

Abstract
The combined treatment with histone deacetylase inhibitors (HDACi) and retinoids has been suggested as a potential epigenetic strategy for the control of cancer. In the present study, we investigated the effects of treatment with butyrate, a dietary HDACi, combined with vitamin A on MCF-7 human breast cancer cells. Cell proliferation was evaluated by the crystal violet staining method. MCF-7 cells were plated at 5 x 104 cells/mL and treated with butyrate (1 mM) alone or combined with vitamin A (10 µM) for 24 to 120 h. Cell proliferation inhibition was 34, 10 and 46% following treatment with butyrate, vitamin A and their combination, respectively, suggesting that vitamin A potentiated the inhibitory activities of butyrate. Furthermore, exposure to this short-chain fatty acid increased the level of histone H3K9 acetylation by 9.5-fold (Western blot), but not of H4K16, and increased the expression levels of p21WAF1 by 2.7-fold (Western blot) and of RARβ by 2.0-fold (quantitative real-time PCR). Our data show that RARβ may represent a molecular target for butyrate in breast cancer cells. Due to its effectiveness as a dietary HDACi, butyrate should be considered for use in combinatorial strategies with more active retinoids, especially in breast cancers in which RARβ is epigenetically altered.
Keywords: Breast cancer, MCF-7 cells, Butyric acid, Vitamin A, Epigenetics
Introduction
Breast cancer is the second leading cause of death by cancer in women globally and each year approximately 1 million new cases of this disease are diagnosed (1). Thus, novel approaches to the prevention and treatment of breast cancer are imperative (2).
Deregulation of epigenetic mechanisms, such as DNA methylation and post-translational modifications of histones, has been implicated in breast carcinogenesis due to the observed silencing of critical tumor suppressor genes (3). In contrast to genetic alterations, epigenetic modifications are potentially reversible and have been identified as relevant molecular targets for cancer prevention and treatment (4). Bioactive food components exhibiting anticancer potential have been shown to modulate DNA methylation and histone acetylation (5,6) in both in vitro and in vivo systems. Reactivation of epigenetically silenced genes by dietary components could potentially have important implications for cancer control strategies (7).
Among the dietary histone deacetylase inhibitors (HDACi), butyrate (BT) is of primary interest because of its anticancer potential (8,9) and because it has already been tested for the treatment of cancer in phase I clinical trials (10,11). This short-chain fatty acid is produced by the fermentation of non-digestible dietary carbohydrates by intestinal bacteria and is also present in honey and milk fat (12). BT represents a promising candidate for the control of breast cancer progression (13,14). Previous studies have demonstrated that BT treatment inhibits the growth of breast cancer cell lines by acting as an HDACi and by inducing the expression of several genes, including cyclin-dependent kinase inhibitor p21 (14-16).
Vitamin A (VA; retinol) is an essential nutrient that regulates several cellular processes, including growth, death and differentiation, and is known to be frequently deregulated during carcinogenesis. A majority of the functions exhibited by VA are exerted by its conversion to retinoic acid, which modulates gene expression by activating retinoic acid receptors (RARs) and retinoid X receptors. Due to these properties, VA has been considered for the treatment and prevention of breast cancer (17). An important limitation of its therapeutic application, however, involves the deregulation of retinol metabolism in breast cancer cells that lack the ability to produce retinyl esters and thus to store VA (18). At the molecular level, this aberrant metabolism has been associated with epigenetic down-regulation of the tumor suppressor genes RARβ and cellular retinol-binding protein I (CRBP-I) (19-21). CRBP-I is responsible for the intracellular transport, esterification and oxidation of retinol (22).
The combination of an HDACi and retinoids represents a potential epigenetic strategy for cancer control (23), although few studies have addressed its efficacy in the context of breast cancer. In a previous study, retinoic acid treatment potentiated the inhibitory effects of the synthetic HDACi trichostatin on breast cancer cells in vitro and in vivo (24). More specifically, reactivation of RARβ expression induced by the synthetic HDACi increased the susceptibility of these cells to the inhibitory activities of retinoic acid (24). Based on these studies, we investigated the effects of the dietary HDACi BT alone and in combination with VA on estrogen receptor-α-positive MCF-7 human breast cancer cells. To assess the efficacy of these treatments, we evaluated several parameters such as cell proliferation, histone H3K9 and H4K16 acetylation patterns, global DNA methylation patterns, RARβ and CRBP-I gene and p21 protein expression, RARβ gene promoter methylation, and intracellular retinoid levels.
