

Abstract
Since its identification nearly 30 years ago, Lyme disease has continued to spread, and there have been increasing numbers of cases in the northeastern and north central US. The Lyme disease agent, Borrelia burgdorferi, causes infection by migration through tissues, adhesion to host cells, and evasion of immune clearance. Both innate and adaptive immune responses, especially macrophage- and antibody-mediated killing, are required for optimal control of the infection and spirochetal eradication. Ecological conditions favorable to the disease, and the challenge of prevention, predict that Lyme disease will be a continuing public health concern.
In the late 20th century, Lyme disease, or Lyme borreliosis, was recognized as an important emerging infection (1). It is now the most commonly reported arthropod-borne illness in the US and Europe and is also found in Asia (2). Since surveillance for Lyme disease was begun in the US by the Centers for Disease Control and Prevention, the number of reported cases has increased steadily, and in the year 2000, more than 18,000 cases were reported (3).
Lyme disease was recognized as a separate entity in 1976 because of geographic clustering of children in the Lyme, Connecticut, area who were thought to have juvenile rheumatoid arthritis (4, S1 ). It then became apparent that Lyme arthritis was a late manifestation of an apparently tick-transmitted, multisystem disease, of which some manifestations had been recognized previously in Europe and America (S2–S6 ). In 1981, Burgdorfer and colleagues discovered a previously unidentified spirochetal bacterium, called Borrelia burgdorferi, in a nymphal Ixodes scapularis (also called Ixodes dammini) tick (S7 ). This spirochete was then cultured from patients with early Lyme disease, and patients’ immune responses were linked conclusively with that organism, proving the spirochetal etiology of the infection (S8, S9).
Based on genotyping of isolates from ticks, animals, and humans, the formerly designated B. burgdorferi has now been subdivided into multiple Borrelia species, including three that cause human infection. In the US, the sole cause is B. burgdorferi (S10 ). Although all three species are found in Europe, most of the disease there is due to Borrelia afzelii or Borrelia garinii, and only these two species seem to be responsible for the illness in Asia (S11, S12 ). During the 20th century, conditions evolved in the northeastern US that were especially favorable for enzootic B. burgdorferi infection (5). In this setting, Lyme disease continues to flourish and spread.
Biology of B. burgdorferi
The agents of Lyme borreliosis belong to the eubacterial phylum of spirochetes, which are vigorously motile, corkscrew-shaped bacteria (Figure 1). The spirochetal cell wall consists of a cytoplasmic membrane surrounded by peptidoglycan and flagella and then by a loosely associated outer membrane. The B. burgdorferi (strain B31) genome has been completely sequenced. It has a small linear chromosome that is just under one megabase (6), and nine circular and 12 linear plasmids that constitute 40% of its DNA (7). Some of these plasmids are indispensable and could be thought of as mini-chromosomes. Although it has been difficult to manipulate the B. burgdorferi genome, progress has recently been made using modified selectable markers and shuttle vectors (8, S13–S18 ).
Figure 1.
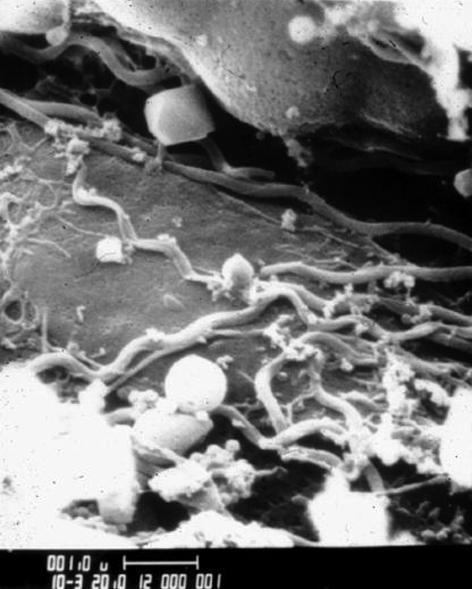
A scanning electron micrograph of B. burgdorferi spirochetes in the midgut of a nymphal I. scapularis tick. The picture is a kind gift of Willy Burgdorfer.
The most remarkable aspect of the B. burgdorferi genome is the large number of sequences encoding predicted or known lipoproteins, including outer-surface proteins (Osp’s) A through F (6). Lipoproteins are found in the outer leaflet of the cytoplasmic membrane, and in both the inner and the outer leaflets of the outer membrane. Some of these proteins are differentially expressed, and one surface-exposed lipoprotein, called VlsE, undergoes extensive antigenic variation (9). In contrast, the genome encodes very few proteins with recognizable biosynthetic activity, and therefore, the organism depends on the host for most of its nutritional requirements. A very unusual feature of B. burgdorferi is that it does not require iron, at least for growth in vitro (10). This may allow the spirochete to circumvent the usual host defense of limiting the availability of iron. Finally, the B. burgdorferi genome encodes no recognizable toxins. Instead, this extracellular pathogen causes infection by migration through tissues, adhesion to host cells, and evasion of immune clearance.
Enzootic cycles of B. burgdorferi infection
The genus Borrelia currently includes three pathogenic species that cause Lyme borreliosis (S10–S12 ) and eight closely related species that rarely if ever cause human infection (S19–S25 ). These spirochetes live in nature in enzootic cycles involving ticks of the Ixodes ricinus complex (also called the Ixodes persulcatus complex) and a wide range of animal hosts (Table 1) (11). These enzootic cycles have evolved somewhat differently in different locations (S26 ). The important vectors of the three pathogenic species of human Lyme borreliosis are the deer tick, I. scapularis, in the northeastern and north central US; Ixodes pacificus in the western US; the sheep tick, I. ricinus, in Europe; and the taiga tick, I. persulcatus, in Asia.
Table 1.
The genospecies of Borrelia burgdorferi and their tick vectors and locations

In the northeastern US from Maine to Maryland and in the north central states of Wisconsin and Minnesota, a highly efficient, horizontal cycle of B. burgdorferi transmission occurs among larval and nymphal I. scapularis ticks and certain rodents, particularly white-footed mice and chipmunks (12, S27 ). This cycle results in high rates of infection among rodents and nymphal ticks and many new cases of human Lyme disease during the late spring and early summer months (Figure 2). White-tailed deer, which are not involved in the life cycle of the spirochete, are the preferred host of adult I. scapularis, and they seem to be critical for the survival of the ticks (S28 ).
Figure 2.

The enzootic cycle of B. burgdorferi infection in the northeastern US and intersection with human Lyme disease. I. scapularis ticks feed once during each of the three stages of their usual 2-year life cycle. Typically, larval ticks take one blood meal in the late summer (A), nymphs feed during the following late spring and early summer (B), and adults feed during the fall (C), after which the female tick lays eggs (D) that hatch the next summer (E). It is critical that the tick feeds on the same host species in both of its immature stages (larval and nymphal), because the life cycle of the spirochete (wavy red line) depends on horizontal transmission: in the early summer, from infected nymphs to certain rodents, particularly mice or chipmunks (B); and in the late summer, from infected rodents to larvae (A), which then molt to become infected nymphs that begin the cycle again in the following year. Therefore, B. burgdorferi spends much of its natural cycle in a dormant state in the midgut of the tick. During the summer months, after transmission to rodents, the spirochete must evade the immune response long enough to be transferred to feeding larval ticks. Although the tick may attach to humans at all three stages, it is primarily the tiny nymphal tick (∼1 mm) that transmits the infection (F). This stage of the tick life cycle has a peak period of questing in the weeks surrounding the summer solstice. Humans are an incidental host and are not involved at all in the life cycle of the spirochete.
