

Abstract
Most drugs function by binding reversibly to specific biological targets, and therapeutic effects generally require saturation of these targets. One means of decreasing required drug concentrations is incorporation of reactive metal centers that elicit irreversible modification of targets. A common approach has been the design of artificial proteases/nucleases containing metal centers capable of hydrolyzing targeted proteins or nucleic acids. However, these hydrolytic catalysts typically provide relatively low rate constants for target inactivation. Recently, various catalysts were synthesized that use oxidative mechanisms to selectively cleave/inactivate therapeutic targets, including HIV RRE RNA or angiotensin converting enzyme (ACE). These oxidative mechanisms, which typically involve reactive oxygen species (ROS), provide access to comparatively high rate constants for target inactivation. Target-binding affinity, co-reactant selectivity, reduction potential, coordination unsaturation, ROS products (metal-associated vs metal-dissociated; hydroxyl vs superoxide), and multiple-turnover redox chemistry were studied for each catalyst, and these parameters were related to the efficiency, selectivity, and mechanism(s) of inactivation/cleavage of the corresponding target for each catalyst. Important factors for future oxidative catalyst development are 1) positioning of catalyst reduction potential and redox reactivity to match the physiological environment of use, 2) maintenance of catalyst stability by use of chelates with either high denticity or other means of stabilization, such as the square planar geometric stabilization of Ni- and Cu-ATCUN complexes, 3) optimal rate of inactivation of targets relative to the rate of generation of diffusible ROS, 4) targeting and linker domains that afford better control of catalyst orientation, and 5) general bio-availability and drug delivery requirements.
Keywords: Catalytic metallodrug, Artificial nuclease, Ribonuclease mimic, Artificial protease, HIV, Angiotensin converting enzyme
Introduction
The current most universally employed paradigm for drug development has a critical limitation. Drug molecules are typically designed to bind reversibly to specific biological targets that are associated with disease states, and therapeutic effects are achieved through saturation of these targets. For these drugs, target recognition operates within a ‘lock and key’ paradigm, and specificity for a given target is dictated by the structural recognition and bonding complementarity of a drug molecule with respect to the target. Following binding, the target is either inhibited or stimulated with regard to its role in disease progression, resulting in the desired therapeutic effect. However, these drugs are critically limited by the reversible nature of the binding interaction with their targets.
The reversible nature of drug/target binding is such that a drug repetitively binds and releases its target (on a microscopic level), and this fact dictates that a high enough concentration of a drug must be present in order to achieve saturation of a sufficient majority of the population of the target (on a macroscopic level), in order to achieve the therapeutic effect. This limitation prevents the use of many drugs, since the extremely high doses required to achieve target saturation also result in binding to non-targeted tissues, causing unacceptable side effects. Accordingly, there is a need for improved paradigms for drug development.
One approach that holds much promise involves the design of catalytic metallodrugs that contain both a catalytic metal center and a targeting domain, which mediates the localization of each reactive catalyst to the target (1, 2). Catalytic metallodrugs have the potential to circumvent the most critical problem outlined above, which is the requirement for high dosage that results from the reversible nature of most drug/target interactions. Catalytic metallodrugs are able to both bind to and irreversibly modify the target in a catalytic process, introducing the potential for multiple turnover function (3) such that substoichiometric concentrations of the catalytic metallodrug may be used to achieve the therapeutic effect. The resulting enzyme-like behavior of catalytic metallodrugs could permit lower doses to be used. Additionally, catalytic metallodrugs possess the potential for enhanced selectivity as a result of a double filter mechanism whereby target selectivity arises from both binding and intrinsic reactivity (1, 2). That is, inactivation of a target by a catalytic metallodrug is achieved only if two criteria are met: 1) the metallodrug binds the target with sufficiently high affinity, and 2) the metallodrug-target complex is such that the catalytic metal center is properly aligned to react with the target (1, 2, 4). The catalytic process thereby adds stringency to the function of catalytic metallodrugs, relative to drugs that merely bind to their targets.
Recent progress in the development of catalytic metallodrugs
Catalytic metallodrugs developed to date typically consist of artificial nucleases and artificial proteases that modify their target, through either hydrolytic or oxidative mechanisms. Artificial proteases, which modify protein targets, have included hydrolytic Co- and Cu-cyclen, oxidative Ni- and Cu-ATCUN, and photo-activated Ru-complexes, as well as a variety of other oxidative M-chelates with various tunable properties, typically conjugated to a targeting molecule, to effect protein modification (1, 2,5- 17). The artificial protease class of catalytic metallodrugs represents an attractive route to therapy, due to the direct role of many protein targets in disease states and the relative ease of delivery of drugs to many proteins (especially extracellular proteins).
Similarly, artificial nucleases, which modify DNA or RNA targets, have included metal-based catalysts with oxidative (1-4,18-27), hydrolytic (28-32), and/or photoreactive properties (33, 34), and conjugation to targeting molecules has been shown to endow such catalysts with specificity toward targeted DNA or RNA sequences (4,18,26,31,32,35). Cisplatin derivatives (36,37) and DNA-alkylating drugs (38-41) provide other means of eliciting DNA damage, although by differing mechanisms of strand breakage and base-alkylation/crosslinking, respectively. The development of artificial nucleases is of significant interest since the information encoded in nucleic acid polymers is central to all biological phenomena, and attaining control of this encoded genetic information is an ultimate goal. However, critical challenges remain that must first be overcome.
Challenges in the development of catalytic metallodrugs
Despite much promise and the remarkable strides made so far, development of catalytic metallodrugs for therapeutic use remains at an early stage, with many factors to be considered. For instance, many of the catalytic metallodrugs developed to date provide insufficient rates of inactivation of their targets, with respect to the in vivo half-lives of both biological targets and drug molecules. Additionally, the in vivo stabilities of many metallodrugs are limited, thus curtailing in vivo use. However, there are countless natural examples of optimized catalytic metal cofactors that perform important catalytic roles with extremely high efficiency, selectivity, and stability, providing researchers with realistic hopes of developing highly efficient catalytic metallodrugs for specific therapeutic targets.
Catalytic metallodrugs function through various mechanisms, some of which are complex and difficult to study, with some catalysts producing an abundance of diffusible reactive oxygen species (ROS) that may damage non-targeted cellular tissues, while other catalysts either produce controllable metal-bound ROS or promote hydrolytic mechanisms that effect modification of targets in a more selective manner. The reactivity of oxidative catalysts toward therapeutic targets depends heavily on catalysts' properties, such as reduction potential, reactivity with redox coreactants, coordination environment, the identity of the active intermediate, catalyst stability, and the metal/ligand combination used to provide target selectivity. Therefore, it has become critical to elucidate the interrelationships among these catalyst parameters and the overall ability of such catalysts to inactivate, damage, and/or cleave therapeutic targets.
Redox-active catalytic metallodrugs
Our laboratory has recently focused on the development of several families of catalytic metallodrugs (1-6,17-24), in which irreversible modification of targeted biomolecules is achieved through conjugation of a redox reactive catalytic metal center to an appropriate targeting domain. Many of the resulting catalytic metallodrugs have been shown to both bind to and selectively inactivate their biological targets, in some cases with very high efficiency and/or selectivity. We have studied several families of catalysts based on combinations of the transition metals Fe2+, Co2+, Ni2+, and Cu2+ with the chelators cyclam, cyclen, CB-TE2A, DOTA, DTPA, EDTA, tripeptide GGH, tetrapeptide KGHK, NTA, and TACN, shown in Figure 1 (19). We have linked many of these catalysts to targeting molecules such as the Rev peptide (4, 18), lisinopril (17), the peptide YrFK (3), and the cyclic peptide cyclo(RGDfK) for the purpose of targeting HIV-1 Rev response element (RRE) RNA, human angiotensin-1 converting enzyme (sACE-1), the internal ribosome entry site RNA of hepatitis C virus, and integrin proteins, respectively, and successful targeted catalytic inactivation and/or cleavage has been demonstrated.
Figure 1. Catalytic transition metal chelates. M = Fe3+, Co2+, Ni2+, Cu2+. The metal chelates shown here possess variability in size, coordination unsaturation, reduction potential, reactivity with coreactants, the ability to generate reactive oxygen species (ROS), the type of ROS produced, and whether the ROS produced are metal-bound or diffusible.
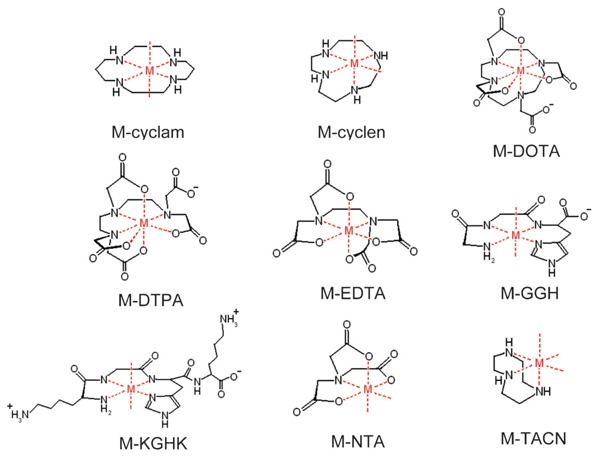
Many of the metal chelates (M-chelates) shown here possess known reactivity with biologically available redox coreactants, including ascorbate, H2O2, and O2, promoting formation of target-damaging ROS, as shown in Scheme 1 (19,42-44). These catalysts provide variability in reduction potential, coordination unsaturation, coreactant reactivity, and size, while correlations between several of these parameters and metallodrug activity have been shown (4,17-19). We have demonstrated that it is possible to tune the reactivity of metallodrugs to match target-specific requirements by varying these catalyst parameters in order to achieve desired effects, by either maximizing reactivity with the available coreactants, maximizing reactivity with the target, or both.
Scheme 1. Metal chelates can undergo multiple turnovers of single-electron reduction of dioxygen or peroxide to form reactive oxygen species. Ascorbate re-reduces oxidized metal centers, allowing multiple turnovers to occur. Alternate pathways, such as 2-electron or 3-electron reduction of O2 are also possible.
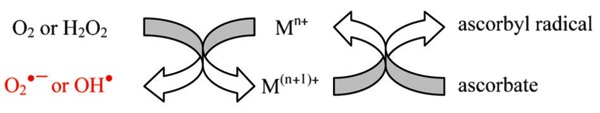
Mechanisms of action
The ability of metallodrugs to catalyze irreversible inactivation and/or cleavage of their respective targets is attributed to the known ability of metal chelates to facilitate conversion of the oxidants O2 or H2O2 to ROS through electron transfer, followed by the reaction of these nascent ROS with the bound target, resulting in inactivation and/or cleavage of the target. Meanwhile, ascorbate, a biologically available single-electron reductant, is known to function as a pro-oxidant by re-reducing oxidized metal centers following generation of ROS, allowing multiple turnovers to occur. The oxidants O2 and/or H2O2 and the reductant ascorbate provide the driving forces responsible for the observed reactivity, while the metal chelate catalyzes single electron transfer from reductant to oxidant, generating ROS in the process, as illustrated in Scheme 1.