Material and Methods
Cell culture and solution preparation
The MCF-7 human breast cancer cell line (HTB 22, American Type Culture Collection, USA) was grown in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (Gibco) at 37°C in a 5% CO2/air atmosphere. VA (retinyl palmitate; BASF, Germany) was dissolved in ethanol to produce 30 mM solutions. Further dilutions were made in culture medium to a final working solution of 10 µM VA containing 0.035% (v/v) ethanol. The VA solution was prepared immediately before use for each experiment. BT (sodium butyrate; Sigma-Aldrich, USA) was dissolved in culture medium to produce stock solutions of 0.5 M. Further dilutions were made in culture medium to achieve a final concentration of 1 mM BT. To control for the residual concentration of ethanol (0.035%, v/v) in the culture medium in the 10-µM VA working solution, ethanol was added to the culture medium, at this same concentration, of both BT-treated and control cells.
Cell proliferation assays
MCF-7 cells were plated at 5 × 104 cells/mL onto 96-well plates and incubated for 24 h. Cells were then treated with BT (0.5, 1, 2, 3, 4, and 5 mM) or VA (1, 5, 10, 15, and 20 µM) and incubated for 24 to 120 h. Next, the combinatorial effects of BT and VA on MCF-7 cell proliferation were evaluated. Cells were treated with 1 mM BT and 10 µM VA, individually or combined, and incubated with the treatments for 24 to 120 h. Following incubation, cell proliferation was evaluated by the crystal violet staining method. Data are reported as means ± SEM.
Cell cycle analysis by cell cytometry
MCF-7 cells were plated at a density of 1 × 105 cells/mL, treated with 1 mM BT and 10 µM VA, individually or combined, and incubated for 24 to 120 h. Following trypsinization, the harvested cells were resuspended with 1 mL chilled phosphate-buffered saline (PBS; 137 mM NaCl; 10 mM Na2HPO4; 2.68 mM KCl; 1.76 mM KH2PO4) and permeabilized with 3 mL chilled 100% methanol overnight. Afterwards the cells were centrifuged at 160 g for 5 min and washed with 1 mL PBS. The pellet was resuspended in 200 µL PBS containing propidium iodide (10 µg/mL) and RNase (10 mg/mL) and incubated for 1 h. The DNA content of the cells was then analyzed with a FACS CANTO II flow cytometer (BD Biosciences, USA).
Western blot analysis
Acetylation of H3K9 and H4K16. MCF-7 cells were plated at a density of 1 x 105 cells/mL, treated with 1 mM BT and 10 µM VA, individually or combined, and incubated for 96 and 120 h. Following these incubation periods, cells were harvested and subjected to histone acid extraction as previously described by Jeong et al. (25). Total histone extracts were separated on a 15% denaturing polyacrylamide gel (SDS-PAGE). Proteins were transferred to Hybond-ECL membranes (GE Healthcare, Sweden) that were blocked with the ECL Advance Chemiluminescence kit (GE Healthcare) for 1 h at room temperature. Incubation with anti-acetyl-histone H3K9 or H4K16 primary antibody (1:5000; Upstate Biotechnology, USA) was carried out at 4°C overnight, and incubation with the secondary antibody conjugated to horseradish peroxidase (1:10,000; GE Healthcare) was carried out at room temperature for 1 h. Subsequently, membranes were incubated with primary anti-H1 antibody (1:5000; Upstate Biotechnology) for 1 h at 37°C and then incubated with the secondary antibody conjugated to horseradish peroxidase (1:10,000; GE Healthcare) at room temperature for 1 h. Membranes were developed using the ECL Advance Chemiluminescence kit (GE Healthcare), and bands were revealed with the ImageQuant 400 capture imaging system (GE Healthcare). Protein bands detected at 11, 17, and 30 kDa corresponded to the expected molecular weight of histone H4, H3, and H1, respectively. A BIO-RAD densitometer (Imaging Densitometer Model GS-700, USA) with specific software (Molecular Analyst, USA) was used to quantify band intensities. To control for unequal protein loading, expression of the proteins of interest was normalized to the H1 signal (26).
p21WAF1 expression. MCF-7 cells were plated at a density of 1 x 105 cells/mL, treated with 1 mM BT and 10 µM VA, individually or combined, and incubated for 96 and 120 h. Cells were harvested and subjected to protein extraction with the T-PER® Tissue Protein Extraction Reagent (Pierce, USA), according to the manufacturer protocol. Western blotting was performed as described for the H3K9 and H4K16 acetylation analyses. Proteins were incubated with primary anti-p21WAF1 antibody (1:1000; Santa Cruz Biotechnology, USA) at 4°C overnight and then incubated with a secondary antibody conjugated to horseradish peroxidase (1:10,000; GE Healthcare) at room temperature for 1 h. Subsequently, membranes were incubated with a primary anti-β-actin antibody (1:5000; Santa Cruz) for 1 h at 37°C and then incubated with a secondary antibody conjugated to horseradish peroxidase (1:10,000; GE Healthcare) at room temperature for 1 h. The resulting bands of 21 and 40 kDa, corresponding to the expected molecular weight of p21WAF1 and β-actin, respectively, were quantified as previously described. To control for unequal protein loading, expression of the protein of interest was normalized to the β-actin signal (14).