The vector ecology of B. burgdorferi is quite different on the West Coast in northern California, where the frequency of Lyme disease is low. There, two intersecting cycles are necessary for disease transmission, one involving the dusky-footed wood rat and Ixodes spinipalpis (also called Ixodes neotomae) ticks, which do not bite humans and which maintain the cycle in nature, and the other involving wood rats and I. pacificus ticks, which are less often infected but do bite humans (S29 ). Similarly, in Colorado, wood rats and I. spinipalpis ticks may be infected with Borrelia bissettii, one of the nonpathogenic species, in a cycle that is not known to cause human infection (13). In the southeastern US, nymphal I. scapularis feed primarily on lizards, which are resistant to B. burgdorferi infection because of complement-mediated killing of the spirochete (14). Therefore, Lyme disease is rare in that part of the country.
In Europe, there is still debate about the preferred animal hosts of I. ricinus. These ticks feed on more than 300 animal species, including large and small mammals, birds, and reptiles (15). In Asia, immature I. persulcatus commonly feed on voles, shrews, and birds, and adult ticks feed on virtually all larger animals, including hares, deer, and cattle (16). Because the Borrelia species differ in their resistance to complement-mediated killing, small rodents are important reservoirs for B. afzelii, B. bissettii, and Borrelia japonica, whereas birds are strongly associated with B. garinii, Borrelia valaisiana, and Borrelia turdi (17).
Emergence of human Lyme borreliosis
The earliest known American cases of Lyme disease occurred in Cape Cod in the 1960s (S30 ). However, B. burgdorferi DNA has been identified by PCR in museum specimens of ticks and mice from Long Island dating from the late 19th and early 20th centuries (S31 ), and the infection has probably been present in North America for millennia (2). During the European colonization of North America, woodland in New England was cleared for farming, and deer were hunted almost to extinction (5). However, during the 20th century, conditions improved in the northeastern US for the ecology of Lyme disease. As farmland reverted to woodland, deer proliferated, white-footed mice were plentiful, and the deer tick thrived. Soil moisture and land cover, as found near rivers and along the coast, were favorable for tick survival (18). Finally, these areas became heavily populated with both humans and deer, as more rural wooded areas became wooded suburbs in which deer were without predators and hunting was prohibited.
During the past 40 years, the infection has continued to spread in the northeastern US (19); it has caused focal outbreaks in some coastal areas (S30, S32, S33 ), and it now affects suburban locations near Boston, New York, Philadelphia, and Baltimore, the most heavily populated parts of the country (S34 ). In the year 2000, the overall incidence of reported cases in Connecticut, the state with the highest reported frequency of Lyme disease, was 111 per 100,000 residents (3). However, most of the cases still clustered in foci, particularly in two counties in the southeastern part of the state where the original epidemiologic investigation took place in the town of Lyme (S1 ). In a large, two-year vaccine trial, such high-risk areas had a yearly incidence of the disease of greater than 1 per 100 participants, and the frequency of seropositivity to B. burgdorferi at study entry was as high as 5 per 100 participants (20).
As in America, the European agents of Lyme borreliosis have probably been present there for many thousands of years. They are now known to be widely established in Europe’s remaining forested areas (15). The highest reported frequencies of the disease are in middle Europe, particularly in Germany, Austria, Slovenia, and Sweden (2). In 1995, the yearly incidence of the disease in Slovenia and Austria was estimated to be 120–130 cases per 100,000 residents (2), similar to the frequency in Connecticut.
Clinical manifestations and disease pathogenesis
To maintain its complex enzootic cycle, B. burgdorferi must adapt to markedly different environments, the tick and the mammalian or avian host. The spirochete survives in a dormant state in the nymphal tick midgut during the fall, winter, and early spring, where it expresses primarily OspA (21). When the tick feeds in the late spring or early summer, the expression of a number of spirochetal proteins is altered (S35 ). For example, OspA is downregulated, and OspC is upregulated (21). OspC expression is required for infection of the mammalian host (22, 23). In addition, the spirochete binds mammalian plasminogen and its activators, present in the blood meal, which facilitates spreading of the organism within the tick (24). Within the salivary gland, OspC expression predominates, but some organisms express only OspE and OspF; OspA and OspB are absent (25).
After transmission of the spirochete, human Lyme disease generally occurs in stages, with remissions and exacerbations and different clinical manifestations at each stage (4). Early infection consists of stage 1, localized infection of the skin, followed within days or weeks by stage 2, disseminated infection, and months to years later by stage 3, persistent infection. However, the infection is variable; some patients have only localized infection of the skin, while others have only later manifestations of the illness, such as arthritis. Moreover, there are regional variations, primarily between the illness found in America and that found in Europe and Asia (1).
Localized infection
After an incubation period of 3–32 days, a slowly expanding skin lesion, called erythema migrans (EM), forms at the site of the tick bite in 70–80% of cases (26, 27). In the US, the skin lesion is frequently accompanied by flu-like symptoms, such as malaise and fatigue, headache, arthralgias, myalgias, and fever, and by signs that suggest dissemination of the spirochete (28). In about 18% of cases (27), these symptoms are the presenting manifestation of the illness (29). In contrast, EM in Europe is more often an indolent, localized infection, and spirochetal dissemination is less common (30).
In addition to the Lyme disease agent, I. scapularis ticks in the US and I. ricinus ticks in Europe may transmit Babesia microti (a red-blood-cell parasite) or Anaplasma phagocytophilum (formerly referred to as “the agent of human granulocytic ehrlichiosis”) (31–33). In a recent prospective study in the US, 4% of patients with culture-proven EM had coinfection with one of these other two tick-borne agents (34). Although these two infections are usually asymptomatic, coinfection may lead to more severe, acute flu-like illness (35).
In most patients, immune cells first encounter B. burgdorferi at the site of the tick bite. Depending on the Borrelia species and the host, complement-mediated lysis of the organism may be the first line of host defense (Figure 3) (36). On histologic examination, the resulting EM skin lesions consist of mild to marked perivascular infiltrates of lymphocytes, DCs, macrophages, and small numbers of plasma cells (37). As a part of the innate immune response, macrophages engulf and kill spirochetes (38–41). Inflammatory cells within the lesion produce primarily proinflammatory cytokines, including TNF-α and IFN-γ (37, 42). B. burgdorferi–stimulated PBMCs from patients with EM produce Th1 proinflammatory cytokines, especially IFN-γ (43). Within days after disease onset, most patients have an IgM antibody response to OspC or the 41-kDa flagellar protein of the spirochete (44). Thus, both innate and adaptive cellular elements are mobilized to fight the infection.
