compared CD25 induction on normal naïve B cells cultured with CD40L alone or with IL-10 or IL-21. Unlike IL-21, IL-10 did not increase CD25 over levels observed with CD40L alone (Figure 3A). It was also possible that IL-21 induced B cells to produce IL-2, which functioned to promote expression of CD25 on B cells, as occurs in T cells.22 However, microarray analysis indicated expression of IL2 by in vitro-stimulated B cells was low/negligible, consistent with previous studies of cytokine production by human B cells.38,39 Thus, the failure of IL-10 to enhance CD25 on naïve B cells, and the lack of production of IL-2 by human B cells, excludes secretion of endogenous IL-10 or IL-2 by IL-21–stimulated B cells as the mechanism by which IL-21 induces STAT3-dependent CD25 expression.
Figure 3.

IL-21, but not IL-10, induces CD25 expression on all activated naïve B cells. (A) Naïve B cells from healthy donors were cultured for 5 days with CD40L alone (blue histogram) or in the presence of IL-10 (orange histogram) or IL-21 (red histogram). Expression of CD25 was determined after 5 days. (B-D) Normal naïve B cells were cultured for 5 days with CD40L alone or together with IL-21 for 5 days. (B) Differential expression of CD27 and CD38 delineates IL-21–induced plasmablasts (CD38hiCD27hi; red gate) and nonplasmablasts (CD38loCD27lo; blue gate). Expression of CD25 (C) or DNAM-1 (D) was determined on all B cells in cultures stimulated with CD40L alone or on nonplasmablasts and plasmablasts present in cultures of CD40L/IL-21–stimulated B cells. Gray histograms represent isotype controls. Numbers represent percentage of live cells within each gate.
The inability of IL-21 to induce CD25 on STAT3MUT naïve B cells is not due to preferential expression by in vitro-generated plasmablasts
Although microarrays detect changes in mRNA expression globally within a population of cells, if the population studied is heterogeneous, they cannot distinguish differing expression levels between subsets of these cells. Following in vitro culture with CD40L/IL-21, a subset of normal naïve B cells differentiates into plasmablasts.12,13 STAT3MUT naïve B cells, however, do not undergo this differentiation program.23 Thus, it was possible that impaired up-regulation of CD25 on IL-21–stimulated STAT3MUT naïve B cells reflected preferential expression on plasmablasts and was a consequence of an absence of such cells in these cultures. To address this, expression of CD25 was determined on plasmablasts (CD38hiCD27hi) and non-plasmablasts (CD38loCD27−)34 in cultures of CD40L/IL-21–stimulated normal naïve B cells (Figure 3B). CD25 was equally expressed on both of these populations of cells (Figure 3C). In contrast, DNAM1 (CD226), which was found to be an IL-21–induced STAT3-dependent gene (Figure 1A), was preferentially induced on plasmablasts (Figure 3D). Thus, functional STAT3 is required for induction of CD25 on all IL-21–stimulated B cells, rather than being selectively acquired by a distinct subset.
IL-2 promotes IL-21–induced plasmablast generation and Ig secretion by naïve B cells
We next investigated the physiological significance of IL-21–induced CD25 expression on naïve B cells. Normal naïve B cells were cultured with CD40L alone or together with IL-2, IL-21, or both cytokines. CD40L alone or CD40L/IL-2 failed to induce plasmablasts (CD38hiCD27hi) from naïve cells (Figure 4A). However, a discrete plasmablast population was detected in cultures of CD40L/IL-21–stimulated naïve B cells, the frequency of which was increased by IL-2 (Figure 4A).
Figure 4.