Our laboratory has elucidated the relative rates of ROS generation (in the form of diffusible radicals), rates of multiple-turnover ascorbate consumption, and reduction potentials for the metal chelates shown in Figure 1 (18, 19). The relative rates of generation of diffusible radicals by combinations of metal chelates and the oxidants O2 or H2O2 were determined by experiments using TEMPO-9-AC (Figure 2B) and rhodamine B as radical traps and monitoring, in real time, the changes in fluorescence and absorbance, respectively, that occur upon reaction of these radical traps with the diffusible radicals produced by each metal chelate (19). TEMPO-9-AC is sensitive to both superoxide and hydroxyl radicals (45-47), and the presence or absence of added H2O2 likely distinguishes whether the radicals observed by reaction with TEMPO-9-AC were primarily hydroxyl or superoxide radicals, respectively (under aerobic conditions); rhodamine B is known to react with only hydroxyl radicals (48). Rates of catalyst-mediated multiple-turnover ascorbate consumption were similarly determined by monitoring the decrease in absorbance of ascorbate over time, following catalyst-mediated conversion of ascorbate to ascorbyl radical, with either O2 or H2O2 as an oxidant (Figure 2A) (19). Reduction potentials for each metal chelate were separately determined by use of square wave voltammetry, and relationships between reduction potential and redox reactivity were observed (18, 19).
Figure 2. Summary of rates of multiple-turnover ascorbate consumption (A) and rates of TEMPO-9-AC monitored radical generation (B) for each M-chelate obtained, under aerobic conditions (19). Data within each subfigure are for reactions with either O2 (front) or H2O2 (rear) as an oxidant. Concentrations were initially 10 µM M-chelate, 10 µM TEMPO-9-AC (when present), 1 mM H2O2 (when present), and/or 1 mM ascorbate (when present). *Fe-TACN/H2O2 was observed to promote very rapid radical generation relative to other catalysts, requiring stopped-flow measurements to determine an observed rate of 25.94 ± 0.02 µM/min with the conditions used.
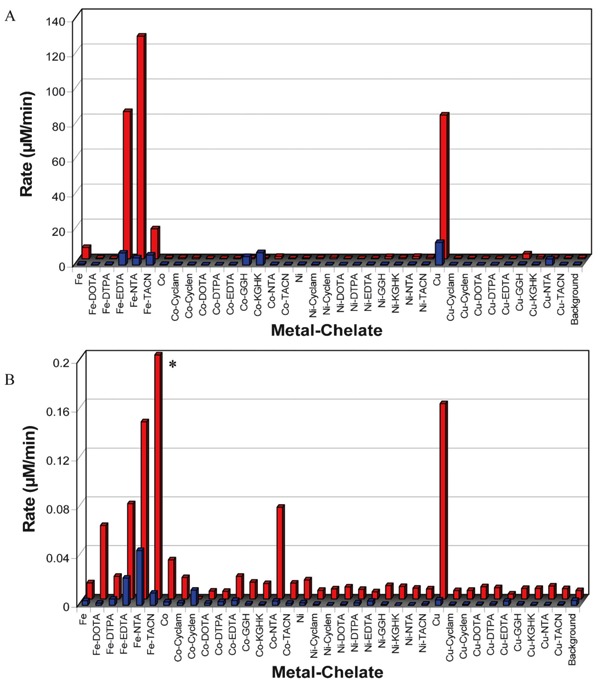
Knowledge of the redox reactivity for these metal chelates has proven useful for the interpretation of the observed reactivities of catalytic metallodrugs that utilize these metal chelates, while certain relationships between catalyst reduction potential, the ability to generate diffusible ROS, and the ability to consume ascorbate with multiple turnovers have been observed. For instance, we observed that the highest rates of both diffusible radical generation and multiple-turnover ascorbate consumption occur when the catalyst reduction potential is poised between the reduction potentials of the relevant coreactant half-reactions (Figure 3), such that multiple turnovers are thermodynamically favored. The reduction potentials for ascorbyl radical/ascorbate and H2O2/OH· at neutral pH are -66 mV and +380 mV, respectively, and M-chelates in this range of reduction potential generally provide the greatest redox reactivity (18, 19). Similar phenomena have been observed elsewhere, especially for superoxide dismutase, which also possesses a reduction potential poised between those of the relevant redox half reactions (49,50).
Figure 3. The highest rates of multiple turnover ascorbate consumption and radical generation were found for M-chelates with biologically relevant reduction potentials (19). A, Ascorbate consumption by M-chelate/H2O2. B, Ascorbate consumption by M-chelate/O2. C, Radical generation by M-chelate/H2O2. D, Radical generation by M-chelate/O2. The reduction potentials for the H2O2/hydroxyl radical (E° = +380 mV) and ascorbyl radical/ascorbate (E° = -66 mV) half reactions are shown by the dashed lines.
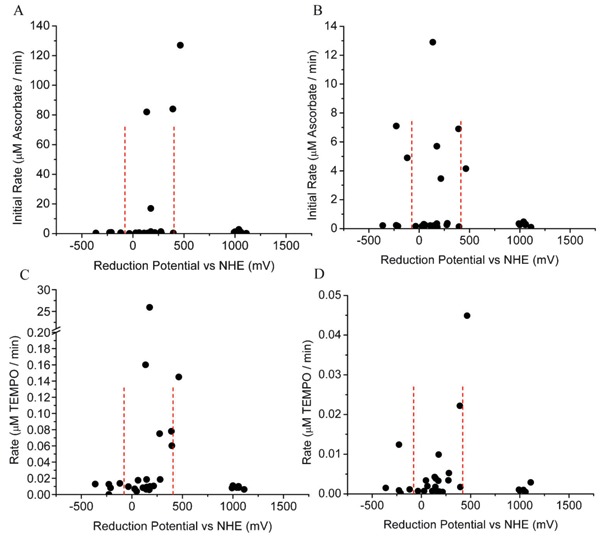
The dependence of redox activity on M-chelate reduction potentials is akin to the role of a middleman or broker in a financial market: the middleman (M-chelate) catalyzes the transfer of money (electrons) from buyer (reductant) to seller (oxidant) - the middleman's operation typically only has long-term viability (thermodynamically favored multiple turnovers) if his price (reduction potential) remains poised between those of the buyer and seller. Similarly, M-chelates with reduction potentials between those of the relevant coreactant half-reactions generally provide the highest reactivity, both at the level of coreactant consumption and ROS generation and also at the level of target inactivation and/or cleavage, although a few exceptions do exist.
Artificial nucleases that oxidatively cleave targeted nucleic acids were found to possess optimal nuclease activity when the reduction potentials for the attached metal chelates were between those of the relevant coreactant half reactions (-66 and +380 mV), as expected (4,18, 19). However, optimal inactivation of protein targets, such as human ACE, by catalytic metallodrugs was found to occur for catalysts with higher reduction potentials (approximately 1000 mV), most likely reflecting the increased stability of protein targets toward oxidation, relative to nucleic acids, as discussed later (5,6,17).
The ability of M-chelates to consume coreactants selectively and the degree to which the ROS produced are metal-bound or diffusible are both key determinants of the mechanisms and efficiency of target inactivation and/or cleavage by catalytic metallodrugs that incorporate these M-chelates. The in vivo concentrations of the redox coreactants O2, H2O2, and ascorbate (and/or glutathione), and the resulting ability of metallodrugs to function, are highly variable and dependent on the in vivo location (i.e., extracellular vs intracellular; cytoplasm vs nucleus) and the redox state of this location (i.e., hypoxic vs normal conditions vs oxidative stress), and so the relative abilities of metallodrugs to function in vivo are expected to be directly related to the ability of each M-chelate to preferentially consume ascorbate in cooperation with either O2 or H2O2. Studies of the relative abilities of M-chelates to consume ascorbate, and the degree to which this reactivity was selectively favored by either O2 or H2O2, are useful for this purpose. Similarly, metallodrugs that generate a plethora of diffusible radicals are expected to be less selective in vivo, since diffusible radicals may harm non-targeted tissues, while metallodrugs that react via more controlled pathways, such as a metal-bound ROS, may be able to elicit cleaner inactivation of the targeted molecule. Comparison of the rates of M-chelate-mediated ascorbate consumption with the rates of diffusible radical generation allows an assessment of the degree to which the ROS produced by each metal center were either metal-bound or diffusible, while other techniques, such as determining the radius of oxidative reactivity relative to a fixed metal center, have also been used to identify the type of ROS produced by each metallodrug.
Applications
The general concept behind the design of redox-active catalytic metallodrugs involves tethering of a redox-active M-chelate to a targeting molecule, such that the targeting molecule mediates the localization of the redox-active M-chelate to the target, with subsequent inactivation and/or cleavage of the target occurring as a result of the proximal formation of ROS. Following synthesis and characterization of metallodrug compounds, the typical set of experiments consists of, first, determining the relative binding affinity of each compound to the corresponding target in the absence of any redox reactivity (no coreactants present). Second, once the relative binding affinities have been established, the catalysts, coreactants, and the target are co-incubated, under conditions with known extents of target-saturation and binding stiochiometry, and the rate constants for irreversible target inactivation are determined, typically in the presence of various combinations of coreactants. Examples are outlined in the following sections.
Targeted cleavage of HIV RRE RNA by M-chelate-Rev catalysts
Catalysts that target and oxidatively cleave/modify viral mRNA represent a particularly ideal class of anti-viral drugs, since oxidative modification of mRNA is an irreversible process because cells lack a general RNA repair mechanism. Furthermore, each copy of mRNA typically precedes multiple copies of translation products critical to the production of active viral particles, and therefore mRNA is a high-value target.
Our laboratory has synthesized and evaluated a series of compounds that target reactive M-chelates to the HIV-1 RRE mRNA (4, 18). These compounds are based on combinations of the transition metals Fe2+, Co2+, Ni2+, and Cu2+ and selected chelators, including DOTA, DTPA, EDTA, NTA, tripeptide GGH, and tetrapeptide KGHK, each of which was chemically linked to the RNA-targeting Rev peptide that promotes high-affinity binding to the RRE stem loop IIB mRNA. The resulting catalysts are summarized in Figure 4 (51, 52). Our laboratory has also recently demonstrated successful inactivation of the hepatitis C virus by several metallodrugs containing complexes between copper and amino terminal copper/nickel-binding motifs (Cu-ATCUN complexes) that target and cleave the internal ribosome entry site of HCV mRNA, as demonstrated by use of both spectroscopic and HCV replicon assays (3).
Figure 4. Summary of the metal-chelate complexes and their modes of conjugation to the Rev peptide (18). M = Fe2+, Co2+, Ni2+, Cu2+. The sequence of the Rev peptide is shown above. The 7-coordinate geometries shown here for M-DOTA-Rev and M-DTPA-Rev are based on the X-ray crystal structures of [Fe-DOTA]1- and [Fe-DTPA]2- (51, 52). Note that Fe2+ complexes of GGH-Rev and KGHK-Rev were not observed and therefore were not used.