Figure 3.

Host mechanisms of spirochetal killing. Complement-mediated lysis of the organism may be the first line of host defense. Spirochetal lipoproteins and other spirochetal signals activate macrophages, leading to the production of strong proinflammatory cytokines, especially TNF-α and IL-1β. Macrophages engulf spirochetes and degrade them in intracellular compartments. Spirochetal lipoproteins, which are B cell mitogens, also stimulate adaptive T cell–independent B cell responses. Humoral immune responses to nonlipidated spirochetal proteins are more likely to be T cell dependent. The primary role of B. burgdorferi–specific CD4+ Th1 cells is to prime T cell–dependent B cell responses, and antigen-specific CD8+ T cells may be a significant source of IFN-γ. Antibody-mediated spirochetal killing occurs by complement fixation and opsonization.
Disseminated infection
Within days to weeks after disease onset, B. burgdorferi often disseminates widely. During this period, the spirochete has been recovered from blood and cerebrospinal fluid (S7, S36, S37 ), and it has been seen in small numbers in specimens of myocardium, retina, muscle, bone, spleen, liver, meninges, and brain (45). Possible clinical manifestations include secondary annular skin lesions, acute lymphocytic meningitis, cranial neuropathy, radiculoneuritis, atrioventricular nodal block, migratory musculoskeletal pain in joints, bursae, tendon, muscle, or bone, and, rarely, eye manifestations (reviewed in ref. 4). Less often, spirochetal dissemination is asymptomatic.
To disseminate, B. burgdorferi binds certain host proteins and adheres to integrins, proteoglycans, or glycoproteins on host cells or tissue matrices. As in the tick, spreading of the spirochete through tissue matrices may be facilitated by the binding of plasminogen and its activators to the surface of the organism (24). A 47-kDa spirochetal protein (BBK32) binds fibronectin, an ECM protein (46). The sequences of OspC vary considerably among strains, and only a few sequences are associated with disseminated infection (47), probably because they bind as-yet unidentified host structures. A 66-kDa outer-surface protein of the spirochete binds the fibrinogen receptor (αIIbβ3) and the vitronectin receptor (αvβ3) (48), which may allow the organism to establish an initial foothold and disseminate in the vasculature. A 26-kDa Borrelia glycosaminoglycan-binding (GAG-binding) protein, Bgp, binds to the GAG side chains of heparan sulfate on endothelial cells, and to both heparan sulfate and dermatan sulfate on neuronal cells (49, 50). Finally, spirochetal decorin-binding proteins A and B (DbpA and DbpB) bind decorin, a proteoglycan that associates with collagen (51). This may explain the alignment of spirochetes with collagen fibrils in the ECM of the heart, nervous system, or joints (45).
Despite an active immune response, B. burgdorferi may survive during dissemination by changing or minimizing antigenic expression of surface proteins and by inhibiting certain critical host immune responses. Two linear plasmids (lp’s) seem to be essential, including lp25, which encodes a nicotinamidase (52), and lp28-1, which encodes the VlsE lipoprotein (53), the protein that undergoes antigenic variation. In addition, the spirochete has a number of families of highly homologous, differentially expressed lipoproteins, including the OspE/F paralogs, which further contribute to antigenic diversity (54). B. burgdorferi may downregulate lipoproteins because of host immune pressure (54, 55). For example, in a mouse model, the development of antibody to OspC, a prominent early response, induces downregulation of OspC; and therefore, this antibody response does not completely clear the infection (56). Finally, B. afzelii and, to a lesser degree, B. burgdorferi have complement regulator–acquiring surface proteins that bind complement factor H and factor H–like protein 1 (57). These complement factors inactivate C3b, which protects the organism from complement-mediated killing (57, S38–S41 ). In contrast, B. garinii is efficiently killed by complement (S42 ).
As shown definitively in mouse models, both innate and adaptive immune responses are required for optimal control of disseminated infection (Figure 3). B. burgdorferi lipoproteins, which are B cell mitogens (S39 ), stimulate adaptive T cell–independent B cell responses (58, S43, S44 ). For example, antibody responses to OspC kill spirochetes (59). In addition, humoral immune responses to nonlipidated spirochetal proteins, which are more likely to be T cell–dependent, aid in spirochetal killing (60, 61). The primary role of B. burgdorferi–specific Th1 cells is to prime these T cell–dependent B cell responses (62). The combination of these responses leads to the production of antibodies against many components of the organism (63, 64), which promote spirochetal killing by complement fixation and opsonization (S45 ). Within several weeks to months, these antibody responses, in conjunction with innate immune mechanisms, control widely disseminated infection even without antibiotic treatment, and generalized symptoms resolve.
Persistent infection
After weeks of disseminated infection, the Lyme disease agents may still survive in localized niches for several years. By this time, systemic symptoms are minimal or absent altogether. Although each of the three pathogenic species may spread to the joints, nervous system, or other skin sites, they seem to vary in the frequency of dissemination to these sites and in their ability to persist there. B. burgdorferi, the sole cause of the infection in the US, seems to be the most arthritogenic. Months after the onset of illness, about 60% of untreated patients with this infection experience intermittent attacks of arthritis, primarily of the large joints, especially the knee (65).
As shown in a mouse model, neutrophil extravasation into the infected joint is a key initial step in the development of joint inflammation (66). In the human infection, CD4+ Th cells are of the proinflammatory Th1 subset (S46, S47 ), and B. burgdorferi–specific CD8+ T cells are found as well (S48 ). Within the joint, B. burgdorferi–specific γδ T cells may aid in the regulation of these inflammatory responses (S49, S50 ). Compared with other inbred strains of mice, C57BL/6 mice are protected from severe arthritis by IL-6 and IL-10, despite large numbers of spirochetes in the joint (67, 68). It is unknown, however, whether certain human patients control joint inflammation in this way. Patients with Lyme arthritis have very high antibody responses to many spirochetal proteins, suggestive of hyperimmunization due to recurrent waves of spirochetal growth (63, 64). Even without antibiotic treatment, the number of patients who continue to have attacks of arthritis decreases by about 10–20% each year, and few patients have had attacks for longer than 5 years (65). Thus, these immune mechanisms seem to succeed eventually in eradicating B. burgdorferi from the joint.
In Europe and Asia, B. afzelii may persist in the skin for decades, resulting in acrodermatitis chronica atrophicans, a skin condition that occurs primarily on sun-exposed surfaces of distal extremities in elderly women (S51 ). Compared with EM lesions, infiltrates of T cells and macrophages in acrodermatitis lesions had a restricted cytokine profile, lacking IFN-γ production (37). Consistent with this finding, ultraviolet B irradiation of B. burgdorferi–infected C3H mice decreased the Th1 response (69). Thus, spirochetal persistence in acrodermatitis skin lesions may involve both spirochetal factors and an ineffective local immune response.