Dot blot analysis
Dot blot analysis was performed as described by Alyaqoub et al. (27). MCF-7 cells were plated at a density of 1 x 105 cells/mL and treated with 1 mM BT and 10 µM VA, individually or combined, for 96 and 120 h. Cells were harvested and subjected to DNA extraction. DNA was isolated from MCF-7 cells by digestion with RNase A and proteinase K followed by organic extraction with phenol and chloroform. Purified DNA (2 µg) was denatured in 200 µL 10 x 0.1 N NaOH/SSC buffer (3 M NaCl; 0.3 M sodium citrate, pH 7.0) at 95°C for 5 min and dotted onto Hybond-ECL membranes (GE Healthcare). Membranes were dried, incubated with 5% milk in a solution of PBS + Tween-20 for 2 h and then incubated with primary anti-5-methylcytosine antibody (1:1000; Megabase Research Products, USA) for 2 h. Following this incubation step, membranes were washed 3 times with the PBS + Tween-20 solution for 10 min and incubated with a secondary antibody conjugated to horseradish peroxidase (1:20,000; GE Healthcare) for 2 h at room temperature. After additional washing with the PBS + Tween-20, membranes were treated with detection reagents of the ECL Chemiluminescence kit (GE Healthcare) and exposed to Kodak autoradiography films. The absorbance of the dots was quantified using a BIO-RAD densitometer (Imaging Densitometer, Model GS-700) with specific software (Molecular Analyst). Previous staining of the nitrocellulose membrane with methylene blue was performed to control for the relative amount of DNA.
Quantitative real-time PCR analysis
MCF-7 cells were plated at a density of 1 x 105 cells/mL, treated with 1 mM BT and 10 µM VA, individually or combined, incubated for 96 and 120 h, and then harvested. RNA extraction was performed with the illustra RNAspin Mini RNA isolation kit (GE Healthcare, Germany), according to the manufacturer protocol. RNA quality was assessed by gel electrophoresis. Approximately 1 µg of total RNA was used for the synthesis of first-strand cDNA using the SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen, USA) according to manufacturer instructions. Quantitative PCR analysis was performed in replicates using TaqMan chemistry and Assays on Demand probes (Applied Biosystems, USA) for RARβ (Hs00233407-m1) and CRBP-I (Hs01011514-m1). GAPDH (Hs99999905-m1) was used as a control. mRNA levels were quantified using the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Amplification reactions were performed with a three-step thermal cycling method consisting of a 2-min step at 50°C, 10-min step at 95°C, and a final step of 50 cycles at 95°C for 15 s and 60°C for 1 min.
Methylation-specific PCR analysis
To analyze the methylation pattern of the RARβ gene promoter region, bisulfite modification of genomic DNA was assessed by methods similar to those described by Goldenberg et al. (28). The primers used for PCR were specific for the methylated and unmethylated DNA sequence. A volume of 2 µL modified DNA was used in the PCR assay with 5 µL 10X RT-PCR buffer (2.5 mM MgCl2 final concentration), 1 µL dNTP mix (10 mM of each dNTP), 1 µL forward and reverse primers (10 mM), 1.25 U Platinum Taq DNA Polymerase and PCR grade water making up a total volume of 50 µL. The primer pairs were previously described by Esteller et al. (19): unmethylated sequence (5′-TTGGGATGTTGAGAATGTGAGTGATTT-3′ sense and 5′-CTTACTCAACCAATCCAACCAAAACAA 3′ antisense) and methylated sequence (5′-TGTCGAG AACGCGAGCGATTC-3′ sense and 5′-CGACCAATCCAA CCGAAACGA-3′ antisense). The PCR conditions were as follows: 95°C for 5 min followed by 40 cycles of 94°C for 30 s, 58°C for 1 min, and 72°C for 30 s, with a final extension of 72°C for 5 min. The PCR products were separated on 2% agarose gels, and DNA was visualized with Blue Green (LGC Biotechnology, Brazil) staining.