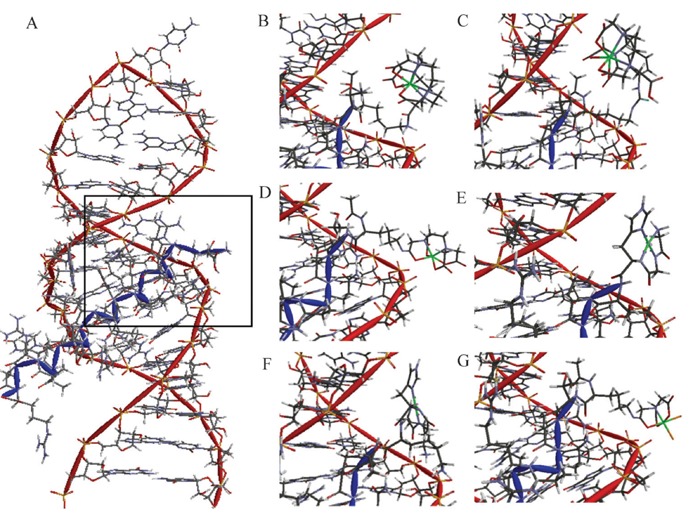
First, dissociation constants for binding of HIV RRE RNA were determined by use of a fluorescence titration assay (Figure 5), using 2-aminopurine-labeled RRE RNA (AP-RRE), originally developed by Lacourciere et al. (53). These experiments revealed high-affinity RRE RNA-binding by all metal-chelate-Rev species, with KD values ranging from ∼0.2-16 nM, indicating little to no loss of RNA affinity due to the coupling of the metal-chelates to the Rev peptide, although a few minor trends related to the size and charge of the attached M-chelate were observed (Table 1). We also observed secondary binding of M-chelate-Rev catalysts to a low-affinity binding site on the RRE RNA (KD2 ∼200-2600 nM). Meanwhile, binding of the M-chelate-Rev peptides to plasmid DNA was found to have a much lower binding affinity (KD,app ∼1700-3600 nM bp DNA; Table 2) (4), and the Rev peptide has previously been shown to bind yeast tRNA with a similarly low affinity (KD ∼2400 nM) (54), confirming the selectivity of RRE binding for the Rev peptide and its utility as an RRE RNA-targeting molecule. Molecular modeling of the complexes formed between each M-chelate-Rev catalyst and the HIV RRE RNA (Figure 6) (18) has been enabled by availability of the NMR solution structure of the complex formed between the Rev peptide and stem loop IIB of the HIV RRE RNA, determined by Battiste et al. (55).
Figure 5. Titration fluorescence response curve for binding of Fe-DOTA-Rev to 2-aminopurine-labeled Rev response element (RRE) RNA (AP-RRE) (18). Titration progress was monitored by use of the fluorescence emission at 371 nm, with excitation at 310 nm. Binding of AP-RRE by all other M-chelate-Rev complexes was studied in the same manner.
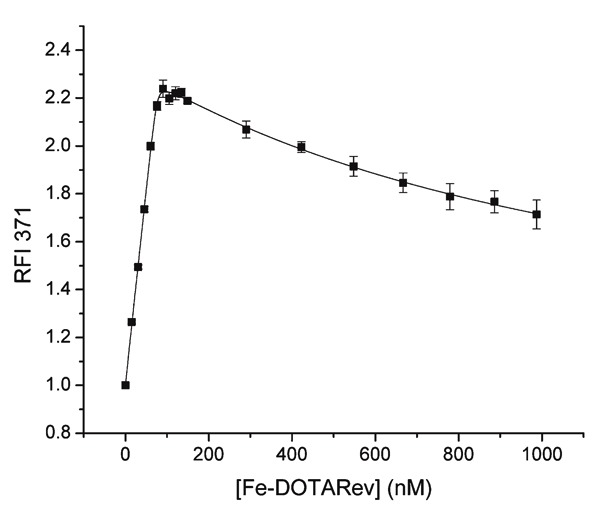

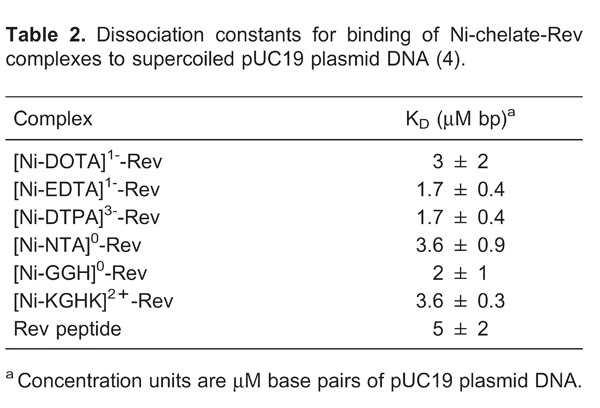
Figure 6. High-affinity binding of Rev response element (RRE) stem loop IIB by A, Rev peptide (NMR structure) (55). Inset: Energy minimized structural models of high-affinity site binding by M-chelate-Rev peptides; B, Fe-DOTA-Rev; C, Fe-DTPA-Rev; D, Fe-EDTA-Rev; E, Cu-GGH-Rev; F, Cu-KGHK-Rev; G, Fe-NTA-Rev, described previously (18).
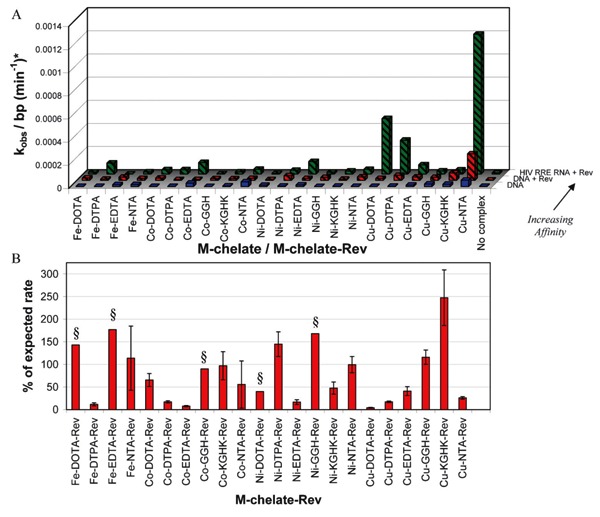
Following elucidation of the RRE-binding affinities for each M-chelate-Rev catalyst, each catalyst was incubated with RRE RNA in the presence of coreactants, with variable time points, and the rates of catalyst-promoted cleavage of the RRE RNA and identities of cleavage products were determined by use of several independent methods: including PAGE analysis, real time fluorimetry (Figure 7), and MALDI-TOF mass spectrometry (MS) analysis (Figure 8). The high-affinity complex formation in the M-chelate-Rev/RRE system allows careful analysis of the nuclease activity for each Rev-coupled M-chelate, such that variability in binding affinities and overall alignment of the Rev peptide with the RRE RNA backbone are not significant. This fact has allowed elucidation of correlations between nuclease activity and catalyst parameters such as reduction potential, coordination unsaturation, coreactant selectivity, the overall charge associated with each M-chelate-Rev complex, and the alignment of metal centers with the RNA backbone. In particular, a clear correlation was noted between the nuclease activity of the M-chelate-Rev catalysts and the reduction potential of the attached M-chelate, such that optimal rates of RNA cleavage were observed for M-chelates with reduction potentials between those of H2O2/OH· (380 mV) and ascorbyl radical/ascorbate -66 mV, consistent with the general trends observed for the general reactivity of M-chelates with redox coreactants (Figure 3).
Figure 7. Rate of Rev response element (RRE) modification by 100 nM M-chelate-Rev vs M-chelate complexes, in the presence of both 1 mM H2O2 and 1 mM ascorbate. The relative rates for each catalyst scale differently at higher concentrations, most likely as a result of differing higher-order reactivity with coreactants. For instance, at 10 µM catalyst/RNA, Fe-EDTA-Rev shows the highest rate of cleavage. Enhanced rates were observed for M-chelates attached to Rev for 100% of the species with above-background rates. The background rate with no complex was 0.28 ± 0.03 nM/min.
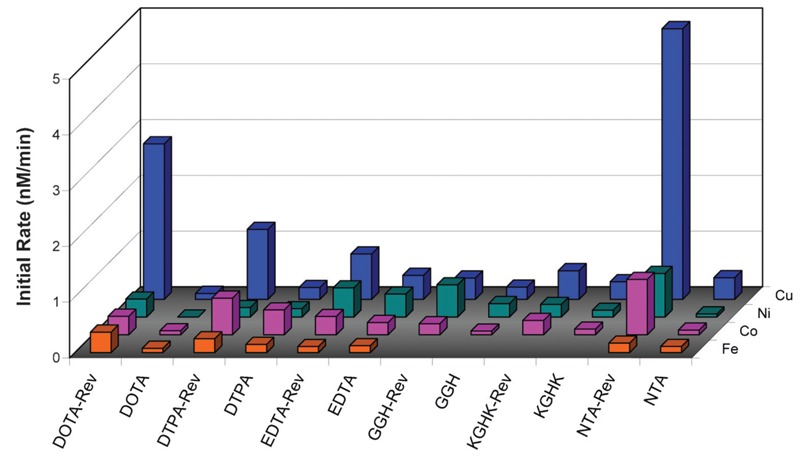
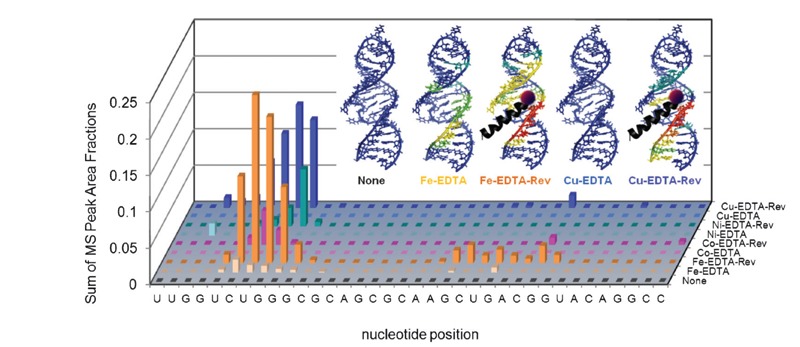
Figure 8. Targeted cleavage of Rev response element (RRE) RNA required attachment of catalysts to the Rev peptide in order to achieve efficient cleavage, as seen by differences in reactivity between the M-chelate-Rev catalysts and M-chelates lacking Rev (56). After 1 h incubation of each catalyst and co-reactants with the Fl-RRE RNA, the apparent abundances of individual RNA cleavage fragments were quantified. The data correspond to the abundances (MS peak area fractions) of oxidation products (cleavage fragments containing nascent terminal 3′-PO4, 3′-PG, or 5′-PO4 overhangs), at positions corresponding mechanistically (adjacent) to each illustrated site of H-abstraction, after 1 h reaction. Inset: The same data were mapped onto the 3D solution structure of the RRE RNA, for several catalysts and the control lacking a catalyst (red corresponds to highest reactivity; blue corresponds to reactivity below background).