B. garinii, which is also found only in Europe and Asia, appears to be the most neurotropic of the three Borrelia species. It may cause an exceptionally wide range of neurologic abnormalities (70), including borrelial encephalomyelitis (S52 ), a multiple sclerosis–like illness. In the US, a rare, late neurologic syndrome has been described, called Lyme encephalopathy or polyneuropathy, which is manifested primarily by subtle cognitive disturbances, spinal radicular pain, or distal paresthesias (71, S53 ). With each of these three late neurologic complications, the possible duration of spirochetal persistence and the pathogenetic mechanisms are unknown.
Putative postinfectious syndromes
Treatment-resistant Lyme arthritis.
About 10% of patients with Lyme arthritis have persistent joint inflammation for months or even several years after standard courses of antibiotic treatment (72), a complication rarely noted in Europe (S54 ). Although B. burgdorferi DNA can often be detected by PCR in the joint fluid of these patients prior to antibiotic treatment (73, S55 ), PCR results are usually negative after antibiotic treatment (73), suggesting that joint swelling may persist after complete or nearly complete eradication of the spirochete from the joint with antibiotic therapy.
To explain this course, it has been hypothesized that these patients may have persistent infection or infection-induced autoimmunity (74). In support of the persistent-infection hypothesis, ex vivo–infected synovial cells contained B. burgdorferi in the cytosol (75), a site that might be protected from antibiotics. However, spirochetes have not been seen in intracellular locations in situ in human or mouse synovia (45). Moreover, PCR results for B. burgdorferi DNA were negative in synovial tissue in all 26 patients with treatment-resistant arthritis who underwent arthroscopic synovectomy a median of 7 months after the completion of antibiotic therapy (76). This methodology may be insufficient to identify rare spirochetes and would not detect retained spirochetal antigens.
In support of the autoimmunity hypothesis, treatment-resistant Lyme arthritis is associated with HLA-DRB1*0401, 0101, and other related alleles (77), and with cellular and humoral immune responses to OspA of B. burgdorferi (78, S56–S58 ). In an epitope-mapping study, 15 of 16 treatment-resistant patients had T cell reactivity with the OspA165–173 epitope, the immunodominant epitope presented by the DRB1*0401 or 0101 molecule, compared with only one of five treatment-responsive patients (78). One homolog of this epitope, human lymphocyte function–associated antigen 1αL332–340 (LFA-1αL332–340), acted as a weak, partial agonist for OspA165–173-reactive T cells in DRB1*0401-positive patients (79, 80), but the LFA-1 peptide did not bind the 0101 molecule (77), which suggests that the LFA-1 peptide is unlikely to be a relevant autoantigen. Although the pathogenesis of this syndrome is incompletely delineated, future technologies may allow the identification of spirochetal components or a relevant autoantigen, or both, in the synovia of these patients.
Post–Lyme disease syndrome.
A small percentage of patients with well-documented Lyme disease may develop disabling musculoskeletal pain, neurocognitive symptoms, or fatigue along with or soon after symptoms of the infection (S59–S62 ). This post–Lyme disease syndrome, or chronic Lyme disease (the terms are used interchangeably), which is similar to chronic fatigue syndrome or fibromyalgia, persists for months or years after standard antibiotic treatment of the infection. In a study of such patients who then received intravenous ceftriaxone for 30 days followed by oral doxycycline for 60 days, or intravenous or oral placebo preparations for the same duration, no significant differences were found between the groups in the percentage of patients who said that their symptoms had improved, gotten worse, or stayed the same (81). Therefore, it is hypothesized that B. burgdorferi may trigger immunologic or neurohormonal processes in the brain that cause persistent pain, neurocognitive, or fatigue symptoms, despite spirochetal killing with antibiotic therapy (82). Among B. burgdorferi–infected patients, a prior history of depression or anxiety seems to be a risk factor for the development of chronic Lyme disease (83).
A counterculture has emerged regarding chronic Lyme disease (84). In contrast with the findings of evidence-based medicine, some people believe that the tests for Lyme disease are often inaccurately negative, and that antibiotic therapy is necessary for months or years to suppress the symptoms of this often incurable illness. A number of investigators at academic medical centers have reported series of patients referred for chronic Lyme disease in which the majority of patients had pain or fatigue syndromes with little or no evidence of past or present B. burgdorferi infection (85–87). Prolonged antibiotic therapy may be harmful. In studies of patients with unsubstantiated Lyme disease, minor side effects were common (86), prolonged ceftriaxone therapy sometimes resulted in biliary complications (88), and in one reported case, the prolonged administration of cefotaxime resulted in death (89). Furthermore, prolonged use of antibiotics was recently associated with an increased risk of breast cancer (90). Although antibiotic use may not be causally related to cancer, this observation reinforces the advisability of prudent use of antibiotics.
Diagnosis and treatment
Algorithms for the diagnosis and treatment of early or late Lyme disease are presented in Figures 4 and 5. Except in those with active EM, the diagnosis is usually based on the recognition of a characteristic clinical picture (S63 ) and a positive antibody response to B. burgdorferi by whole-cell sonicate ELISA and Western blot, interpreted according to the criteria of the Centers for Disease Control and Prevention (S64 ). Evidence-based treatment recommendations are incorporated from those presented by the Infectious Disease Society of America (91).
Figure 4.

An algorithm for the diagnosis and treatment of the early events surrounding Lyme disease in the summer months. Serologic testing for Lyme disease has limited utility during the first 1 or 2 weeks of infection, and early treatment, without serologic testing, is recommended. If serologic testing is done, acute and convalescent samples should be obtained. GI, gastrointestinal.
Figure 5.

An algorithm for the diagnosis and treatment of late organ-system involvement in Lyme disease. By the time that organ-system involvement is present, which is at least several weeks after the onset of infection, almost all patients have a positive IgG response to B. burgdorferi. Depending on the manifestation, treatment with either oral or intravenous antibiotic therapy is recommended.
Every summer, the lay public and physicians in endemic areas deal with the early events surrounding Lyme disease, including tick bites, early infection, and coinfection (Figure 4). Since 24–72 hours of tick attachment is necessary before transmission of the spirochete occurs, removal of the tick within 24 hours of attachment is usually sufficient to prevent Lyme disease (54, S65, S66 ). If an engorged nymphal I. scapularis tick is found, a single, 200-mg dose of doxycycline usually prevents the infection (92). Serologic tests are insensitive during the first 1 or 2 weeks of infection and depend largely on detection of a positive IgM response, which may still represent a false-positive response (63, S67 ). Because of these limitations, treatment is recommended for 10 to 20 days, without serologic testing, for presumed EM, most commonly with doxycycline in adults or amoxicillin in children (93, S68 ). If serologic testing is done, both acute and convalescent samples should be obtained, since most patients have a positive IgM or IgG response by convalescence at the conclusion of antibiotic treatment, and the demonstration of seroconversion provides better serologic support for the diagnosis. Reinfection may occur in patients who are treated with antibiotics early in the illness (94).