HPLC analysis
MCF-7 cells were plated at a density of 1 x 105 cells/mL, treated with 1 mM BT or 10 µM VA, or the combination of both compounds, incubated for 96 and 120 h, and then harvested. HPLC was carried out according to Murray et al. (29). Samples were analyzed by chromatography (Shimadzu model LC9A, Japan) using a C18 reversephase column (CLC-ODS; 4.6 mm x 25 cm x 0.5 mm). The chromatograph consisted of a multisolvent pump system, an auto-injector (model SIL-6B) and a photodiode array UV-VIS detector (model SPD-M6A). The substances were eluted at a flow rate of 1.5 mL/min in a mobile phase consisting of acetonitrile, dichloromethane and methanol at a 20:20:10 ratio, respectively. Retinoids (retinol and retinyl palmitate) were detected at 325 nm and were identified on the basis of comparisons of their retention times and those of authentic standards. These compounds were quantified on the basis of peak areas.
Statistical analysis
Data are reported as means ± SEM, and all analyses were carried out with the Statistica 8.0 software (Statsoft, USA). Depending on the type of data, either one-way analysis of variance followed by the Tukey or Duncan post hoc test or the Kruskal Wallis test was applied. For all analyses, the level of significance was set at P ≤ 0.05.
Results
Cell proliferation
Figures 1A and B depict the BT and VA dose-response effects on MCF-7 cell proliferation, respectively. Relative to controls, 5 mM BT-treated cells demonstrated significantly lower (P ≤ 0.05) cell proliferation from 48 to 120 h. The other BT concentrations (0.5-4 mM) showed lower (P ≤ 0.05) MCF-7 cell proliferation only after 120 h. Relative to control, 20 µM VA-treated cells demonstrated lower (P ≤ 0.05) cell proliferation after 120 h. The other VA concentrations (1-15 µM) did not reduce MCF-7 cell proliferation at any time.
Figure 1. Effect of treatment with butyrate and/or vitamin A on MCF-7 cell proliferation evaluated by the crystal violet staining method (absorbance at 570 nm). A, MCF-7 cells treated with butyrate (BT; 0.5-5 mM); B, MCF-7 cells treated with vitamin A (VA; 1-20 µM); C, MCF-7 cells treated with butyrate (BT; 1 mM) and/or vitamin A (VA; 10 µM). Data are reported as means ± SEM. Three independent experiments were performed with N = 6 for each treatment. aP ≤ 0.5 for 5 mM BT treatment in A, 20 µM VA treatment in B and BT+VA treatment in C compared to respective controls; bP ≤ 0.5 for 0.5-4.0 mM BT treatments in A and BT treatment alone in C compared to respective controls (ANOVA followed by the Tukey test).

For the evaluation of the effects of combined treatment with BT and VA on MCF-7 cells, we selected doses that yielded intermediate cell proliferation inhibition in order to allow potential interactions between the dietary HDACi and the retinoid. Figure 1C depicts the results obtained after treatment with 1 mM BT and/or 10 µM VA. After 24 h of treatment, no differences was observed between control cells and cells subjected to the different treatments. Relative to controls, BT-treated cells showed lower (P ≤ 0.05) cell proliferation only after 72 h of treatment, whereas VA-treated cells did not differ from control at any time point. Compared to control, BT+VA-treated cells showed lower (P ≤ 0.05) cell proliferation after 48, 72, 96, and 120 h of treatment. Following 120 h of BT, VA and BT+VA treatments, cell proliferation was inhibited by 34, 10 and 46%, respectively. These data suggest that VA may have potentiated the inhibitory effects of BT on MCF-7 cell proliferation.
Cell cycle profile
Figure 2 illustrates the cell cycle profile of MCF-7 cells treated with 1 mM BT and/or 10 µM VA for 48 h. BT+VA treatment decreased (P ≤ 0.05) and increased (P ≤ 0.05) the percentage of cells in the G0/G1 and G2/M phases, respectively. No differences were observed between treatments during the other periods (24, 72, 96, and 120 h; data not shown).
Figure 2. Effect of 48-h treatment with butyrate (BT) and/or vitamin A (VA) on MCF-7 cell cycle phases. Cell cycle profiles were analyzed by flow cytometry and the percentages of the cells in the G0/G1, S, and G2/M phases were calculated. The data are representative of three independent experiments. aP ≤ 0.5 for BT+VA treatment compared to control (ANOVA followed by the Tukey test).