IL-21–induced CD25 expression enables naïve B cells to respond to IL-2 with enhanced plasmablast generation and Ig secretion. Naïve B cells were sorted from normal spleens and then cultured with CD40L alone in the presence or absence of IL-2 and/or IL-21. The proportion of plasmablasts (CD27hiCD38hi) (A,C) and expression of CD25 (B) were determined by flow cytometry after 5 days. The data depicted are representative of 4 (A) or 3 (B) separate experiments using naïve B cells isolated from different donor spleens. C represents the mean ± SEM from 4 separate experiments; *P < .05, comparing cultures with and without IL-2. Secretion of IgM and IgG (D) or IgG subclasses (E) was determined after 10 to 12 days by enzyme-linked immunosorbent assay. Statistical analysis using 2-way ANOVA with Bonferroni post-test analysis confirmed a statistically significant difference between cultures with or without IL-2 (P < .005 for both IgM and IgG) and between different concentrations of IL-21 (P < .005 for IgM; P < .001 for IgG). Data in D show mean ± SEM from 3 independent experiments on 3 different donor spleens, each performed in triplicate. Data in E show mean ± SEM from a single experiment performed in triplicate but is representative of 2 independent experiments performed on different normal donor spleens. (F) Naïve and memory B cells were isolated from a normal donor and a CD25-deficient patient and then cultured with CD40L/IL-21 alone or together with IL-2. Secretion of IgM and IgG was determined after 10 days. Values represent the mean ± SEM of triplicate cultures for normal B cells and the mean of single cultures for CD25-deficient B cells. (G) Carboxyfluorescein diacetate succinimidyl ester profiles of splenic naïve B cells from normal donors cultured for 5 days with CD40L alone or with varying concentrations of IL-21 in the absence or presence of IL-2. Results are representative of 4 independent experiments using different normal donor spleens.
Induction of CD25 by IL-21 on CD40L-stimulated naïve B cells was dose dependent, and addition of IL-2 further enhanced CD25 expression above that induced by IL-21 (Figure 4B). Consistent with these dose-dependent changes in CD25 expression, IL-2 significantly augmented IL-21–induced plasmablast generation from, and IgM and IgG secretion by, naïve (Figure 4C-D) and memory (not shown) B cells at most IL-21 concentrations tested. As IL-21 induces isotype switching to IgG1 and IgG3,11,12,16 we assessed the effect of IL-2 on IgG subclasses. The IL-2–mediated increase in total IgG secretion by CD40L/IL-21–stimulated naïve B cells predominantly resulted from enhanced IgG1 and IgG3 (Figure 4E). Thus, IL-2 amplifies the response induced by IL-21 rather than skewing IgG production to different subclasses. To further demonstrate the physiological significance of IL-2–mediated enhancement of IL-21–induced B-cell differentiation, we examined responses of B cells isolated from an individual with an IL2RA mutation.32 Whereas IL-2 increased IgM and IgG production by IL-21–stimulated normal naïve and memory B cells by >2-fold, IL-2 failed to augment IL-21–induced differentiation of CD25-deficient B cells (Figure 4F). Thus, through a positive feedback whereby IL-21 induces CD25 expression on naïve B cells and IL-2 further amplifies its level of expression, IL-21–stimulated naïve B cells acquire responsiveness to the stimulatory effects of IL-2. Importantly, this requires expression of the high-affinity IL-2 receptor, as evidenced by unresponsiveness of B cells that lack CD25 but continue to express components of the low and intermediate affinity IL-2 receptor.32
IL-2 promotes proliferation of many cell types, including human B cells.6 As various facets of lymphocyte differentiation are linked to cell division,15,16,19 we established whether the effect of IL-2 on IL-21–stimulated B cells resulted from enhanced proliferation or differentiation. Carboxyfluorescein diacetate succinimidyl ester-labeled naïve B cells were cultured for 5 days with varying concentrations of IL-21 in the absence or presence of IL-2. IL-2 had no significant effect on proliferation of CD40L-stimulated B cells regardless of the IL-21 concentration (Figure 4G). Thus, IL-2 enhances plasmablast generation and Ig secretion by IL-21–stimulated naïve human B cells by increasing the rate of differentiation of these cells rather than promoting proliferation or survival.