Flu-like illness during summer is a more difficult issue, since most cases are not caused by B. burgdorferi infection. However, if a patient from a highly endemic area has a febrile illness with headache and joint or muscle pain, without respiratory or gastrointestinal symptoms, in the weeks surrounding the summer solstice, antibiotic treatment may be indicated (Figure 4). For such patients, a second-generation serologic test, an IgG ELISA that uses a peptide in the sixth invariant region of the VlsE lipoprotein of B. burgdorferi, may be valuable, since this test typically becomes positive before five IgG bands are present on Western blot (29, 95). Although both babesiosis and anaplasmosis are usually asymptomatic, coinfection should be considered in a patient with more severe flu-like symptoms, including high fever, particularly if the patient is very young or old or asplenic (Figure 4). Fortunately, Lyme disease and anaplasmosis can both be treated with doxycycline. For severe cases of babesiosis, intravenous clindamycin and oral quinine, or oral atovaquone and azithromycin, may be effective (S69 ).
By the time that organ-system involvement is present in Lyme disease, which is at least several weeks after the onset of infection, almost all patients have a positive IgG response to B. burgdorferi (63) (Figure 5). Objective neurologic abnormalities require treatment with intravenous antibiotic therapy, usually intravenous ceftriaxone (S70 ), with the possible exception of facial palsy alone, without other neurologic manifestations. Lyme arthritis may be treated with either oral or intravenous therapy (72, S70 ), but oral therapy is easier to administer, is associated with fewer side effects, and is considerably less expensive (S71 ). Reinfection has not been reported in patients with the expanded immune response associated with Lyme arthritis.
After antibiotic treatment, antibody titers fall slowly, but IgG and even IgM responses may persist for years (96), as may the IgG response to the VlsE peptide (97). Moreover, asymptomatic IgG seroconversion to B. burgdorferi occurs in about 7% of patients in the US (98). If patients with asymptomatic seroconversion or past infection have symptoms caused by another illness, the danger is to attribute them incorrectly to Lyme disease, and therefore, the clinical picture must always be considered with the serologic result.
If patients with Lyme arthritis have persistent joint inflammation after 2 months of oral antibiotics or 1 month of intravenous antibiotics and the results of PCR testing are negative, we treat them with nonsteroidal anti-inflammatory agents, disease-modifying antirheumatic drugs, or arthroscopic synovectomy. In those with post–Lyme disease syndrome, we follow the guidelines for treating chronic fatigue syndrome or fibromyalgia (82).
Prevention
Ecological conditions favorable to Lyme disease, the steady increase in the number of cases, and the challenge of prevention predict that the infection will be a continuing public health concern. Personal protection measures, including protective clothing, repellents or acaricides, tick checks, and landscape modifications in or near residential areas, may be helpful (99). However, these measures are difficult to perform regularly throughout the summer. Attempts to control the infection on a larger scale by the eradication of deer or widespread use of acaricides, which may be effective, have had limited public acceptance (99). New methods of tick control, including host-targeted acaricides against rodents and deer, are being developed and may provide help in the future.
In the 1990s, recombinant OspA vaccines were developed and shown to be safe and effective for the prevention of Lyme disease in the US (20, S72, S73 ). Although one of the vaccines was licensed commercially, its acceptance by the public and by physicians was also limited, and it was withdrawn by the manufacturer in 2002 (100). Some of the reasons why its acceptance was limited included the low risk of Lyme disease in most parts of the country, the need for booster injections every year or every other year, and the relatively high cost of this preventive approach compared with antibiotic treatment of early infection (S74, S75 ). In addition, there was a theoretical, though never proven, concern that in rare cases, vaccination might trigger autoimmune arthritis.
For now, control of Lyme disease depends primarily on public and physician education about personal protection measures, signs and symptoms of the disease, and appropriate antibiotic therapy (99). However, if the risk of the infection continues to increase or if public perceptions change, vaccine development may again become a priority. Experience gained in the last ten years has proven the feasibility of vaccination for the prevention of this complex, tick-transmitted infection.
Supplementary Material
Acknowledgments
This work was supported in part by a grant from the NIH (AR20358), a Centers for Disease Control and Prevention Cooperative Agreement (CCU110291), the English, Bonter, Mitchell Foundation, the Lyme/Arthritis Research Fund, and the Eshe Fund.
Due to space constraints, a number of important references could not be included in this article. Interested readers can find a supplementary reading list at http://www.jci.org/cgi/content/full/113/8/1093/DC1.
Footnotes
Nonstandard abbreviations used: outer-surface protein (Osp); erythema migrans (EM); linear plasmid (lp); lymphocyte function–associated antigen 1 (LFA-1).
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Steere AC. Lyme disease. N. Engl. J. Med. 2001;345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- 2.Dennis, D.T., and Hayes, E.B. 2002. Epidemiology of Lyme Borreliosis. In Lyme borreliosis: biology, epidemiology and control. O. Kahl, J.S. Gray, R.S. Lane, and G. Stanek, editors. CABI Publishing. Oxford, United Kingdom. 251–280.
- 3.Lyme disease: United States, 2000. Morb. Mortal. Wkly. Rep. 2002;51:29–31. [PubMed] [Google Scholar]
- 4.Steere AC. Lyme disease. N. Engl. J. Med. 1989;321:586–596. doi: 10.1056/NEJM198908313210906. [DOI] [PubMed] [Google Scholar]
- 5.Spielman A. The emergence of Lyme disease and human babesiosis in a changing environment. Ann. N. Y. Acad. Sci. 1994;740:146–156. doi: 10.1111/j.1749-6632.1994.tb19865.x. [DOI] [PubMed] [Google Scholar]
- 6.Fraser CM, et al. Genomic sequence of a Lyme disease spirochete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 7.Casjens S, et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- 8.Cabello FC, Sartakova ML, Dobrikova EY. Genetic manipulation of spirochetes. Light at the end of the tunnel. Trends Microbiol. 2001;9:245–248. doi: 10.1016/s0966-842x(01)02046-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J-R, Norris SJ. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversation. Infect. Immun. 1998;66:3698–3704. doi: 10.1128/iai.66.8.3698-3704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Posey JE, Gherardini FC. Lack of a role for iron in the Lyme disease pathogen. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- 11.Xu G, Fang QQ, Keirans JE, Durden LA. Molecular phylogenetic analyses indicate that the Ixodes ricinus complex is a paraphyletic group. J. Parasitol. 2003;89:452–457. doi: 10.1645/0022-3395(2003)089[0452:MPAITT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 12.LoGiudice K, Ostfeld RS, Schmidt KA, Keesing F. The ecology of infectious disease: effects of host diversity and community composition on Lyme disease risk. Proc. Natl. Acad. Sci. U. S. A. 2003;100:567–571. doi: 10.1073/pnas.0233733100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider BS, Zeidner NS, Burkot TR, Maupin GO, Piesman J. Borrelia isolates in northern Colorado identifies Borrelia bissettii. J. Clin. Microbiol. 2000;38:3103–3105. doi: 10.1128/jcm.38.8.3103-3105.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo MM, Lane RS, Giclas PC. A comparative study of mammalian and reptilian alternative pathway of complement-mediated killing of the Lyme disease spirochete Borrelia burgdorferi. J. Parasitol. 2000;86:1223–1228. doi: 10.1645/0022-3395(2000)086[1223:ACSOMA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 15.Gern, L., and Pierre-Francois, H. 2002. Ecology of Borrelia burgdorferi sensu lato in Europe. In Lyme borreliosis: biology, epidemiology and control. O. Kahl, J.S. Gray, R.S. Lane, and G. Stanek, editor. CABI Publishing. Oxford, United Kingdom. 149–174.