Acetylation of H3K9 and H4K16
Figure 3 illustrates the results of H3K9 acetylation in MCF-7 cells treated with 1 mM BT and/or 10 µM VA for 96 and 120 h. The 96-h treatment of MCF-7 cells with BT alone or in combination with VA resulted in a 9.5- and 8.0-fold increase (P ≤ 0.05), respectively, in H3K9 acetylation relative to control. This increase was not observed when cells were treated with VA alone. No differences in H3K9 acetylation were observed between BT-and BT+VA-treated cells. The 120-h treatment with BT and/or VA did not alter H3K9 acetylation compared to control. None of the treatments assessed resulted in altered H4K16 acetylation (data not shown).
Figure 3. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on H3K9 acetylation. Western blot analysis of H3K9 acetylation for MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (A) and 120 h (B). Quantitation of histone H3K9 acetylation in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (C) and 120 h (D). The data are representative of three independent experiments and are reported as means ± SEM. aP ≤ 0.5 compared to control; bP ≤ 0.5 compared to treatment with VA (ANOVA followed by the Tukey test).

p21WAF1 expression
Figure 4 depicts p21WAF1 protein expression in MCF-7 cells treated with 1 mM BT and/or 10 µM VA for 96 and 120 h. Following 96 h of exposure, none of the treatment groups expressed altered p21WAF1 compared to control. The 120-h treatment with BT increased p21WAF1 expression by 3.8-fold (P ≤ 0.05) and 2.7-fold (P = 0.054) relative to VA-treated and control cells, respectively. VA and BT+VA treatments did not result in altered p21WAF1 expression compared to control.
Figure 4. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on p21WAF expression. Western blot analysis of p21WAF expression was carried out in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (A) and 120 h (B). Quantitation of p21WAF expression in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (C) and 120 h (D). The data are representative of four independent experiments and are reported as means ± SEM. aP ≤ 0.5 compared to control; bP ≤ 0.5 compared to treatment with VA (ANOVA followed by the Duncan test).

Global DNA methylation patterns
Figure 5 illustrates the result of global DNA methylation pattern for MCF-7 cells treated with 1 mM BT and/or 10 µM VA for 96 and 120 h. Relative to control, 96- and 120-h treatments with VA and BT+VA did not alter the global DNA methylation patterns.
Figure 5. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on global DNA methylation. Dot-blot analyses for global DNA methylation patterns were determined in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (A) and 120 h (B). Membrane staining with methylene blue to control for unequal loading of total DNA of MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (C) and 120 h (D). Quantitation of DNA methylation patterns in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 (E) and 120 h (F). The data are representative of three independent experiments conducted in duplicate and are reported as means ± SEM. No statistically significant differences were detected according to the Kruskal Wallis test (P > 0.05).

RARβ and CRBP-I expression
Figure 6 provides RARβ expression data for MCF-7 cells treated for 96 and 120 h with 1 mM BT and/or 10 µM VA. Relative to control, BT and/or VA treatments did not alter RARβ gene expression after 96 h. Compared to treatment with VA, BT treatment resulted in a 1.6-fold increase in the expression of this gene (P ≤ 0.05). Following 120 h of exposure, treatments with BT alone or in combination with VA resulted in increased RARβ gene expression by 2.0- (P ≤ 0.05) and 1.7-fold (P ≤ 0.05), respectively, compared to control. The same increase was not observed in cells treated with VA alone. Treatment with BT resulted in increased RARβ gene expression by almost 2.0-fold (P ≤ 0.05) relative to VA-treated cells. No significant differences in RARβ gene expression were observed between the BT and BT+VA treatment groups. Following 96 and 120 h of exposure, CRBP-I gene expression was not altered in any treatment group compared to control (data not shown).
Figure 6. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on RARβ expression. qPCR analyses of RARβ expression were carried out in MCF-7 cells treated with or without BT and VA, individually or combined, for 96 (A) and 120 h (B). The data are representative of four independent experiments and are reported as means ± SEM. aP ≤ 0.5 compared to controls; bP ≤ 0.5 compared to treatment with VA (ANOVA followed by the Tukey test).

Methylation of the RARβ promoter
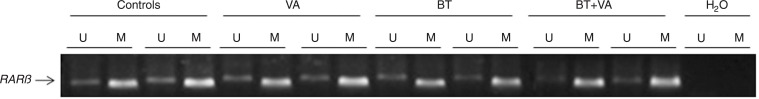
Figure 7 depicts the methylation pattern of the RARβ promoter in MCF-7 cells treated with 1 mM BT and/or 10 µM VA for 96 h. The RARβ promoter region was predominantly methylated in control MCF-7 cells and in cells treated with BT and/or VA.