IL-2 alone can promote plasmablast generation from IL-21–primed naïve B cells
To assess whether the effects of IL-2 required the continual presence of IL-21, naïve B cells were first cultured for 3 days with CD40L/IL-21, then washed and recultured in media alone or with IL-2. The proportion of plasmablasts was determined by flow cytometry after a further 3 days and Ig secretion after 7 days. The frequency of plasmablasts and amounts of secreted IgM, IgG, and IgA were increased 2- to 5-fold in cultures of IL-21–primed/IL-2–stimulated secondary cultures compared with cells primed with IL-21 but recultured without cytokines (Figure 5A-B). IL-2 failed to induce plasmablasts and Ig secretion by cells not primed with IL-21 (not shown), highlighting the dependence of IL-2 on the expression of its high-affinity receptor, induced by IL-21/STAT3 signaling, for these effects to occur. Thus, the initial up-regulation of CD25 by IL-21 enabled IL-2 to promote B-cell differentiation.
Figure 5.

Initial upregulation of CD25 by IL-21 licenses IL-2 to maintain plasmablast generation in the absence of IL-21. Naive B cells from normal human spleens were initially cultured with CD40L together with IL-21. After 3 days, cells were harvested, washed, and recultured either in media alone or with IL-2. (A) After a further 3 days of culture, the proportion of plasmablasts (CD27hiCD38hi) was determined by flow cytometry. Each symbol corresponds to an individual experiment that used naïve B cells from a different normal donor spleen; the horizontal line represents the mean. (B) Secretion of IgM, IgG, and IgA was determined after 7 days of secondary culture in media alone or with IL-2 and Ig secretion was determined by enzyme-linked immunosorbent assay. Results are expressed as fold-change in Ig secretion relative to the “no cytokine” secondary culture (set to equal 1.0). The results for IgM represent mean ± SEM (n = 3); IgG secretion was detected in only 1 of 3 experiments; IgA was detected in 2 of 3 experiments.
IL-2 enhances IL-21–induced STAT3 phosphorylation in human B-cell lines
Binding of IL-21 to IL-21R on B cells activates STAT1, STAT3, and STAT5.23,24 Correspondingly, IL-21 induced phosphorylation of STAT1, STAT3, and to a lesser extent STAT5 in normal LCLs, with a maximal response after 30 min and return to baseline by 120 min (Figure 6A). IL-2 predominantly signals through STAT5.22 To determine whether the combination of IL-2 and IL-21 alters phospho-(p)STAT signaling, LCLs from normal donors or patients with mutations in IL2RA32 or IL21R33 (Figure 6B) were stimulated with IL-2 or IL-21 alone or together and intracellular pSTAT1, pSTAT3, and pSTAT5 were determined. The magnitude of STAT1 and STAT3 phosphorylation induced by IL-21, or of STAT5 induced by IL-2, was similar to that induced by the combination of both IL-2 and IL-21 (Figure 6C). Consistent with this, the frequency of cells expressing pSTAT3 and pSTAT5 was unchanged with IL-2/IL-21 stimulation compared with IL-2 or IL-21 alone (Figure 6D). However, in the presence of IL-2 and IL-21, a greater proportion of B cells coexpressed pSTAT3 and pSTAT5 than with either cytokine alone (Figure 6D). The effect of IL-21 was specific, as it did not induce STAT phosphorylation in IL-21RMUT LCLs, whereas the residual induction of pSTAT5 in CD25MUT LCLs most likely results from IL-2 signaling through the IL-2Rβ/γc complex (Figure 6C). Thus, IL-2 and IL-21 are likely to achieve their complementary effect on B-cell differentiation to plasmablasts by independent signaling pathways in a common subset of activated cells.
Figure 6.

Effect of IL-2 and IL-21 on STAT phosphorylation in human B-cell lines. (A) LCLs from normal donors were incubated for varying times in the absence or presence of IL-21. Phosphorylation of STAT1, STAT3, and STAT5 was then determined by intracellular staining and flow cytometry. Data are depicted as fold change in mean fluorescence intensity of pSTAT in the presence of IL-21 over media alone. Results represent mean ± SEM of experiments using LCLs from 6 different donors. (B) Expression of CD25 and IL-21R was determined on LCLs derived from different normal donors (red histogram) or CD25-deficient (blue histogram) or IL21R-deficient (gray histogram) patients (n = 2/group). (C-D) Normal, CD25-deficient (CD25MUT), and IL-21R–deficient (IL-21RMUT) LCLs were incubated in the absence (Nil) or presence of IL-2, IL-21, or IL-2/IL-21. Expression of pSTAT1, pSTAT3, and pSTAT5 was determined after 30 minutes. Results in C are expressed as fold change above pSTAT expression in cells cultured with no cytokine and represent mean ± SEM of experiments using LCLs from 2 different donors or patients. D shows representative contour plots demonstrating coexpression of pSTAT3 and pSTAT5 in LCLs cultured in the absence (no cytokine) or presence of IL-2, IL-21, or IL-2/IL-21.