- 16.Korenberg, E.I., Gorelova, N.B., and Kovalevskii, Y.V. 2002. Ecology of Borrelia burgdorferi sensu lato in Russia. In Lyme borreliosis: biology, epidemiology and control. O. Kahl, J.S. Gray, R.S. Lane, and G. Stanek, editors. CABI Publishing. Oxford, United Kingdom. 175–200.
- 17.Kurtenbach K, et al. Host association of Borrelia burgdorferi sensu lato: the key role of host complement. Trends Microbiol. 2002;10:74–79. doi: 10.1016/s0966-842x(01)02298-3. [DOI] [PubMed] [Google Scholar]
- 18.Guerra M, et al. Predicting the risk of Lyme disease: habitat suitability for Ixodes scapularis in the north central United States. Emerg. Infect. Dis. 2002;8:289–297. doi: 10.3201/eid0803.010166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glavanakov S, et al. Lyme disease in New York State: spatial pattern at a regional scale. Am. J. Trop. Med. Hyg. 2001;65:538–545. doi: 10.4269/ajtmh.2001.65.538. [DOI] [PubMed] [Google Scholar]
- 20.Steere AC, et al. Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant. N. Engl. J. Med. 1998;339:209–215. doi: 10.1056/NEJM199807233390401. [DOI] [PubMed] [Google Scholar]
- 21.Schwan TG, Piesman J. Temporal changes in outer surface proteins A and C of the Lyme disease-associated spirochete, Borrelia burgdorferi, during the chain of infection in ticks and mice. J. Clin. Microbiol. 2000;38:382–388. doi: 10.1128/jcm.38.1.382-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal U, et al. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J. Clin. Invest. 2004;113:220–230. doi:10.1172/JCI200419894. doi: 10.1172/JCI19894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimm D, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman JL, et al. Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell. 1997;89:1111–1119. doi: 10.1016/s0092-8674(00)80298-6. [DOI] [PubMed] [Google Scholar]
- 25.Hefty PS, et al. OspE-related, OspF-related, and Elp lipoproteins are immunogenic in baboons experimentally infected with Borrelia burgdorferi and in human Lyme disease patients. J. Clin. Microbiol. 2002;40:4256–4265. doi: 10.1128/JCM.40.11.4256-4265.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith RP, et al. Clinical characteristics and treatment outcome of early Lyme disease in patients with microbiologically confirmed erythema migrans. Ann. Intern. Med. 2002;136:421–428. doi: 10.7326/0003-4819-136-6-200203190-00005. [DOI] [PubMed] [Google Scholar]
- 27.Steere AC, Sikand VK. The presenting manifestations of Lyme disease and the outcomes of treatment. N. Engl. J. Med. 2003;348:2472–2474. doi: 10.1056/NEJM200306123482423. [DOI] [PubMed] [Google Scholar]
- 28.Steere AC, et al. The early clinical manifestations of Lyme disease. Ann. Intern. Med. 1983;99:76–82. doi: 10.7326/0003-4819-99-1-76. [DOI] [PubMed] [Google Scholar]
- 29.Steere AC, et al. Systemic symptoms without erythema migrans as the presenting picture of early Lyme disease. Am. J. Med. 2003;114:58–62. doi: 10.1016/s0002-9343(02)01440-7. [DOI] [PubMed] [Google Scholar]
- 30.Strle F, et al. Comparison of culture-confirmed erythema migrans caused by Borrelia burgdorferi sensu stricto in New York state and by Borrelia afzelii in Slovenia. Ann. Intern. Med. 1999;130:32–36. doi: 10.7326/0003-4819-130-1-199901050-00006. [DOI] [PubMed] [Google Scholar]
- 31.Krause PJ, et al. Disease-specific diagnosis of coinfecting tickborne zoonoses: babesiosis, human granulocytic ehrlichiosis, and Lyme disease. Clin. Infect. Dis. 2002;34:1184–1191. doi: 10.1086/339813. [DOI] [PubMed] [Google Scholar]
- 32.Hunfeld KP, et al. Seroprevalence of Babesia infections in humans exposed to ticks in midwestern Germany. J. Clin. Microbiol. 2002;40:2431–2436. doi: 10.1128/JCM.40.7.2431-2436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjoersdorff A, Wittesjo B, Berglun J, Massung RF, Eliasson I. Human granulocytic ehrlichiosis as a common cause of tick-associated fever in Southeast Sweden: report from a prospective clinical study. Scand. J. Infect. Dis. 2002;34:187–191. doi: 10.1080/00365540110080061. [DOI] [PubMed] [Google Scholar]
- 34.Steere AC, et al. Prospective study of coinfection in patients with erythema migrans. Clin. Infect. Dis. 2003;36:1078–1081. doi: 10.1086/368187. [DOI] [PubMed] [Google Scholar]
- 35.Krause PJ, et al. Concurrent Lyme disease and babesiosis: evidence for increased severity and duration of illness. JAMA. 1996;275:1657–1660. [PubMed] [Google Scholar]
- 36.Breitner-Ruddock S, Wurzner R, Schulze J, Brade V. Heterogeneity in the complement-dependent bacteriolysis within the species of Borrelia burgdorferi. Med. Microbiol. Immunol. (Berl.) 1997;185:253–260. doi: 10.1007/s004300050038. [DOI] [PubMed] [Google Scholar]
- 37.Mullegger RR, et al. Differential expression of cytokine mRNA in skin specimens from patients with erythema migrans or acrodermatitis chronica atrophicans. J. Invest. Dermatol. 2000;115:1115–1123. doi: 10.1046/j.1523-1747.2000.00198.x. [DOI] [PubMed] [Google Scholar]
- 38.Wooten RM, et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 39.Talkington J, Nickell SP. Role of Fc gamma receptors in triggering host cell activation and cytokine release by Borrelia burgdorferi. Infect. Immun. 2001;69:413–419. doi: 10.1128/IAI.69.1.413-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modolell M, Schaible UE, Rittig M, Simon MM. Killing of Borrelia burgdorferi by macrophages is dependent on oxygen radicals and nitric oxide and can be enhanced by antibodies to outer surface proteins of the spirochete. Immunol. Lett. 1994;40:139–146. doi: 10.1016/0165-2478(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 41.Montgomery RR, Lusitani D, de Boisfleury Chevance A, Malawista SE. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J. Infect. Dis. 2002;185:1773–1779. doi: 10.1086/340826. [DOI] [PubMed] [Google Scholar]
- 42.Salazar JC, et al. Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. J. Immunol. 2003;171:2660–2670. doi: 10.4049/jimmunol.171.5.2660. [DOI] [PubMed] [Google Scholar]