Figure 7. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on RARβ promoter methylation. MS-PCR analysis of RARβ promoter methylation was carried out in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 h. The presence of the “U” product indicates the unmethylated RARβ gene. The presence of the “M” product indicates the methylated RARβ gene. H2O served as the negative control.
Cellular levels of retinoids
Figure 8 indicates the concentrations of retinol in MCF-7 cells treated with 1 mM BT and/or 10 µM VA. Following 96 h of exposure, no statistical differences in cellular concentrations of this retinoid were detected between treatments. After 120 h of exposure, only cells treated with VA alone demonstrated increased retinol concentrations (3.4-fold) (P ≤ 0.05) relative to control. Retinyl palmitate was not detected in control MCF-7 cells or in cells treated with BT and/or VA for 96 and 120 h.
Figure 8. Effect of treatment with butyrate (BT) and/or vitamin A (VA) on retinoid levels. HPLC quantitation of retinol in MCF-7 cells treated or not with BT and VA, individually or combined, for 96 and 120 h. The data are representative of four independent experiments and are reported as means ± SEM. aP ≤ 0.05 compared to control (ANOVA followed by the Tukey test).

Discussion
Despite the anticancer potential of treatment with an HDACi combined with retinoids (23), few studies have addressed the potential of such interventions, specifically for the control of breast cancer. Efficacy of the combination of retinoic acid with the synthetic HDACis trichostatin (24) and valproic acid (30) was shown in breast cancer cells. Interestingly, our data showed that although VA did not inhibit MCF-7 cell proliferation, it seemed to potentiate inhibition of cell proliferation by BT. This combinatorial effect could involve arrest of MCF-7 cells in the G2/M phase. This reinforces the potential of combining the dietary HDACi BT with retinoids in the context of breast cancer. However, the BT and VA association did not present any combinatorial action on inhibition of proliferation of ER-negative MDA-MB-231 human breast cancer cells (data not shown). This suggests that the type of breast cancer is a relevant issue that should be considered in anticancer combinatorial strategies with butyrate and retinoids. Treatment with BT alone or combined with VA resulted in similar increases in H3K9 acetylation, while H4K16 was not affected by either treatment. BT effects as an HDACi could involve modulation of specific histone residues. Importantly acetylated H3K9 seems to be a preferential site for histone acetyltransferase (31). Among HDACi targets, the cyclindependent kinase inhibitor p21WAF1 has been of primary focus (32). As previously described (16), we observed that treatment with BT resulted in increased p21WAF1 expression in MCF-7 cells, suggesting that its HDACi activities may play a relevant role in its anticancer properties.
Global DNA hypomethylation leads to genomic instability and activation of oncogenes (4). MCF-7 cells express a global hypomethylation compared to normal breast cells (MCF-10-2A) (33,34). In the present study, treatment with BT alone or combined with VA did not alter global DNA methylation patterns. Thus, BT anticancer actions do not seem to involve interference with this epigenetic process.
Aberrant VA signaling has been associated with the progressive loss of RARβ2 expression (21) through epigenetic silencing by promoter hypermethylation (24,35). This epigenetic deregulation is known to contribute to breast cancer development and progression (36,37). In the present study, we observed that, despite the predominantly hypermethylated state of the RARβ promoter in MCF-7 cells, treatment with BT alone or combined with VA could moderately induce the expression of this gene, suggesting that RARβ may represent a molecular target for BT in breast cancer cells. Due to its effectiveness as a dietary HDACi, BT should be considered for use in combinatorial strategies with more active retinoids, especially in breast cancers in which RARβ is epigenetically altered. The possibility of achieving more synergistic anticancer interactions with these interventions remains to be further elucidated.
We confirmed further that CRBP-I is down-regulated in MCF-7 cells (19,20,38). Contrary to what was hypothesized, however, the combination of BT with VA could not reactivate CRBP-I expression and this could provide a molecular explanation of why this intervention could not restore VA metabolism in MCF-7 cells, as evidenced by lack of storage of retinol as retinyl palmitate.
Acknowledgments
Research supported by FAPESP (#2008/58697-9 and #2008/51742-9).