Targeting of the HIV RRE RNA has been demonstrated by three distinct methods: 1) time-dependent cleavage of RRE RNA was studied for both M-chelate-Rev catalysts and for M-chelates lacking attached Rev, and rates of cleavage were generally much higher for the Rev-coupled catalysts than for M-chelates lacking Rev (Figures 7 and 8); 2) PAGE-monitored cleavage of RRE RNA by both M-chelate-Rev catalysts and M-chelates lacking Rev was tested in the presence of variable concentrations of competing tRNA, and attachment to Rev was generally required to maintain the ability to cleave RRE RNA in the presence of elevated tRNA concentrations (Table 3) as evidenced by higher IC50 values for the Rev-coupled catalysts; 3) dissociation constants for binding of M-chelate-Rev catalysts to RRE RNA are ∼1 nM, while dissociation constants for binding to other RNA/DNA targets are approximately 1000-fold higher.

We have recently used MALDI-TOF MS to observe cleavage products resulting from oxidative cleavage of HIV RRE RNA by each M-chelate-Rev catalyst and M-chelate catalyst, with various combinations of coreactants, and these experiments have provided a high level of detail regarding the identities and sites of origin of cleavage products, as well as the general mechanisms of oxidative RNA strand scission employed by each catalyst (56). MALDI-TOF MS analysis of cleavage products reveals strand scission at a distribution of positions within the RNA sequence, with the highest rates of scission occurring at positions nearest to the predicted location of each metal center.
Using MALDI-TOF MS to analyze cleavage products, we observed at least six distinct overhang types produced at nucleotides adjacent to cleavage positions: 3′-hydroxyl (3′-OH), 2′,3′-cyclic phosphate (2′,3′-cPO4), 3′-phosphate (3′-PO4), and 3′-phosphoglycolate (3′-PG) overhangs at nascent 3′ termini; 5′-OH and 5′-PO4 overhangs at nascent 5′ termini, following cleavage (56, 57). Catalysts that promoted rapid oxidative scission gave primarily 3′-PO4 and 3′-PG overhangs, and these products were shown to arise primarily as a result of oxidative hydrogen abstraction (56, 57). Furthermore, following oxidative cleavage, we observed a correlation between the relative concentrations of 3′-PG overhangs and base 2-hydroxypropenal cleavage products, each of which is predicted to result from H-4 abstraction from RNA (base 2-hydroxypropenals are the RNA analogues of the base-propenals formed from H-4 abstraction from DNA) (57, 58); this correlation validates H-4 abstraction as a major pathway of oxidative strand scission for the M-chelate-Rev/RRE RNA system. H-5 abstraction is another likely major pathway, consistent with the similarly high rate of formation of 3′-phosphate overhangs during oxidative cleavage of the RRE RNA, but not under hydrolytic conditions (57). MALDI-TOF MS-monitored oxidative scission of RRE RNA by M-EDTA-Rev and M-EDTA complexes is illustrated in Figure 8.
Mass spectrometric analysis of the cleavage products generated by M-chelate-Rev catalysts revealed highly focused regions of cleavage within the RRE RNA, such that cleavage by each M-chelate-Rev catalyst happened most rapidly in close proximity to the attached metal center (Figures 8 and9). Conversely, the cleavage products observed after reaction with M-chelates lacking Rev were typically present at far lower concentrations and originated over wider, less focused regions of the RRE RNA (Figures 8 and 9), although a few coordinatively unsaturated copper complexes were also observed to bind and cleave within the bulge of stem loop IIB even without attachment to Rev, but only in the presence of H2O2.
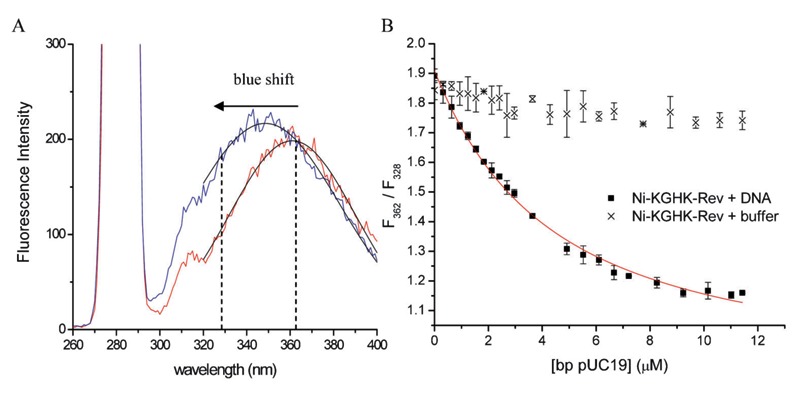
Figure 9. Fluorescence-monitored titration of M-chelate-Rev catalysts with added supercoiled plasmid DNA (4). A, Fluorescence emission of the internal Trp residue of Ni-KGHK-Rev, both before and after the addition of plasmid DNA, illustrating the blue shift in Trp fluorescence that occurred upon binding of Ni-KGHK-Rev to DNA. B, Titrations were monitored by following the decrease in the ratio of fluorescence intensities at 362 nm and 328 nm, resulting from the blue shift in Trp fluorescence emission that occurred upon binding to DNA. Control experiments lacking DNA did not provide a response.
MALDI-TOF MS methods of analysis of M-chelate-Rev/RRE RNA reactions have provided a unique technique by which to probe whether the ROS responsible for RNA cleavage, produced by each catalyst, were primarily metal-bound or diffusible. A combination of the relative abundances of 3′-phosphoglycolate overhang products that resulted from H-4 hydrogen abstraction events at various nucleotide positions within the RRE RNA, as monitored by MALDI-TOF MS, and the cartesian coordinates of all H-4 atoms in the solution structure of the Rev/RRE complex allowed a 3D radius of oxidative reactivity to be established for each M-chelate-Rev catalyst, providing an assessment of whether the catalyst-generated ROS responsible for H-4 abstraction by each catalyst were primarily diffusible or metal-bound (57). A low radius of reactivity was attributed to a metal-bound ROS (hydroxyl or superoxide radical) as the reactive intermediate responsible for the observed hydrogen abstraction from RNA by each catalyst, whereas a high radius of reactivity was attributed to a diffusible ROS (hydroxyl or superoxide radical) as the reactive intermediate. Certain catalysts, such as Fe-EDTA-Rev and Fe-DTPA-Rev, appeared to cleave the RRE RNA by diffusible radical intermediates (high radius of reactivity), as expected, whereas others, such as Cu-GGH-Rev, appeared to cleave the RNA by metal-bound radicals (low radius of reactivity). These data complement the basic studies of diffusible radical generation by each M-chelate, described earlier (Figure 2B).
In summary, these M-chelate-Rev compounds are promising reagents for irreversible inactivation of the RRE RNA, due to the RRE-binding selectivity and irreversible nature of their nuclease activity, and represent a remarkable improvement over the majority of current drugs, which operate reversibly and thus require higher concentrations in order to attain effectiveness. Studies of targeted cleavage of HIV RRE RNA by the family of M-chelate-Rev catalysts shown herein have provided a wealth of information and have revealed important relationships between target-binding affinity, nuclease activity, catalyst reduction potential, and redox activity. Therefore, these studies provide a benchmark for further development of reagents that function by irreversibly cleaving mRNA targets.
DNA nuclease activity of M-chelate-Rev and M-chelate complexes
DNA nucleases represent an important approach to treatment of certain diseases, such as cancer and HIV, that ultimately stem from genetic sources, since eradication of the genes of such diseases is required to effect a cure (4,19, 21, 25, 26, 28, 31, 32, 59). Current anticancer strategies include the use of DNA-binding cisplatin derivatives (36, 37, 60, 61), DNA intercalators (62, 63), and alkylating drugs (40, 41) as antineoplastic agents as well as the use of retroviral gene therapy, and these current techniques are quite limited, since they either lack selectivity, cause unintended insertional mutagenesis, and/or cause secondary cancers (64, 65). A more selective approach is required, and one such possibility is the use of metallodrugs that contain both a reactive metal center and a gene-targeting domain, such that only targeted genes are inactivated.
The M-chelate-Rev catalysts, which were originally designed to target and cleave HIV RRE RNA (18), were also tested for their ability to bind to and cleave double- stranded DNA, since the positively charged Arg-rich Rev peptide possesses moderate DNA-binding affinity (54), endowing the M-chelate-Rev catalysts with moderate DNA-binding affinity (Table 2) and enhanced DNA nuclease activity, relative to the M-chelates lacking any targeting domain (4). Although the Rev peptide is not known to bind specifically to any DNA sequence used in our experiments, comparison of the nuclease activities for the M-chelate-Rev catalysts and the M-chelates lacking attached Rev reveals the ability to enhance the nuclease activity of the reactive M-chelates to DNA by electrostatic targeting (attachment to positively charged Rev peptide). Therefore, if such enhancements can be caused by mere electrostatic targeting between negatively charged DNA and positively charged M-chelate-Rev catalysts, then far greater enhancements to nuclease activity and gene-targeted therapy are expected if the targeting domains instead consisted of more specific gene-targeting sequences, such as anti-sense PNA or oligonucleotide sequences.
Our experiments consisted of, first, determining the binding affinities between the Rev peptide and supercoiled plasmid DNA, and second, comparing the DNA nuclease activities of the M-chelate-Rev catalysts with those of the M-chelates lacking Rev (4). DNA-binding affinities were determined by fluorescence-monitored titrations of M-chelate-Rev catalysts with DNA, for which DNA binding resulted in a blue shift in the fluorescence emission of a Trp residue within the Rev peptide (Figure 9). The M-chelate-Rev catalysts were shown to bind supercoiled plasmid DNA with moderate binding affinity (KD,app ∼1700-3600 nM; Table 2), although the observed DNA-binding affinity was ∼3 orders of magnitude weaker than the RRE RNA-binding by the M-chelate-Rev catalysts (Table 1).
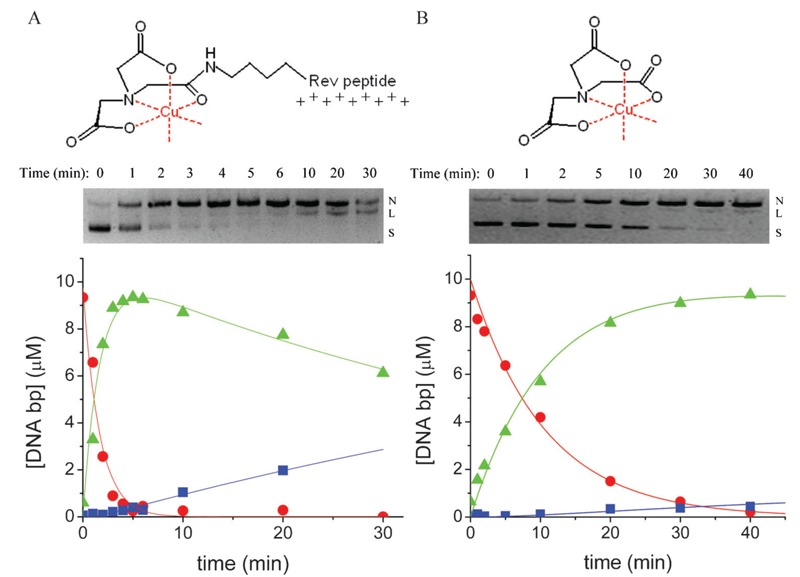
Following elucidation of the plasmid DNA-binding affinities for each of the Ni-chelate-Rev catalysts, the nuclease activities of the M-chelate-Rev catalysts and M-chelates lacking Rev were studied. Knowledge of the DNA-binding affinities for the M-chelate-Rev catalysts allowed the design of experiments with concentrations of DNA (10 µM bp) and catalyst (100 nM) such that ∼73-86% of each M-chelate-Rev catalyst was initially DNA-bound, whereas M-chelates lacking Rev, which possess DNA-binding affinities that are several orders of magnitude worse, were ∼0% DNA-bound. Under these conditions, significantly higher rates of DNA cleavage were observed for the Rev-coupled M-chelates than for the M-chelates lacking Rev, as shown for Cu-NTA-Rev vs Cu-NTA in Figure 10.