- 43.Glickstein L, et al. Inflammatory cytokine production predominates in early Lyme disease in patients with erythema migrans. Infect. Immun. 2003;71:6051–6053. doi: 10.1128/IAI.71.10.6051-6053.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaz A, et al. Cellular and humoral immune responses to Borrelia burgdorferi antigens in patients with culture-positive early Lyme disease. Infect. Immun. 2001;69:7437–7444. doi: 10.1128/IAI.69.12.7437-7444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duray PH, Steere AC. Clinical pathologic correlations of Lyme disease by stage. Ann. N. Y. Acad. Sci. 1988;539:65–79. doi: 10.1111/j.1749-6632.1988.tb31839.x. [DOI] [PubMed] [Google Scholar]
- 46.Probert WS, Johnson BJB. Identification of a 47kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol. Microbiol. 1998;30:1003–1015. doi: 10.1046/j.1365-2958.1998.01127.x. [DOI] [PubMed] [Google Scholar]
- 47.Seinost G, et al. Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect. Immun. 1999;67:3518–3524. doi: 10.1128/iai.67.7.3518-3524.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coburn J, Chege W, Magoun L, Bodary SC, Leong JM. Characterization of a candidate Borrelia burgdorferi β3-chain integrin ligand identified using a phage display library. Mol. Microbiol. 1999;34:926–940. doi: 10.1046/j.1365-2958.1999.01654.x. [DOI] [PubMed] [Google Scholar]
- 49.Parveen N, Leong JM. Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 2000;35:1220–1234. doi: 10.1046/j.1365-2958.2000.01792.x. [DOI] [PubMed] [Google Scholar]
- 50.Leong JM, et al. Different classes of proteglycans contribute to the attachment of Borrelia burgdorferi to cultured endothelial and brain cells. Infect. Immun. 1998;66:994–999. doi: 10.1128/iai.66.3.994-999.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo BP, Brown EL, Dorward DW, Rosenberg LC, Hook M. Decorin-binding adhesins from Borrelia burgdorferi. Mol. Microbiol. 1998;30:711–723. doi: 10.1046/j.1365-2958.1998.01103.x. [DOI] [PubMed] [Google Scholar]
- 52.Purser JE, et al. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol. Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- 53.Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hefty PS, Jolliff SE, Caimano MJ, Wikel SK, Akins DR. Changes in temporal and spatial patterns of outer surface lipoprotein expression generate population heterogeneity and antigenic diversity in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 2002;70:3468–3478. doi: 10.1128/IAI.70.7.3468-3478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang FT, Nelson FK, Fikrig E. Molecular adaptation of Borrelia burgdorferi in the murine host. J. Exp. Med. 2002;196:275–280. doi: 10.1084/jem.20020770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang FT, Jacobs MB, Bowers LC, Philipp MT. An immune evasion mechanism for spirochetal persistence in Lyme borreliosis. J. Exp. Med. 2002;195:415–422. doi: 10.1084/jem.20011870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kraiczy P, et al. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 2004;279:2421–2429. doi: 10.1074/jbc.M308343200. [DOI] [PubMed] [Google Scholar]
- 58.McKisic MD, Barthold SW. T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of Lyme disease. Infect. Immun. 2000;68:5190–5197. doi: 10.1128/iai.68.9.5190-5197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rousselle JC, et al. Borreliacidal antibody production against outer surface protein C of Borrelia burgdorferi. J. Infect. Dis. 1998;178:733–741. doi: 10.1086/515382. [DOI] [PubMed] [Google Scholar]
- 60.Fikrig E, et al. Borrelia burgdorferi P35 and P37 proteins, expressed in vivo, elicit protective immunity. Immunity. 1997;6:531–537. doi: 10.1016/s1074-7613(00)80341-6. [DOI] [PubMed] [Google Scholar]
- 61.Hanson MS, et al. Active and passive immunity against Borrelia burgdorferi decorin binding protein A (DbpA) protects against infection. Infect. Immun. 1998;66:2143–2153. doi: 10.1128/iai.66.5.2143-2153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keane-Myers A, Nickell SP. T cell subset-dependent modulation of immunity to Borrelia burgdorferi in mice. J. Immunol. 1995;154:1770–1776. [PubMed] [Google Scholar]
- 63.Dressler F, Whalen JA, Reinhardt BN, Steere AC. Western blotting in the serodiagnosis of Lyme disease. J. Infect. Dis. 1993;167:392–400. doi: 10.1093/infdis/167.2.392. [DOI] [PubMed] [Google Scholar]
- 64.Akin E, McHugh GL, Flavell RA, Fikrig E, Steere AC. The immunoglobin (IgG) antibody response to OspA and OspB correlates with severe and prolonged Lyme arthritis and the IgG response to P35 correlates with mild and brief arthritis. Infect. Immun. 1999;67:173–181. doi: 10.1128/iai.67.1.173-181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann. Intern. Med. 1987;107:725–731. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 66.Brown CR, Blaho VA, Loiacono CM. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J. Immunol. 2003;171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- 67.Anguita J, et al. Borrelia burgdorferi-infected, interleukin-6-deficient mice have decreased Th2 responses and increased Lyme arthritis. J. Infect. Dis. 1998;178:1512–1515. doi: 10.1086/314448. [DOI] [PubMed] [Google Scholar]
- 68.Brown JP, Zachary JF, Teuscher C, Weis JJ, Wooten RM. Dual role of interleukin-10 in murine Lyme disease: regulation of arthritis severity and host defense. Infect. Immun. 1999;67:5142–5150. doi: 10.1128/iai.67.10.5142-5150.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown EL, et al. Modulation of immunity to Borrelia burgdorferi by ultraviolet irradiation: differential effect on Th1 and Th2 immune responses. Eur. J. Immunol. 1995;25:3017–3022. doi: 10.1002/eji.1830251105. [DOI] [PubMed] [Google Scholar]
- 70.Oschmann P, et al. Stages and syndromes of neuroborreliosis. J. Neurol. 1998;245:262–272. doi: 10.1007/s004150050216. [DOI] [PubMed] [Google Scholar]
- 71.Logigian EL, Kaplan RF, Steere AC. Chronic neurologic manifestations of Lyme disease. N. Engl. J. Med. 1990;323:1438–1444. doi: 10.1056/NEJM199011223232102. [DOI] [PubMed] [Google Scholar]
- 72.Steere AC, et al. Treatment of Lyme arthritis. Arthritis Rheum. 1994;37:878–888. doi: 10.1002/art.1780370616. [DOI] [PubMed] [Google Scholar]
- 73.Nocton JJ, et al. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid in Lyme arthritis. N. Engl. J. Med. 1994;330:229–234. doi: 10.1056/NEJM199401273300401. [DOI] [PubMed] [Google Scholar]