References
- 1.WHO - World Health Organization . World Health Statistics. Geneva: World Health Organization; 2008. [Google Scholar]
- 2.Sun J, Xu X, Liu J, Liu H, Fu L, Gu L. Epigenetic regulation of retinoic acid receptor beta2 gene in the initiation of breast cancer. Med Oncol. 2011;28:1311–1318. doi: 10.1007/s12032-010-9685-y. [DOI] [PubMed] [Google Scholar]
- 3.Veeck J, Esteller M. Breast cancer epigenetics: from DNA methylation to microRNAs. J Mammary Gland Biol Neoplasia. 2010;15:5–17. doi: 10.1007/s10911-010-9165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross SA. Nutritional genomic approaches to cancer prevention research. Exp Oncol. 2007;29:250–256. [PubMed] [Google Scholar]
- 6.Kuroiwa-Trzmielina J, de Conti A, Scolastici C, Pereira D, Horst MA, Purgatto E, et al. Chemoprevention of rat hepatocarcinogenesis with histone deacetylase inhibitors: efficacy of tributyrin, a butyric acid prodrug. Int J Cancer. 2009;124:2520–2527. doi: 10.1002/ijc.24212. [DOI] [PubMed] [Google Scholar]
- 7.Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol. 2010;80:1771–1792. doi: 10.1016/j.bcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delage B, Dashwood RH. Dietary manipulation of histone structure and function. Annu Rev Nutr. 2008;28:347–366. doi: 10.1146/annurev.nutr.28.061807.155354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Conti A, Kuroiwa-Trzmielina J, Horst MA, Bassoli BK, Chagas CE, Purgatto E, et al. Chemopreventive effects of the dietary histone deacetylase inhibitor tributyrin alone or in combination with vitamin A during the promotion phase of rat hepatocarcinogenesis. J Nutr Biochem. 2012;23:860–866. doi: 10.1016/j.jnutbio.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Miller AA, Kurschel E, Osieka R, Schmidt CG. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol. 1987;23:1283–1287. doi: 10.1016/0277-5379(87)90109-x. [DOI] [PubMed] [Google Scholar]
- 11.Conley BA, Egorin MJ, Tait N, Rosen DM, Sausville EA, Dover G, et al. Phase I study of the orally administered butyrate prodrug, tributyrin, in patients with solid tumors. Clin Cancer Res. 1998;4:629–634. [PubMed] [Google Scholar]
- 12.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 13.Belobrajdic DP, McIntosh GH. Dietary butyrate inhibits NMU-induced mammary cancer in rats. Nutr Cancer. 2000;36:217–223. doi: 10.1207/S15327914NC3602_11. [DOI] [PubMed] [Google Scholar]
- 14.De los Santos M, Martinez-Iglesias O, Aranda A. Anti-estrogenic actions of histone deacetylase inhibitors in MCF-7 breast cancer cells. Endocr Relat Cancer. 2007;14:1021–1028. doi: 10.1677/ERC-07-0144. [DOI] [PubMed] [Google Scholar]
- 15.Walker GE, Wilson EM, Powell D, Oh Y. Butyrate, a histone deacetylase inhibitor, activates the human IGF binding protein-3 promoter in breast cancer cells: molecular mechanism involves an Sp1/Sp3 multiprotein complex. Endocrinology. 2001;142:3817–3827. doi: 10.1210/endo.142.9.8380. [DOI] [PubMed] [Google Scholar]
- 16.Chopin V, Toillon RA, Jouy N, Le Bourhis X. P21(WAF1/CIP1) is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in MCF-7 breast cancer cells. Oncogene. 2004;23:21–29. doi: 10.1038/sj.onc.1207020. [DOI] [PubMed] [Google Scholar]
- 17.Simeone AM, Tari AM. How retinoids regulate breast cancer cell proliferation and apoptosis. Cell Mol Life Sci. 2004;61:1475–1484. doi: 10.1007/s00018-004-4002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayden LJ, Satre MA. Alterations in cellular retinol metabolism contribute to differential retinoid responsiveness in normal human mammary epithelial cells versus breast cancer cells. Breast Cancer Res Treat. 2002;72:95–105. doi: 10.1023/a:1014815112078. [DOI] [PubMed] [Google Scholar]
- 19.Esteller M, Guo M, Moreno V, Peinado MA, Capella G, Galm O, et al. Hypermethylation-associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Res. 2002;62:5902–5905. [PubMed] [Google Scholar]
- 20.Mira YL, Zheng WL, Kuppumbatti YS, Rexer B, Jing Y, Ong DE. Retinol conversion to retinoic acid is impaired in breast cancer cell lines relative to normal cells. J Cell Physiol. 2000;185:302–309. doi: 10.1002/1097-4652(200011)185:2<302::AID-JCP15>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 22.Farias EF, Ong DE, Ghyselinck NB, Nakajo S, Kuppumbatti YS, Lopez R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. J Natl Cancer Inst. 2005;97:21–29. doi: 10.1093/jnci/dji004. [DOI] [PubMed] [Google Scholar]