Figure 10. Time-dependent cleavage of plasmid DNA by A, Cu-NTA-Rev and B, Cu-NTA lacking Rev (4). Conversion of plasmid DNA from supercoiled (S) to nicked (N), and then to linear (L) forms by the respective metal complexes was monitored by agarose gel electrophoresis. Variation of the relative concentrations of supercoiled, nicked, and linear form DNA were fit to a first-order consecutive reaction model (S = circles; N = triangles; L = squares), and rate constants of DNA nicking (knick) and subsequent linearization (klin) were determined from fitting. Attachment of M-chelates to the Rev peptide (positively charged) results in increased electrostatic attraction to DNA (negatively charged), and consequently, M-chelate-Rev complexes generally react with DNA more quickly than do M-chelates lacking Rev. The apparent loss of linearized DNA seen at 30 min for the Cu-NTA-Rev reaction is a result of cleavage of linearized DNA to form smaller DNA fragments (smearing); this was evident only at higher time points (≥30 min) and only for the Cu-NTA-Rev reaction, and as a result, linearized DNA at the 30-min time point was not quantified.
Enhancements in the rates of DNA cleavage that occurred upon attachment of M-chelates to Rev presumably occurred as a result of electrostatic attraction between the positively charged Rev peptide and negatively charged DNA, but were not nearly as high as those expected from attachment of a targeting ligand with much greater target-binding affinity, such as observed for binding of the M-chelate-Rev catalysts to HIV RRE RNA. To illustrate this point, nuclease activities for three systems are summarized in Figure 11, each with different target-binding affinities, under otherwise similar reaction conditions. The comparison includes, first, targeted cleavage of HIV RRE RNA by M-chelate-Rev complexes with KD values ∼0.2-16 nM (100% saturation of catalyst); second, semi-targeted cleavage of pUC19 plasmid DNA by M-chelate-Rev complexes with KD values ∼1700-3600 nM (73-86% saturation of catalyst), and third, non-targeted cleavage of pUC19 plasmid DNA by M-chelates (∼0% saturation of catalyst expected). There is a clear correlation between binding affinity and nuclease activity when low concentrations of catalyst (100 nM) are used.
Figure 11. A, Comparison of first-order rate constants for three nuclease/nucleic acid systems: nicking of plasmid DNA by M-chelates lacking any targeting sequence (front), nicking of plasmid DNA by M-chelate-Rev complexes (middle), and targeted nicking of HIV RRE RNA by M-chelate-Rev complexes (rear) (4, 18, 19). B, Percentage of the expected rate constant for DNA nicking by M-chelate-Rev complexes, expressed as a percentage of the analogous rate constant for targeted nicking of HIV RRE RNA, and normalized using the corresponding percentage of catalyst that is DNA-bound (Table 2); rate constants less than 100% of the expected rate constant imply a suboptimal orientation of the catalytic metal center with the DNA backbone, while a value greater than 100% suggests improved reactivity through more optimal orientation. All reactions were performed under similar conditions with 100 nM catalyst, 1 mM ascorbate and 1 mM H2O2. *The normalized rate constants shown here were obtained by dividing each kobs by the number of base pairs present in each species (2686 bp for pUC19 plasmid, 17 bp for the HIV RRE RNA construct), as required, to normalize against the impact of nucleic acid polymer length on the rate of nicking. §Greater than or equal to the value shown, since the corresponding rate of HIV RRE RNA cleavage was below the limit of detection.
Comparison of the reactivity of M-chelate-Rev catalysts toward either plasmid DNA or HIV RRE RNA clearly reveals the importance of target-binding affinity, especially when compared with the reactivities of M-chelates lacking Rev. However, another important conclusion may be drawn from these experiments: since the relative rates of DNA cleavage and RNA cleavage by each M-chelate-Rev catalyst do not consistently match those expected based on the percentage of catalyst that is target-bound (Figure 11B), it is evident that other factors, such as alignment of the catalytic metal center with the nucleic acid backbone, the degree of geometric constraint, and/or the relative ease of oxidation of DNA relative to RNA targets, contribute significantly to the observed nuclease activities. Therefore, one major conclusion of this study is that the alignment between catalyst and the nucleic acid backbone is a critical parameter that must be considered in the future design of artificial nucleases.
Targeted catalytic inactivation of human angiotensin converting enzyme
Human sACE-1 is an important therapeutic target for the treatment of hypertension and heart failure due to its critical physiological roles in cardiovascular function, primarily the hydrolysis of the peptides angiotensin I and bradykinin; both of these processes result in overall vasoconstriction (66-69). Several inhibitors of sACE-1, such as lisinopril (currently marketed as Zestril and Prinivil), successfully alleviate hypertension through a reversible mode of competitive inhibition of sACE-1, blocking binding of the natural substrates of sACE-1 (66, 67, 70-75). However, the reversible mode of inhibition by lisinopril requires high doses (5 to 40 mg/day) in order to achieve stoichiometric saturation of sACE-1 (66).
There is significant interest in the development of multiple-turnover drugs that irreversibly modify critical protein/enzyme targets, allowing substoichiometric concentrations of drug to be used. Several research groups have recently developed various artificial proteases that use target-selective metal catalysts, as described in an earlier section. However, many of the catalytic metallodrugs developed previously for the purposes of enzyme inactivation (especially hydrolytic metallodrugs) function with relatively slow second-order chemistry, as a result of either low enzyme-binding affinity or sluggish inactivation chemistry once bound to the targeted enzyme, limiting the practicality of clinical use.
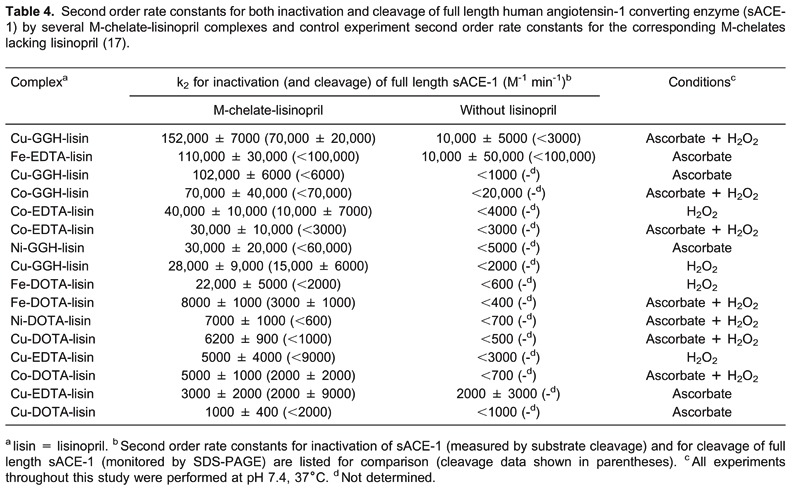
In an effort to overcome these limitations, we have developed a class of catalytic metallodrugs (Figure 12) based on combinations of the transition metals Fe3+, Co2+, Ni2+, and Cu2+ and the chelators DOTA, EDTA, NTA, and tripeptide GGH, which were chemically linked to the lysine side chain of lisinopril, such that the inhibitor lisinopril targeted each reactive M-chelate to the active sites of sACE-1 (17). The resulting M-chelate-lisinopril catalysts both bind to sACE-1 with high affinity (Figure 13) and also irreversibly inactivate sACE-1 with high selectivity (Table 4). The oxidative nature of these M-chelate-lisinopril catalysts distinguishes them from the relatively slow hydrolytic catalysts that have been developed previously, while lisinopril is a particularly potent targeting ligand (IC50 ∼1.9 nM), resulting in a high level of selectivity for sACE-1 relative to non-targeted proteins.
Figure 12. Metal-chelate-lisinopril catalysts (17). M = Fe3+, Co2+, Ni2+, Cu2+.
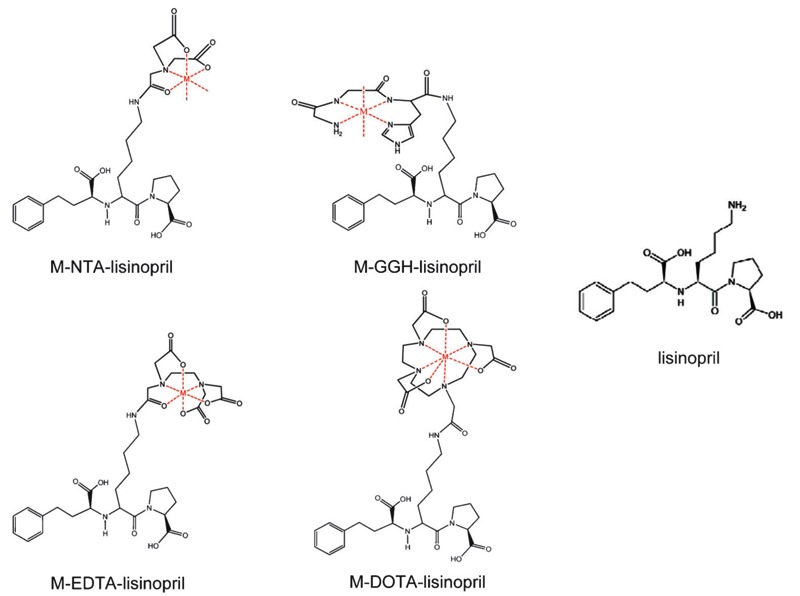
Figure 13. Concentration-dependent inactivation of human angiotensin-1 converting enzyme (sACE-1) by Fe-NTA-lisinopril (17). A, sACE-1 (1 nM) was preincubated with variable concentrations of Fe-NTA-lisinopril, and subsequently sACE-1 activity was measured from the initial cleavage rates of fluorogenic substrate. B, Dependence of sACE-1 activity on Fe-NTA-lisinopril concentration. IC50 values were determined for all synthesized M-chelate-lisinopril complexes in the same manner.