IL-2 and IL-21 produced by Tfh cells cooperate to induce Ig secretion by cocultured naïve B cells
B-cell differentiation into plasma cells and memory cells occurs predominantly in GCs.1,2 Much of the IL-21 that stimulates B cells is derived from Tfh cells co-localizing with B cells in GCs. On the other hand, IL-2 is produced by most subsets of activated CD4+ T cells.35 For IL-2 to best promote the effects of IL-21 in vivo, both cytokines would need to be produced by the same cell type. To address this, naïve, CXCR5lo, CXCR5intermediate, and CXCR5hi Tfh cells were isolated from human tonsils,35 stimulated for 5 days, and cytokine expression determined. While IL-2 was expressed by 50% to 80% of these CD4+ T-cell subsets, expression of IL-21 was greatest for CXCR5intermediate and CXCR5hi Tfh cells35 (Figure 7A). Importantly, 70% to 90% of IL-21–expressing CD4+ T cells coexpressed IL-2 (Figure 7A).
Figure 7.

IL-2 and IL-21 produced by Tfh cells cooperate to induce Ig secretion by co-cultured naïve B cells. (A) Naïve, CXCR5lo, CXCR5intermediate, and CXCR5hi Tfh CD4+ T cells were sort-purified from human tonsils and then stimulated in vitro with TAE beads. After 5 days, expression of IL-2 and IL-21 in these different populations following restimulation with PMA/ionomycin was determined. The values represent the proportions of cytokine-expressing cells. (B) CD4+CXCR5intermediate T cells and CXCR5hi Tfh cells were sorted from tonsil and co-cultured with autologous naïve B cells either in the absence (Nil) or presence of TAE beads (blue). Endogenous IL-2 and/or IL-21 were neutralized by the addition of anti-IL-2 mAb (red), IL-21R-Fc (pink), or anti-IL-2 mAb plus IL-21R-Fc (yellow), respectively. An isotype control mAb (green) was also included. IgM secretion was measured after 9 days. Results represent 2 independent experiments performed using cells from different donor tonsils. *P < .05; **P < .01; ***P < .0001. (C) IL-21, secreted from Tfh cells, promotes B-cell maturation by inducing Blimp-1. IL-21 also enhances CD25 expression on naïve B cells, sensitizing them to the effects of IL-2, which is also secreted by Tfh cells. IL-2 then enhances the effects of IL-21 on B cells. IL-21 and IL-2 thus work cooperatively to induce plasma cell development and Ig secretion.
Coexpression of these cytokines by Tfh cells could facilitate their combined effect on B-cell differentiation. To test this, naïve B cells and CXCR5+CD4+ T cells were co-cultured in media alone or with TAE beads in the absence or presence of neutralizing Ab against IL-2, IL-21R-Fc, or both. IgM secretion was measured after 9 days. Activated, but not resting, CD4+CXCR5intermediate T cells and CXCR5hi Tfh cells induced substantial IgM secretion by co-cultured B cells, and this was unaffected by an isotype control mAb (Figure 7B). However, blocking endogenous IL-2 or IL-21 reduced IgM secretion by ∼35-80% and ∼70%, respectively. Importantly, there was a greater reduction in IgM secretion (>80%) when both IL-2 and IL-21 were concurrently blocked, confirming that IL-2 and IL-21 derived from CD4+CXCR5+ T cells cooperate to induce Ig secretion. Expression of IL-2, IL-21, and ICOS by CXCR5+CD4+ T cells was equivalent regardless of IL-2 or IL-21 neutralization (data not shown), suggesting that the reduction in IgM secretion was a direct effect of cytokine blockade on B-cell function rather than a consequence of impaired T-cell activation.