- 74.Steere AC, Glickstein L. Elucidation of Lyme arthritis. Nat. Rev. Immunol. 2004;4:143–152. doi: 10.1038/nri1267. [DOI] [PubMed] [Google Scholar]
- 75.Girschick HJ, Huppertz HI, Rüssmann H, Krenn V, Karch H. Intracellular persistence of Borrelia burgdorferi in human synovial cells. Rheumatol. Int. 1996;16:125–132. doi: 10.1007/BF01409985. [DOI] [PubMed] [Google Scholar]
- 76.Carlson D, et al. Lack of Borrelia burgdorferi DNA in synovial samples in patients with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 1999;42:2705–2709. doi: 10.1002/1529-0131(199912)42:12<2705::AID-ANR29>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 77.Steere AC, et al. Binding of outer surface protein A and human lymphocyte function-associated antigen 1 peptides to HLA-DR molecules associated with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 2003;48:534–540. doi: 10.1002/art.10772. [DOI] [PubMed] [Google Scholar]
- 78.Chen J, et al. Association of antibiotic treatment-resistant Lyme arthritis with T cell responses to dominant epitopes of outer-surface protein A (OspA) of Borrelia burgdorferi. Arthritis Rheum. 1999;42:1813–1822. doi: 10.1002/1529-0131(199909)42:9<1813::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 79.Gross DM, et al. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science. 1998;281:703–706. doi: 10.1126/science.281.5377.703. [DOI] [PubMed] [Google Scholar]
- 80.Trollmo C, Meyer AL, Steere AC, Hafler DA, Huber BT. Molecular mimicry in Lyme arthritis demonstrated at the single cell level: LFA-1α(L) is a partial agonist for outer surface protein A-reactive T cells. J. Immunol. 2001;166:5286–5291. doi: 10.4049/jimmunol.166.8.5286. [DOI] [PubMed] [Google Scholar]
- 81.Klempner MS, et al. Two controlled trials of antibiotic treatment in patients with persistent symptoms and a history of Lyme disease. N. Engl. J. Med. 2001;345:85–92. doi: 10.1056/NEJM200107123450202. [DOI] [PubMed] [Google Scholar]
- 82.Steere AC. A 58-year-old man with a diagnosis of chronic Lyme disease. JAMA. 2002;288:1002–1010. doi: 10.1001/jama.288.8.1002. [DOI] [PubMed] [Google Scholar]
- 83.Solomon SP, Hilton E, Weinschel BS, Pollack S, Grolnick E. Psychological factors in the prediction of Lyme disease course. Arthritis Care Res. 1998;11:419–426. doi: 10.1002/art.1790110514. [DOI] [PubMed] [Google Scholar]
- 84.Sigal LH, Hassett AL. Contributions of societal and geographical environments to “chronic Lyme disease”: the psychopathogenesis and aporology of a new “medically unexplained symptoms” syndrome. Environ. Health Perspect. 2002;110(Suppl. 4):607–611. doi: 10.1289/ehp.02110s4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steere AC, Taylor E, McHugh GL, Logigian EL. The overdiagnosis of Lyme disease. JAMA. 1993;269:1812–1816. [PubMed] [Google Scholar]
- 86.Reid MC, Schoen RT, Evans J, Rosenberg JC, Horwitz RI. The consequences of overdiagnosis and overtreatment of Lyme disease: an observational study. Ann. Intern. Med. 1998;128:354–362. doi: 10.7326/0003-4819-128-5-199803010-00003. [DOI] [PubMed] [Google Scholar]
- 87.Qureshi MZ, New D, Zulqarni NJ, Nachman S. Overdiagnosis and overtreatment of Lyme disease in children. Pediatr. Infect. Dis. J. 2002;21:12–14. doi: 10.1097/00006454-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 88.Ettestad PJ, et al. Biliary complications in the treatment of unsubstantiated Lyme disease. J. Infect. Dis. 1995;171:356–361. doi: 10.1093/infdis/171.2.356. [DOI] [PubMed] [Google Scholar]
- 89.Patel R, Grogg KL, Edwards WD, Wright AJ, Schwenk NM. Death from inappropriate therapy for Lyme disease. Clin. Infect. Dis. 2000;31:1107–1109. doi: 10.1086/318138. [DOI] [PubMed] [Google Scholar]
- 90.Velicer CM, et al. Antibiotic use in relation to the risk of breast cancer. JAMA. 2004;291:827–835. doi: 10.1001/jama.291.7.827. [DOI] [PubMed] [Google Scholar]
- 91.Wormser GP, et al. Practice guidelines for the treatment of Lyme disease. The Infectious Diseases Society of America. Clin. Infect. Dis. 2000;31(Suppl. 1):S1–S14. doi: 10.1086/314053. [DOI] [PubMed] [Google Scholar]
- 92.Nadelman RB, et al. Prophylaxis with single-dose doxycycline for the prevention of Lyme disease after an Ixodes scapularis tick bite. N. Engl. J. Med. 2001;345:79–84. doi: 10.1056/NEJM200107123450201. [DOI] [PubMed] [Google Scholar]
- 93.Wormser GP, et al. Duration of antibiotic therapy for early Lyme disease. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2003;138:697–704. doi: 10.7326/0003-4819-138-9-200305060-00005. [DOI] [PubMed] [Google Scholar]
- 94.Bennet L, Berglund J. Reinfection with Lyme borreliosis: a retrospective follow-up study in southern Sweden. Scand. J. Infect. Dis. 2002;34:183–186. doi: 10.1080/00365540110080070. [DOI] [PubMed] [Google Scholar]
- 95.Bacon RM, et al. Serodiagnosis of Lyme disease by kinetic enzyme-linked immunosorbent assay using recombinant VlsE1 or peptide antigens of Borrelia burgdorferi compared with 2-tiered testing using whole-cell lysates. J. Infect. Dis. 2003;187:1187–1199. doi: 10.1086/374395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kalish RA, et al. Persistence of immunoglobulin M or immunoglobulin G antibody responses to Borrelia burgdorferi 10-20 years after active Lyme disease. Clin. Infect. Dis. 2001;33:780–785. doi: 10.1086/322669. [DOI] [PubMed] [Google Scholar]
- 97.Peltomaa M, McHugh G, Steere AC. Persistence of antibody response to the VlsE sixth invariant region (IR6) peptide of Borrelia burgdorferi after successful antibiotic treatment of Lyme disease. J. Infect. Dis. 2003;187:1178–1186. doi: 10.1086/374376. [DOI] [PubMed] [Google Scholar]
- 98.Steere AC, Sikand VJ, Schoen RT, Nowakowski J. Asymptomatic infection with Borrelia burgdorferi. Clin. Infect. Dis. 2003;37:528–532. doi: 10.1086/376914. [DOI] [PubMed] [Google Scholar]
- 99.Hayes EB, Piesman J. How can we prevent Lyme disease? N. Engl. J. Med. 2003;348:2424–2430. doi: 10.1056/NEJMra021397. [DOI] [PubMed] [Google Scholar]
- 100.Hanson MS, Edelman R. Progress and controversy surrounding vaccines against Lyme disease. Expert Rev. Vaccines. 2003;2:683–703. doi: 10.1586/14760584.2.5.683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.