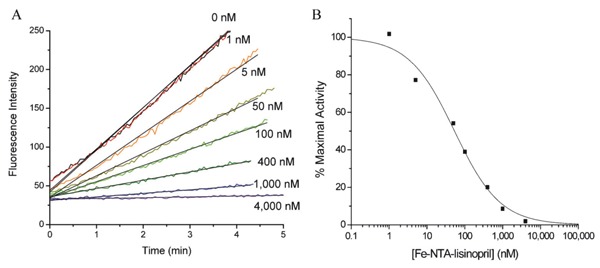
As with the M-chelate-Rev catalysts discussed in previous sections, the variety of lisinopril-coupled M-chelates used provided variability in catalyst size, coordination unsaturation, reduction potential, and charge, such that the effect of these catalyst parameters on the ability of M-chelate-lisinopril catalysts to inactivate sACE-1 could be assessed. The ability of each M-chelate-lisinopril complex to both reversibly inhibit and catalytically inactivate sACE-1 was assessed, and varying binding affinities and rates of targeted irreversible inactivation (and cleavage) of sACE-1 by M-chelate-lisinopril complexes were observed.
The relative ACE-binding affinity of each catalyst was assessed by incubating the enzyme with variable concentrations of each catalyst (in the absence of coreactants) and following the change in rate for sACE-1-mediated cleavage of the fluorogenic substrate Mca-RPPGFSAFK(Dnp)-OH, shown in Figure 13. IC50 values for sACE-1 inhibition were determined for each species. Lisinopril was found to have an IC50 of 1.9 nM. Following attachment of M-chelates to lisinopril, IC50 values were found in the range 44-4500 nM, with IC50 values increasing with the size of the attached M-chelate and, to a lesser extent, the negative charge of the attached M-chelate. The inverse correlation between ACE-binding affinity and M-chelate size, illustrated in Figure 14, is consistent with energy-minimized models of active site sACE-1 binding by M-chelate-lisinopril catalysts (Figure 15), for which the attached M-chelate of largest species (M-DOTA-lisinopril) had the most difficulty settling into the active site.
Figure 14. Human angiotensin-1 converting enzyme (sACE-1) binding affinity of M-chelate-lisinopril complexes was inversely correlated with the size and negative charge of the species attached to the lysine side chain of lisinopril (17). Lisinopril and all M-chelate-lisinopril species (metal-bound and metal-free) are shown: lozenges = lisinopril; squares = NTA-lisinopril; triangles = GGH-lisinopril; circles = EDTA-lisinopril; inverted triangles = DOTA-lisinopril. Orange = Fe; pink = Co; cyan = Ni; blue = Cu; black = no metal. The charge of the modified lysine side chain of lisinopril is listed for each attachment; the charge for each Fe3+ complex was 1+ higher than for each corresponding M2+ complex.
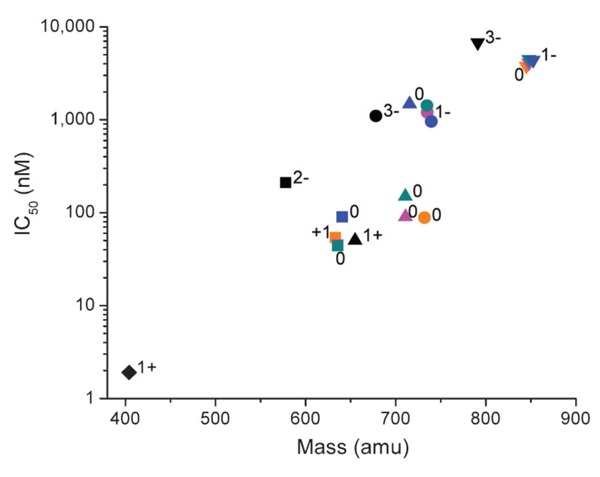
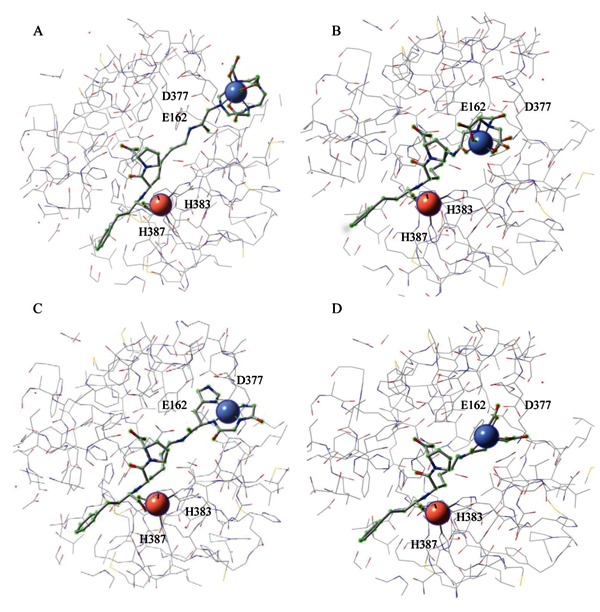
Figure 15. Energy-minimized structural models of tACE active site (equivalent to the active site of C-domain of sACE except for 36 additional residues at the N-terminus of tACE) (17). The models show binding by A, Fe-DOTA-lisinopril, B, Fe-EDTA-lisinopril, C, Cu-GGH-lisinopril, and D, Fe-NTA-lisinopril. The redox-active metal for each metal-chelate-lisinopril complex is shown as a blue sphere (upper right within each model), and the active site Zn2+ is shown as a pink sphere (bottom left within each model). Residues E162 and D377 can interact with the lysine side chain of unmodified lisinopril, while H383 and H387 form part of the conserved HEXXH Zn2+-binding motif.
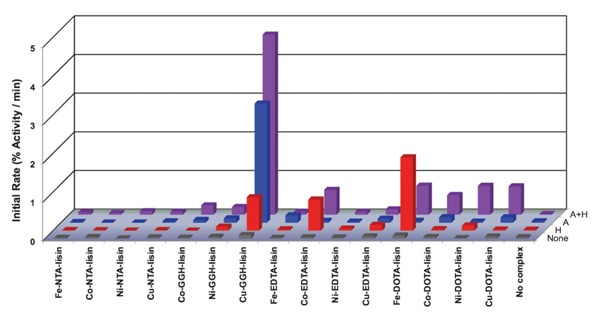
Once the IC50 values for binding of each M-chelate-lisinopril to sACE-1 were established, time-dependent catalytic inactivation of sACE-1 by each M-chelate-lisinopril complex was studied by incubating each complex with sACE-1 in the presence of coreactants and assaying the substrate cleavage activity of sACE-1 at various time intervals during each incubation (Figure 16). Catalytic inactivation of sACE-1 promoted by each M-chelate-lisinopril complex was tested aerobically under a variety of coreactant conditions that included 1 mM ascorbate + 1 mM H2O2, 1 mM ascorbate, 1 mM H2O2, and with no added coreactants. The initial rates of inactivation of sACE-1 by each M-chelate-lisinopril catalyst (with each combination of coreactants) are shown in Figure 17.
Figure 16. Time-dependent inactivation (A) and cleavage (B) of full length sACE-1 by Cu-GGH-lisinopril with the coreactants 1 mM ascorbate and 1 mM H2O2 (circles), 1 mM ascorbate (triangles), 1 mM H2O2 (inverted triangles), or no coreactant (lozenges) (17). Time-dependent cleavage of sACE-1 was quantified by SDS-PAGE (C), shown here for the reaction of Cu-GGH-lisinopril with ascorbate and H2O2.
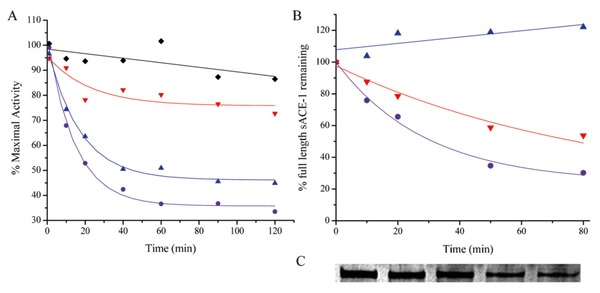
Figure 17. Initial rates for inactivation of sACE-1 by M-chelate-lisinopril complexes with the coreactants 1 mM ascorbate and 1 mM H2O2 (A + H), 1 mM ascorbate (A), 1 mM H2O2 (H), or no coreactant (None) (17).
Control experiments were also performed for M-chelates lacking attached lisinopril. Comparison of the second-order rate constants for inactivation of sACE-1 by M-chelate-lisinopril catalysts and by M-chelates lacking lisinopril confirms the critical importance of the attachment to lisinopril in order for each catalyst to irreversibly inactivate sACE-1 (Table 4). The second-order rate constants for sACE-1 inactivation were typically increased by at least 10-fold upon attachment to lisinopril, reflecting the requirement for active-site binding in order for each catalyst to efficiently inactivate sACE-1.
Initial rates of cleavage of the backbone of sACE-1 by each catalyst were separately monitored by SDS-PAGE, under similar conditions, so that the degree to which the observed inactivation of sACE-1 arose from either modification of amino acid side chains or cleavage of the sACE-1 backbone could be assessed. In general, it was found that inactivation was mediated primarily by modification of amino acid side chains, since the rate of backbone cleavage was typically only a fraction of the total rate of inactivation by each catalyst (Table 4). Following 14-h incubations of sACE-1 with each catalyst and coreactants, comparison of the overall decrease in kcat/KM (for substrate turnover by sACE-1) and the overall portion of sACE-1 that was cleaved (monitored by SDS-PAGE) further confirmed that modification of sACE-1 amino acid side chains was the primary mechanism of inactivation for each catalyst, consistent with other studies of metal-catalyzed oxidation of proteins (6, 76, 77).
As observed for the nuclease activity of M-chelate-Rev catalysts, certain relationships were observed between the reduction potential of the attached M-chelate and the ability of each M-chelate-lisinopril catalyst to mediate inactivation of sACE-1 (Figure 18B). However, whereas the nuclease activity of Rev-coupled M-chelates was highest for M-chelates with reduction potentials between -66 and +380 mV, the most rapid inactivation of sACE-1 by M-chelate-lisinopril complexes was observed for M-chelates with reduction potentials near +1000 mV (Cu-GGH-lisinopril and Ni-GGH-lisinopril). This contrast most likely arises due to the increased difficulty of oxidation of protein side chains, relative to the (deoxy)ribose rings of DNA and RNA, and suggests a unique mechanism of oxidation for the catalysts containing metal complexes of the naturally occurring ATCUN motifs such as M-GGH. The natural in vivo abundance of ATCUN motifs in albumins (78-81), histatins (82, 83), and neuromedin C (84), typically used for Cu- and Ni transport, as well as selective oxidation chemistry, most likely reflects both the placement of the Cu-ATCUN and Ni-ATCUN reduction potentials outside of the typical physiological window and the lack of diffusible ROS produced by these complexes (Figure 2B).
Figure 18. The relationship between the rate of catalyst-mediated inactivation and/or cleavage on the catalyst reduction potential depended on both the nature of the target (nucleic acid vs protein) and reactivity with the principle redox coreactants (17, 18). A, Cleavage of HIV Rev response element (RRE) RNA by M-chelate-Rev catalysts. B, Inactivation of sACE-1 by M-chelate-lisinopril catalysts. For RNA and DNA cleavage, rates were highest for reduction potentials (low potential region) between those of H2O2/hydroxyl radical (E° = +380 mV) and ascorbyl radical/ascorbate (E° = -66 mV), shown by the dashed lines, for which multiple turnovers (and generation of reactive oxygen species) were thermodynamically favored. For inactivation of the enzyme sACE-1, rates were highest for M-chelates with reduction potentials near 1000 mV (high region).