Discussion
Previous studies have suggested a role for IL-2 during human B-cell differentiation. However, its exact role in the complex network of cytokines influencing B cells has not been fully elucidated. IL-2 receptors have long been identified on normal and malignant human B-cell subsets,19,40 and CD25 can be induced by T-derived and T-independent stimuli, including CD40L,41 IL-10,19,42 BCR engagement,41,43 and TLR ligands.41 When combined with IL-2, most of these stimuli enable Ab secretion by activated B cells.41,44,45 We have identified a novel role for IL-21 in regulating human B-cell differentiation. By inducing CD25, IL-21 sensitizes activated B cells to the differentiation-promoting effects of IL-2, thereby enabling cooperative interplay between IL-2 and IL-21 to amplify plasmablast generation and Ab secretion. Thus, IL-2 may play an adjunctive role in IL-21–induced B-cell differentiation.
By examining B cells from individuals with specific genetic mutations, we revealed the critical requirement for the IL-21R/γc complex as well as STAT3, but not STAT1, in this process. These findings identify an additional defect contributing to the humoral impairment in STAT3-deficient AD-HIES patients, who are predisposed to pyogenic infections with Staphylococcus aureus and encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae), frequently culminating in parenchymal lung damage such as bronchiectasis and pneumatocoeles. This may reflect impaired Ag-specific Ab responses after immunization with T-dependent Ag.29-31 Previous studies have suggested that the humoral impairment in AD-HIES arises predominantly from intrinsic B-cell abnormalities, thereby highlighting the dependence of normal B-cell function on STAT3. Thus, although serum levels of IgM, IgG, and IgA are normal in AD-HIES, STAT3-deficient individuals fail to generate normal levels of Ag-specific Ab or a normal pool of Ag-specific memory B cells in vivo23 and their naïve B cells fail to differentiate into Ab-secreting cells in response to IL-10 or IL-21 due to an inability to undergo the molecular changes required for commitment to the plasma cell lineage.23 Our work extends these observations by demonstrating that STAT3MUT naïve B cells fail to up-regulate CD25 in response to IL-21, rendering them unable to respond to the stimulatory effects of IL-2. This secondary defect would exacerbate the limited response of STAT3MUT B cells to the direct effects of IL-21 on B-cell maturation.
An inability to respond to IL-2 due to a lack of IL-21–induced priming of naïve B cells may also contribute to the humoral immunodeficiency in γc-deficient X-SCID patients, whose impaired response to IL-21 causes their profound Ab deficiency,21 as well as in patients with loss-of-function mutations in IL21R who manifest a memory B-cell deficiency and an inability to elicit normal Ab responses following vaccination and infection.33 Because the consequences of humoral immunodeficiencies are often severe, necessitating ongoing intravenous immunoglobulin therapy to prevent recurrent infections and bronchiectasis, an improved understanding of the molecular basis for their immune dysfunction may pave the way for more targeted therapies.
Our data revealed that IL-2 and IL-21 are coexpressed by a substantial proportion of Tfh cells, which would enable cooperation between these cytokines in promoting B-cell maturation in GCs (Figure 7C). The requirement for IL-21 or other exogenous stimuli such as BCR engagement41 to prime B cells to the effects of IL-2 through enhanced CD25 expression ensures that the B-cell response to IL-2 is regulated to occur only “on demand,” as B cells are sensitive to only IL-2 in the presence of specific Ag or Tfh-derived cytokines. Our data may also provide a mechanism for the positive correlation between the frequency of IL-2–producing Ag-specific memory CD4+ T cells and IgG titers in vivo.46 It is plausible that the IL-2+ memory CD4+ T cells examined46 contained IL-21–producing Tfh cells.
IL-2, IL-21, and their receptors share several structural and functional similarities. Both are type I cytokines whose receptors not only require γc for signalling, but IL-21R exhibits substantial amino-acid homology to IL-2Rβ.47 Both IL-2 and IL-21 are secreted by activated CD4+ T cells4,5,35,48 and act as T-cell growth factors.22,48 BLIMP-1, which influences terminal differentiation of B and T lymphocytes,49 is induced by IL-21 in B cells1,2,23,24 and IL-2 in T cells.50 Although this list is not exhaustive, these similarities support the evolution of a concurrent role for IL-2 in B-cell maturation, given the essential role of IL-21 in this process.