- 23.Spurling CC, Suhl JA, Boucher N, Nelson CE, Rosenberg DW, Giardina C. The short chain fatty acid butyrate induces promoter demethylation and reactivation of RARbeta2 in colon cancer cells. Nutr Cancer. 2008;60:692–702. doi: 10.1080/01635580802008278. [DOI] [PubMed] [Google Scholar]
- 24.Sirchia SM, Ren M, Pili R, Sironi E, Somenzi G, Ghidoni R, et al. Endogenous reactivation of the RARbeta2 tumor suppressor gene epigenetically silenced in breast cancer. Cancer Res. 2002;62:2455–2461. [PubMed] [Google Scholar]
- 25.Jeong J, Adamson LK, Hatam R, Greenhalgh DG, Cho K. Alterations in the expression and modification of histones in the liver after injury. Exp Mol Pathol. 2003;75:256–264. doi: 10.1016/s0014-4800(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 26.Druesne-Pecollo N, Chaumontet C, Pagniez A, Vaugelade P, Bruneau A, Thomas M, et al. In vivo treatment by diallyl disulfide increases histone acetylation in rat colonocytes. Biochem Biophys Res Commun. 2007;354:140–147. doi: 10.1016/j.bbrc.2006.12.158. [DOI] [PubMed] [Google Scholar]
- 27.Alyaqoub FS, Tao L, Kramer PM, Steele VE, Lubet RA, Gunning WT, et al. Prevention of mouse lung tumors and modulation of DNA methylation by combined treatment with budesonide and R115777 (Zarnestra MT) Carcinogenesis. 2007;28:124–129. doi: 10.1093/carcin/bgl136. [DOI] [PubMed] [Google Scholar]
- 28.Goldenberg D, Harden S, Masayesva BG, Ha P, Benoit N, Westra WH, et al. Intraoperative molecular margin analysis in head and neck cancer. Arch Otolaryngol Head Neck Surg. 2004;130:39–44. doi: 10.1001/archotol.130.1.39. [DOI] [PubMed] [Google Scholar]
- 29.Murray M, Butler AM, Martini R. Inhibition of microsomal 17 beta-hydroxysteroid oxidoreduction activities in rat liver by all-trans-, 9-cis- and 13-cis-retinoic acid. Biochim Biophys Acta. 1994;1222:227–233. doi: 10.1016/0167-4889(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 30.Mongan NP, Gudas LJ. Valproic acid, in combination with all-trans retinoic acid and 5-aza-2′-deoxycytidine, restores expression of silenced RARbeta2 in breast cancer cells. Mol Cancer Ther. 2005;4:477–486. doi: 10.1158/1535-7163.MCT-04-0079. [DOI] [PubMed] [Google Scholar]
- 31.Rahim R, Strobl JS. Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anticancer Drugs. 2009;20:736–745. doi: 10.1097/CAD.0b013e32832f4e50. [DOI] [PubMed] [Google Scholar]
- 32.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–1374. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Tryndyak VP, Kovalchuk O, Pogribny IP. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol Ther. 2006;5:65–70. doi: 10.4161/cbt.5.1.2288. [DOI] [PubMed] [Google Scholar]
- 34.Shann YJ, Cheng C, Chiao CH, Chen DT, Li PH, Hsu MT. Genome-wide mapping and characterization of hypomethylated sites in human tissues and breast cancer cell lines. Genome Res. 2008;18:791–801. doi: 10.1101/gr.070961.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arapshian A, Kuppumbatti YS, Lopez R. Methylation of conserved CpG sites neighboring the beta retinoic acid response element may mediate retinoic acid receptor beta gene silencing in MCF-7 breast cancer cells. Oncogene. 2000;19:4066–4070. doi: 10.1038/sj.onc.1203734. [DOI] [PubMed] [Google Scholar]
- 36.Cho YH, Shen J, Gammon MD, Zhang YJ, Wang Q, Gonzalez K, et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res Treat. 2012;131:197–205. doi: 10.1007/s10549-011-1712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Widschwendter M, Berger J, Muller HM, Zeimet AG, Marth C. Epigenetic downregulation of the retinoic acid receptorbeta2 gene in breast cancer. J Mammary Gland Biol Neoplasia. 2001;6:193–201. doi: 10.1023/a:1011360724350. [DOI] [PubMed] [Google Scholar]
- 38.Arapshian A, Bertran S, Kuppumbatti YS, Nakajo S, Lopez R. Epigenetic CRBP downregulation appears to be an evolutionarily conserved (human and mouse) and oncogene-specific phenomenon in breast cancer. Mol Cancer. 2004;3:13. doi: 10.1186/1476-4598-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]