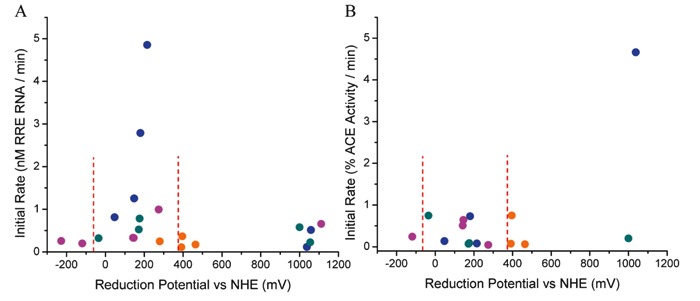
These M-chelate-lisinopril catalysts possess the advantage of irreversible catalytic inactivation of sACE-1, in contrast to the reversible mode of inhibition for lisinopril and other reversible inhibitors, and demonstrate how catalyst parameters such as reduction potential, redox reactivity, coordination unsaturation, geometric alignment, size, and charge may be tuned to optimize catalytic efficiency. These catalysts provide a useful starting point in the development of more efficient catalytic metallodrugs for inactivation of sACE-1 as well as for catalysts directed toward other therapeutic targets.
Footnotes
First published online July 3, 2013.
Presented at the XLI Annual Meeting of the Sociedade Brasileira de Bioquímica e Biologia Molecular, Foz do Iguaçu, PR, Brazil, May 19-22, 2012.
References
- 1.Hocharoen L, Cowan JA. Metallotherapeutics: novel strategies in drug design. Chemistry. 2009;15:8670–8676. doi: 10.1002/chem.200900821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowan JA. Catalytic metallodrugs. Pure Appl Chem. 2008;80:1799–1810. doi: 10.1351/pac200880081799. [DOI] [Google Scholar]
- 3.Bradford S, Cowan JA. Catalytic metallodrugs targeting HCV IRES RNA. Chem Commun. 2012;48:3118–3120. doi: 10.1039/c2cc17377h. [DOI] [PubMed] [Google Scholar]
- 4.Joyner JC, Keuper KD, Cowan JA. DNA nuclease activity of Rev-coupled transition metal chelates. Dalton Trans. 2012;41:6567–6578. doi: 10.1039/c2dt00026a. [DOI] [PubMed] [Google Scholar]
- 5.Gokhale NH, Cowan JA. Metallopeptide-promoted inactivation of angiotensin-converting enzyme and endothelin-converting enzyme 1: Toward dual-action therapeutics. J Biol Inorg Chem. 2006;11:937–947. doi: 10.1007/s00775-006-0145-2. [DOI] [PubMed] [Google Scholar]
- 6.Gokhale NH, Bradford S, Cowan JA. Catalytic inactivation of human carbonic anhydrase I by a metallopeptide-sulfonamide conjugate is mediated by oxidation of active site residues. J Am Chem Soc. 2008;130:2388–2389. doi: 10.1021/ja0778038. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Udugamasooriya DG, Lim HS, Kodadek T. Potent and selective photo-inactivation of proteins with peptoid-ruthenium conjugates. Nat Chem Biol. 2010;6:258–260. doi: 10.1038/nchembio.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallagher J, Zelenko O, Walts AD, Sigman DS. Protease activity of 1,10-phenanthroline-copper(I). Targeted scission of the catalytic site of carbonic anhydrase. Biochemistry. 1998;37:2096–2104. doi: 10.1021/bi971565j. [DOI] [PubMed] [Google Scholar]
- 9.Suh J, Yoo SH, Kim MG, Jeong K, Ahn JY, Kim MS, et al. Cleavage agents for soluble oligomers of amyloid beta peptides. Angew Chem Int Ed Engl. 2007;46:7064–7067. doi: 10.1002/anie.200702399. [DOI] [PubMed] [Google Scholar]
- 10.Kim MG, Kim MS, Lee SD, Suh J. Peptide-cleaving catalyst selective for melanin-concentrating hormone: Oxidative decarboxylation of N-terminal aspartate catalyzed by Co(III)cyclen. J Biol Inorg Chem. 2006;11:867–875. doi: 10.1007/s00775-006-0139-0. [DOI] [PubMed] [Google Scholar]
- 11.Suh J, Chei WS. Metal complexes as artificial proteases: toward catalytic drugs. Curr Opin Chem Biol. 2008;12:207–213. doi: 10.1016/j.cbpa.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 12.Jeong K, Chung WY, Kye YS, Kim D. Cu(II) cyclen cleavage agent for human islet amyloid peptide. Bioorg Med Chem. 2010;18:2598–2601. doi: 10.1016/j.bmc.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 13.Chei WS, Ju H, Suh J. New chelating ligands for Co(III)-based peptide-cleaving catalysts selective for pathogenic proteins of amyloidoses. J Biol Inorg Chem. 2011;16:511–519. doi: 10.1007/s00775-010-0750-y. [DOI] [PubMed] [Google Scholar]
- 14.Suh J, Chei WS, Lee TY, Kim MG, Yoo SH, Jeong K, et al. Cleavage agents for soluble oligomers of human islet amyloid polypeptide. J Biol Inorg Chem. 2008;13:693–701. doi: 10.1007/s00775-008-0354-y. [DOI] [PubMed] [Google Scholar]
- 15.Meggers E. Targeting proteins with metal complexes. Chem Commun. 2009:1001–1010. doi: 10.1039/b813568a. [DOI] [PubMed] [Google Scholar]
- 16.Brown KC, Yang SH, Kodadek T. Highly specific oxidative cross-linking of proteins mediated by a nickel-peptide complex. Biochemistry. 1995;34:4733–4739. doi: 10.1021/bi00014a030. [DOI] [PubMed] [Google Scholar]
- 17.Joyner JC, Hocharoen L, Cowan JA. Targeted catalytic inactivation of angiotensin converting enzyme by lisinopril-coupled transition-metal chelates. J Am Chem Soc. 2012;134:3396–3410. doi: 10.1021/ja208791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joyner JC, Cowan JA. Targeted cleavage of HIV RRE RNA by Rev-coupled transition metal chelates. J Am Chem Soc. 2011;133:9912–9922. doi: 10.1021/ja203057z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joyner JC, Reichfield J, Cowan JA. Factors influencing the DNA nuclease activity of iron, cobalt, nickel, and copper chelates. J Am Chem Soc. 2011;133:15613–15626. doi: 10.1021/ja2052599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradford S, Kawarasaki Y, Cowan JA. Copper.Lys-Gly-His-Lys mediated cleavage of tRNA(Phe): studies of reaction mechanism and cleavage specificity. J Inorg Biochem. 2009;103:871–875. doi: 10.1016/j.jinorgbio.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Jin Y, Cowan JA. DNA cleavage by copper-ATCUN complexes. Factors influencing cleavage mechanism and linearization of dsDNA. J Am Chem Soc. 2005;127:8408–8415. doi: 10.1021/ja0503985. [DOI] [PubMed] [Google Scholar]
- 22.Jin Y, Cowan JA. Targeted cleavage of HIV rev response element RNA by metallopeptide complexes. J Am Chem Soc. 2006;128:410–411. doi: 10.1021/ja055272m. [DOI] [PubMed] [Google Scholar]
- 23.Jin Y, Cowan JA. Cellular activity of Rev response element RNA targeting metallopeptides. J Biol Inorg Chem. 2007;12:637–644. doi: 10.1007/s00775-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 24.Jin Y, Lewis MA, Gokhale NH, Long EC, Cowan JA. Influence of stereochemistry and redox potentials on the single- and double-strand DNA cleavage efficiency of Cu(II) and Ni(II) Lys-Gly-His-derived ATCUN metallopeptides. J Am Chem Soc. 2007;129:8353–8361. doi: 10.1021/ja0705083. [DOI] [PubMed] [Google Scholar]
- 25.Sigman DS. Chemical nucleases. Biochemistry. 1990;29:9097–9105. doi: 10.1021/bi00491a001. [DOI] [PubMed] [Google Scholar]
- 26.Sigman DS, Bruice T, Mazumder A, Sutton CL. Targeted chemical nucleases. Acc Chem Res. 1993;26:98–104. doi: 10.1021/ar00027a004. [DOI] [Google Scholar]
- 27.Joyner JC, Hodnick WF, Cowan AS, Tamuly D, Boyd R, Cowan JA. Antimicrobial metallopeptides with broad nuclease and ribonuclease activity. Chem Commun. 2013;49:2118–2120. doi: 10.1039/c3cc38977d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzsimons MP, Barton JK. Design of a synthetic nuclease: DNA hydrolysis by a zinc-binding peptide pethered to a rhodium intercalator. J Am Chem Soc. 1997;119:3379–3380. doi: 10.1021/ja9633981. [DOI] [Google Scholar]
- 29.Jeung CS, Kim CH, Min K, Suh SW, Suh J. Hydrolysis of plasmid DNA catalyzed by Co(III) complex of cyclen attached to polystyrene. Bioorg Med Chem Lett. 2001;11:2401–2404. doi: 10.1016/S0960-894X(01)00439-5. [DOI] [PubMed] [Google Scholar]
- 30.Matsumura K, Komiyama M. Enormously fast RNA hydrolysis by lanthanide(III) ions under physiological conditions: eminent candidates for novel tools of biotechnology. J Biochem. 1997;122:387–394. doi: 10.1093/oxfordjournals.jbchem.a021765. [DOI] [PubMed] [Google Scholar]
- 31.Pei D, Corey DR, Schultz PG. Site-specific cleavage of duplex DNA by a semisynthetic nuclease via triple-helix formation. Proc Natl Acad Sci U S A. 1990;87:9858–9862. doi: 10.1073/pnas.87.24.9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pei D, Schultz PG. Artificial nucleases. In: Linn SM, editor. Nucleases. New York: Cold Spring Harbor Laboratory Press; 1993. pp. 317–340. [Google Scholar]
- 33.Joyce LE, Aguirre JD, Angeles-Boza AM, Chouai A, Fu PK, Dunbar KR, et al. Photophysical properties, DNA photocleavage, and photocytotoxicity of a series of dppn dirhodium(II,II) complexes. Inorg Chem. 2010;49:5371–5376. doi: 10.1021/ic100588d. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y, Joyce LE, Dickson NM, Turro C. DNA photocleavage by an osmium(II) complex in the PDT window. Chem Commun. 2010;46:6759–6761. doi: 10.1039/c0cc02571b. [DOI] [PubMed] [Google Scholar]
- 35.Magda D, Wright M, Crofts S, Lin A, Sessler JL. Metal complex conjugates of antisense DNA which display ribozyme-like activity. J Am Chem Soc. 1997:6947–6948. doi: 10.1021/ja9711223. [DOI] [Google Scholar]
- 36.Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. (Review) Oncol Rep. 2003;10:1663–1682. [PubMed] [Google Scholar]
- 37.Go RS, Adjei AA. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J Clin Oncol. 1999;17:409–422. doi: 10.1200/JCO.1999.17.1.409. [DOI] [PubMed] [Google Scholar]
- 38.Crook TR, Souhami RL, McLean AE. Cytotoxicity, DNA cross-linking, and single strand breaks induced by activated cyclophosphamide and acrolein in human leukemia cells. Cancer Res. 1986;46:5029–5034. [PubMed] [Google Scholar]