Overall, our study highlights the utility of examining B cells from patients with monogenic primary immunodeficiencies to not only clarify the roles of particular cytokines in the immune response of normal individuals but also identify mechanisms underlying the humoral defects characteristic of these conditions. In recent years, genetic mutations underlying many primary immunodeficiencies have been identified.1,27,28,33,37 The phenotypic abnormalities in these “Experiments of Nature” demonstrate the nonredundant role for these genes. By examining B cells from patients with loss-of-function mutations in STAT1, STAT3, IL2RG, IL21R, and IL2RA, we have further elucidated the roles of γc cytokines, particularly IL-21 and IL-2, in human B-cell differentiation. The complete loss of IL-21–induced CD25 expression on γc-deficient and IL-21R–deficient B cells and substantial impairment on STAT3-deficient B cells confirms the nonredundant nature of the IL-21R/γc/STAT3 signaling pathway during B-cell differentiation. These patients with rare immunodeficiencies therefore provide a unique opportunity to study molecular requirements underlying human cytokine signaling pathways in health and disease and reveal potential targets for modulating B-cell responses in both immunodeficiency and autoimmunity.
Acknowledgments
The authors thank Rob Salomon, David Snowden, Nikki Alling, and Chris Brownlee for cell sorting, Drs. Warren Kaplan and Mark Cowley for bioinformatics advice, the Ramaciotti Centre (University of New South Wales) for GeneChip hybridization and scanning, Dr. Rene de Waal Malefyt (Merck, formerly DNAX Research Institute, Palo Alto, CA) for providing IL-10, the Australian Red Cross for providing donor spleens, Dr. Ron Bova for providing human tonsils, Lynn Corcoran for assistance with ChIP assays, Drs. Talil Chatilla and Luigi Notarangelo for facilitating access to CD25-deficient patients, and all the patients and their families for participating in this project.
This work was supported by research grants and fellowships awarded by the National Health and Medical Research Council of Australia (L.J.B., C.S.M., E.K.D., M.C.C., S.G.T.) and a Postdoctoral Fellowship from Research Foundation-Flanders (FWO), Belgium (L.M).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.J.B. designed the research, performed experiments, analyzed and interpreted results, and wrote the manuscript; D.T.A., C.S.M., and L.M. performed experiments; C.S.M. and E.K.D. assisted with experimental design; S.B.D., J.B., M.W., S.A., P.D.A., R.B., L.B., H.D., C.M.R., D.A.F., J.B.Z., J.M.S., M.K., C.P., A.D., M.C.C., J.L.C., and G.U. provided patient samples or B-cell lines and clinical details; G.U. provided input into data interpretation; and S.G.T. designed the research, analyzed and interpreted results, and wrote the manuscript; all authors commented on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stuart Tangye, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst 2010, NSW, Australia; e-mail: s.tangye@garvan.org.au.