- 39.Mattes WB, Hartley JA, Kohn KW. DNA sequence selectivity of guanine-N7 alkylation by nitrogen mustards. Nucleic Acids Res. 1986;14:2971–2987. doi: 10.1093/nar/14.7.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boger DL, Garbaccio RM. Shape-dependent catalysis: insights into the source of catalysis for the CC-1065 and duocarmycin DNA alkylation reaction. Ac Chem Res. 1999;32:1043–1052. doi: 10.1021/ar9800946. [DOI] [Google Scholar]
- 41.Minoshima M, Bando T, Shinohara K, Sugiyama H. Molecular design of sequence specific DNA alkylating agents. Nucleic Acids Symp Ser. 2009:69–70. doi: 10.1093/nass/nrp035. [DOI] [PubMed] [Google Scholar]
- 42.Graf E, Mahoney JR, Bryant RG, Eaton JW. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J Biol Chem. 1984;259:3620–3624. [PubMed] [Google Scholar]
- 43.Inoue S, Kawanishi S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987;47:6522–6527. [PubMed] [Google Scholar]
- 44.Oberley LW, Buettner GR. The production of hydroxyl radical by bleomycin and iron(II) FEBS Lett. 1979;97:47–49. doi: 10.1016/0014-5793(79)80049-6. [DOI] [PubMed] [Google Scholar]
- 45.Pou S, Bhan A, Bhadti VS, Wu SY, Hosmane RS, Rosen GM. The use of fluorophore-containing spin traps as potential probes to localize free radicals in cells with fluorescence imaging methods. FASEB J. 1995;9:1085–1090. doi: 10.1096/fasebj.9.11.7649408. [DOI] [PubMed] [Google Scholar]
- 46.Samuni A, Goldstein S, Russo A, Mitchell JB, Krishna MC, Neta P. Kinetics and mechanism of hydroxyl radical and OH-adduct radical reactions with nitroxides and with their hydroxylamines. J Am Chem Soc. 2002;124:8719–8724. doi: 10.1021/ja017587h. [DOI] [PubMed] [Google Scholar]
- 47.Matko J, Ohki K, Edidin M. Luminescence quenching by nitroxide spin labels in aqueous solution: studies on the mechanism of quenching. Biochemistry. 1992;31:703–711. doi: 10.1021/bi00118a010. [DOI] [PubMed] [Google Scholar]
- 48.Yu F, Xu D, Lei R, Li N, Li K. Free-radical scavenging capacity using the fenton reaction with rhodamine B as the spectrophotometric indicator. J Agric Food Chem. 2008;56:730–735. doi: 10.1021/jf072383r. [DOI] [PubMed] [Google Scholar]
- 49.Azab HA, Banci L, Borsari M, Luchinat C, Sola M, Viezzoli MS. Redox chemistry of superoxide dismutase. cyclic voltammetry of wild-type enzymes and mutants on functionally relevant residues. Inorg Chem. 1992;31:4649–4655. doi: 10.1021/ic00048a037. [DOI] [Google Scholar]
- 50.Barrette WC, Jr, Sawyer DT, Fee JA, Asada K. Potentiometric titrations and oxidation - reduction potentials of several iron superoxide dismutases. Biochemistry. 1983;22:624–627. doi: 10.1021/bi00272a015. [DOI] [PubMed] [Google Scholar]
- 51.Finnen D, Pinkerton A, Dunham WR, Sands RH, Funk MO. Structures and spectroscopic characteristics of iron(III) diethylenetriaminepentaacetic acid complexes. A non-heme iron(III) complex with relevance to the iron environment in lipoxygenases. Inorg Chem. 1990;30:3960–3964. doi: 10.1021/ic00020a034. [DOI] [Google Scholar]
- 52.Viola-Villegas N, Doyle R. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N′,N?,N′?-tetraacetic acid (H4DOTA): Structural overview and analyses on structure-stability relationships. Coordin Chem Rev. 2009;253:1906–1925. doi: 10.1016/j.ccr.2009.03.013. [DOI] [Google Scholar]
- 53.Lacourciere KA, Stivers JT, Marino JP. Mechanism of neomycin and Rev peptide binding to the Rev responsive element of HIV-1 as determined by fluorescence and NMR spectroscopy. Biochemistry. 2000;39:5630–5641. doi: 10.1021/bi992932p. [DOI] [PubMed] [Google Scholar]
- 54.Luedtke NW, Tor Y. Fluorescence-based methods for evaluating the RNA affinity and specificity of HIV-1 Rev-RRE inhibitors. Biopolymers. 2003;70:103–119. doi: 10.1002/bip.10428. [DOI] [PubMed] [Google Scholar]
- 55.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, et al. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 56.Joyner JC, Keuper KD, Cowan JA. Analysis of RNA cleavage by MALDI-TOF mass spectrometry. Nucleic Acids Res. 2013;41:e2. doi: 10.1093/nar/gks811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joyner JC, Keuper KD, Cowan JA. Kinetics and mechanisms of oxidative cleavage of HIV RRE RNA by Rev-coupled transition metal-chelates. Chem Sci. 2013;4:1707–1718. doi: 10.1039/c3sc22135k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pogozelski WK, Tullius TD. Oxidative Strand Scission of Nucleic Acids: Routes Initiated by Hydrogen Abstraction from the Sugar Moiety. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 59.Sreedhara A, Freed JD, Cowan JA. Efficient inorganic deoxyribonucleases. Greater than 50-million-fold rate enhancement in enzyme-like DNA cleavage. J Am Chem Soc. 2000;122:8814–8824. doi: 10.1021/ja994411v. [DOI] [Google Scholar]
- 60.Cowan JA. Inorganic biochemistry. An introduction. New York: Wiley-VCH; 1997. [Google Scholar]
- 61.Reedijk J. Mechanistic studies of Pt and Ru compounds with antitumor properties. Med Inorg Chem. 2005:80–109. [Google Scholar]
- 62.Liu HK, Sadler PJ. Metal complexes as DNA intercalators. Acc Chem Res. 2011;44:349–359. doi: 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
- 63.Wheate NJ, Brodie CR, Collins JG, Kemp S, Aldrich-Wright JR. DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev Med Chem. 2007;7:627–648. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]
- 64.Baum C, von Kalle C, Staal FJ, Li Z, Fehse B, Schmidt M, et al. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol Ther. 2004;9:5–13. doi: 10.1016/j.ymthe.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 65.Hacein-Bey-Abina S, von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 66.Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97:1411–1420. doi: 10.1161/01.CIR.97.14.1411. [DOI] [PubMed] [Google Scholar]
- 67.Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdos EG. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1-7) and keto-ACE. Hypertension. 1998;31:912–917. doi: 10.1161/01.HYP.31.4.912. [DOI] [PubMed] [Google Scholar]
- 68.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension. 1998;31:362–367. doi: 10.1161/01.HYP.31.1.362. [DOI] [PubMed] [Google Scholar]
- 69.Georgiadis D, Beau F, Czarny B, Cotton J, Yiotakis A, Dive V. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ Res. 2003;93:148–154. doi: 10.1161/01.RES.0000081593.33848.FC. [DOI] [PubMed] [Google Scholar]
- 70.Lonn EM, Yusuf S, Jha P, Montague TJ, Teo KK, Benedict CR, et al. Emerging role of angiotensin-converting enzyme inhibitors in cardiac and vascular protection. Circulation. 1994;90:2056–2069. doi: 10.1161/01.CIR.90.4.2056. [DOI] [PubMed] [Google Scholar]
- 71.Bernstein KE, Welsh SL, Inman JK. A deeply recessed active site in angiotensin-converting enzyme is indicated from the binding characteristics of biotin-spacer-inhibitor reagents. Biochem Biophys Res Commun. 1990;167:310–316. doi: 10.1016/0006-291X(90)91766-L. [DOI] [PubMed] [Google Scholar]
- 72.Corradi HR, Schwager SL, Nchinda AT, Sturrock ED, Acharya KR. Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J Mol Biol. 2006;357:964–974. doi: 10.1016/j.jmb.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 73.Ehlers MR, Riordan JF. Angiotensin-converting enzyme: zinc- and inhibitor-binding stoichiometries of the somatic and testis isozymes. Biochemistry. 1991;30:7118–7126. doi: 10.1021/bi00243a012. [DOI] [PubMed] [Google Scholar]
- 74.Michaud A, Williams TA, Chauvet MT, Corvol P. Substrate dependence of angiotensin I-converting enzyme inhibition: captopril displays a partial selectivity for inhibition of N-acetyl-seryl-aspartyl-lysyl-proline hydrolysis compared with that of angiotensin I. Mol Pharmacol. 1997;51:1070–1076. doi: 10.1124/mol.51.6.1070. [DOI] [PubMed] [Google Scholar]
- 75.Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 76.Amici A, Levine RL, Tsai L, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- 77.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 78.Bradshaw RA, Peters T., Jr The amino acid sequence of peptide (1-24) of rat and human serum albumins. J Biol Chem. 1969;244:5582–5589. [PubMed] [Google Scholar]
- 79.Camerman N, Camerman A, Sarkar B. Molecular design to mimic the copper (II) transport site of human albumin. Can J Chem. 1976 [Google Scholar]
- 80.Lau SJ, Sarkar B. Ternary coordination complex between human serum albumin, copper (II), and L-histidine. J Biol Chem. 1971;246:5938–5943. [PubMed] [Google Scholar]
- 81.Lau SJ, Kruck TP, Sarkar B. A peptide molecule mimicking the copper(II) transport site of human serum albumin. A comparative study between the synthetic site and albumin. J Biol Chem. 1974;249:5878–5884. [PubMed] [Google Scholar]
- 82.Cabras T, Patamia M, Melino S, Inzitari R, Messana I, Castagnola M, et al. Pro-oxidant activity of histatin 5 related Cu(II)-model peptide probed by mass spectrometry. Biochem Biophys Res Commun. 2007;358:277–284. doi: 10.1016/j.bbrc.2007.04.121. [DOI] [PubMed] [Google Scholar]
- 83.Grogan J, McKnight CJ, Troxler RF, Oppenheim FG. Zinc and copper bind to unique sites of histatin 5. FEBS Lett. 2001;491:76–80. doi: 10.1016/S0014-5793(01)02157-3. [DOI] [PubMed] [Google Scholar]
- 84.Harford C, Sarkar B. Neuromedin C binds Cu(II) and Ni(II) via the ATCUN motif: implications for the CNS and cancer growth. Biochem Biophys Res Commun. 1995;209:877–882. doi: 10.1006/bbrc.1995.1580. [DOI] [PubMed] [Google Scholar]