References
- 1.Tangye SG, Tarlinton DM. Memory B cells: effectors of long-lived immune responses. Eur J Immunol. 2009;39(8):2065–2075. doi: 10.1002/eji.200939531. [DOI] [PubMed] [Google Scholar]
- 2.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11(8):681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 3.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 4.Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 5.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209(7):1241–1253. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banchereau J, Rousset F. Human B lymphocytes: phenotype, proliferation, and differentiation. Adv Immunol. 1992;52:125–262. doi: 10.1016/s0065-2776(08)60876-7. [DOI] [PubMed] [Google Scholar]
- 7.Defrance T, Vanbervliet B, Brière F, Durand I, Rousset F, Banchereau J. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med. 1992;175(3):671–682. doi: 10.1084/jem.175.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubois B, Massacrier C, Vanbervliet B, et al. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J Immunol. 1998;161(5):2223–2231. [PubMed] [Google Scholar]
- 9.Punnonen J, Aversa G, Cocks BG, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA. 1993;90(8):3730–3734. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armitage RJ, Macduff BM, Eisenman J, Paxton R, Grabstein KH. IL-15 has stimulatory activity for the induction of B cell proliferation and differentiation. J Immunol. 1995;154(2):483–490. [PubMed] [Google Scholar]
- 11.Pène J, Gauchat JF, Lécart S, et al. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J Immunol. 2004;172(9):5154–5157. doi: 10.4049/jimmunol.172.9.5154. [DOI] [PubMed] [Google Scholar]
- 12.Ettinger R, Sims GP, Fairhurst AM, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175(12):7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 13.Bryant VL, Ma CS, Avery DT, et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol. 2007;179(12):8180–8190. doi: 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]
- 14.Tangye SG, Ferguson A, Avery DT, Ma CS, Hodgkin PD. Isotype switching by human B cells is division-associated and regulated by cytokines. J Immunol. 2002;169(8):4298–4306. doi: 10.4049/jimmunol.169.8.4298. [DOI] [PubMed] [Google Scholar]
- 15.Tangye SG, Avery DT, Hodgkin PD. A division-linked mechanism for the rapid generation of Ig-secreting cells from human memory B cells. J Immunol. 2003;170(1):261–269. doi: 10.4049/jimmunol.170.1.261. [DOI] [PubMed] [Google Scholar]
- 16.Avery DT, Bryant VL, Ma CS, de Waal Malefyt R, Tangye SG. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol. 2008;181(3):1767–1779. doi: 10.4049/jimmunol.181.3.1767. [DOI] [PubMed] [Google Scholar]
- 17.Brière F, Servet-Delprat C, Bridon JM, Saint-Remy JM, Banchereau J. Human interleukin 10 induces naive surface immunoglobulin D+ (sIgD+) B cells to secrete IgG1 and IgG3. J Exp Med. 1994;179(2):757–762. doi: 10.1084/jem.179.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avery DT, Ma CS, Bryant VL, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood. 2008;112(5):1784–1793. doi: 10.1182/blood-2008-02-142745. [DOI] [PubMed] [Google Scholar]
- 19.Tangye SG, Avery DT, Deenick EK, Hodgkin PD. Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J Immunol. 2003;170(2):686–694. doi: 10.4049/jimmunol.170.2.686. [DOI] [PubMed] [Google Scholar]
- 20.Arpin C, Banchereau J, Liu YJ. Memory B cells are biased towards terminal differentiation: a strategy that may prevent repertoire freezing. J Exp Med. 1997;186(6):931–940. doi: 10.1084/jem.186.6.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Recher M, Berglund LJ, Avery DT, et al. IL-21 is the primary common γ chain-binding cytokine required for human B-cell differentiation in vivo. Blood. 2011;118(26):6824–6835. doi: 10.1182/blood-2011-06-362533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1(3):200–208. doi: 10.1038/35105066. [DOI] [PubMed] [Google Scholar]
- 23.Avery DT, Deenick EK, Ma CS, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207(1):155–171. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diehl SA, Schmidlin H, Nagasawa M, et al. STAT3-mediated up-regulation of BLIMP1 is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol. 2008;180(7):4805–4815. doi: 10.4049/jimmunol.180.7.4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozaki K, Spolski R, Ettinger R, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol. 2004;173(9):5361–5371. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 26.Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23(5):341–349. doi: 10.1016/j.smim.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 28.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 29.Dreskin SC, Goldsmith PK, Gallin JI. Immunoglobulins in the hyperimmunoglobulin E and recurrent infection (Job’s) syndrome. Deficiency of anti-Staphylococcus aureus immunoglobulin A. J Clin Invest. 1985;75(1):26–34. [Google Scholar]
- 30.Leung DY, Ambrosino DM, Arbeit RD, Newton JL, Geha RS. Impaired antibody responses in the hyperimmunoglobulin E syndrome. J Allergy Clin Immunol. 1988;81(6):1082–1087. doi: 10.1016/0091-6749(88)90873-1. [DOI] [PubMed] [Google Scholar]
- 31.Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J Allergy Clin Immunol. 1991;87(4):803–811. doi: 10.1016/0091-6749(91)90126-9. [DOI] [PubMed] [Google Scholar]
- 32.Goudy K, Aydin D, Barzaghi F, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146(3):248–261. doi: 10.1016/j.clim.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kotlarz D, Ziętara N, Uzel G, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. 2013;210(3):433–443. doi: 10.1084/jem.20111229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avery DT, Ellyard JI, Mackay F, Corcoran LM, Hodgkin PD, Tangye SG. Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J Immunol. 2005;174(7):4034–4042. doi: 10.4049/jimmunol.174.7.4034. [DOI] [PubMed] [Google Scholar]
- 35.Ma CS, Suryani S, Avery DT, et al. Early commitment of naïve human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. 2009;87(8):590–600. doi: 10.1038/icb.2009.64. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu Y, Meunier L, Hendershot LM. pERp1 is significantly up-regulated during plasma cell differentiation and contributes to the oxidative folding of immunoglobulin. Proc Natl Acad Sci USA. 2009;106(40):17013–17018. doi: 10.1073/pnas.0811591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boisson-Dupuis S, Kong XF, Okada S, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012;24(4):364–378. doi: 10.1016/j.coi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butch AW, Chung GH, Hoffmann JW, Nahm MH. Cytokine expression by germinal center cells. J Immunol. 1993;150(1):39–47. [PubMed] [Google Scholar]
- 39.Matthes T, Werner-Favre C, Tang H, Zhang X, Kindler V, Zubler RH. Cytokine mRNA expression during an in vitro response of human B lymphocytes: kinetics of B cell tumor necrosis factor alpha, interleukin (IL)6, IL-10, and transforming growth factor beta 1 mRNAs. J Exp Med. 1993;178(2):521–528. doi: 10.1084/jem.178.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldmann TA, Goldman CK, Robb RJ, et al. Expression of interleukin 2 receptors on activated human B cells. J Exp Med. 1984;160(5):1450–1466. doi: 10.1084/jem.160.5.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 requirement for human plasma cell generation: coupling differentiation and proliferation by enhancing MAPK-ERK signaling. J Immunol. 2012;189(1):161–173. doi: 10.4049/jimmunol.1200301. [DOI] [PubMed] [Google Scholar]
- 42.Fluckiger AC, Garrone P, Durand I, Galizzi JP, Banchereau J. Interleukin 10 (IL-10) upregulates functional high affinity IL-2 receptors on normal and leukemic B lymphocytes. J Exp Med. 1993;178(5):1473–1481. doi: 10.1084/jem.178.5.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mittler R, Rao P, Olini G, et al. Activated human B cells display a functional IL 2 receptor. J Immunol. 1985;134(4):2393–2399. [PubMed] [Google Scholar]
- 44.Teranishi T, Hirano T, Lin BH, Onoue K. Demonstration of the involvement of interleukin 2 in the differentiation of Staphylococcus aureus Cowan I-stimulated B cells. J Immunol. 1984;133(6):3062–3067. [PubMed] [Google Scholar]
- 45.Depper JM, Leonard WJ, Robb RJ, Waldmann TA, Greene WC. Blockade of the interleukin-2 receptor by anti-Tac antibody: inhibition of human lymphocyte activation. J Immunol. 1983;131(2):690–696. [PubMed] [Google Scholar]
- 46.Litjens NH, Huisman M, Hijdra D, Lambrecht BM, Stittelaar KJ, Betjes MG. IL-2 producing memory CD4+ T lymphocytes are closely associated with the generation of IgG-secreting plasma cells. J Immunol. 2008;181(5):3665–3673. doi: 10.4049/jimmunol.181.5.3665. [DOI] [PubMed] [Google Scholar]
- 47.Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci USA. 2000;97(21):11439–11444. doi: 10.1073/pnas.200360997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 49.Kallies A, Nutt SL. Terminal differentiation of lymphocytes depends on Blimp-1. Curr Opin Immunol. 2007;19(2):156–162. doi: 10.1016/j.coi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. 2007;178(1):242–252. doi: 10.4049/jimmunol.178.1.242. [DOI] [PubMed] [Google Scholar]