

Abstract
Chemoresistance due to heterogeneity of the tumor microenvironment (TME) hampers the long-term efficacy of frontline therapies for lung cancer. Current combination therapies for lung cancer provide only modest improvement in survival, implicating necessity for novel approaches that suppress malignant growth and stimulate long-term anti-tumor immunity. Oxidative stress in the TME promotes immunosuppression by tumor infiltrating myeloid-derived suppressor cells (MDSC), which inhibit host protective anti-tumor immunity. Using a murine model of lung cancer, we demonstrate that a combination treatment with gemcitabine and a superoxide dismutase mimetic targets immunosuppressive MDSC in the TME and enhances the quantity and quality of both effector and memory CD8+ T cell responses. At the effector cell function level, the unique combination therapy targeting MDSC and redox signaling greatly enhanced cytolytic CD8+ T cell response and further decreased T regulatory cell infiltration. For long-term anti-tumor effects, this therapy altered the metabolism of memory cells with self-renewing phenotype and provided a preferential advantage for survival of memory subsets with long-term efficacy and persistence. Adoptive transfer of memory cells from this combination therapy prolonged survival of tumor-bearing recipients. Furthermore, the adoptively-transferred memory cells responded to tumor re-challenge exerting long-term persistence. This approach offers a new paradigm to inhibit immunosuppression by direct targeting of MDSC function, generate effector and persistent memory cells for tumor eradication, and prevent lung cancer relapse.
Keywords: Lung cancer, MDSC, Memory response, Combination therapy
INTRODUCTION
Lung cancer is the primary cause of cancer-related death in both men and women with an overall 5-year survival rate of 15% (1, 2). Current front-line therapies often involve chemotherapeutic combinations that target the heterogeneity of signaling pathways active among malignant cell populations. Nevertheless, chemoresistance of tumor cells continues to pose a significant challenge for these strategies to be efficacious in prolonging patient survival. Hence, multi-modal therapies that also stimulate anti-tumor immune responses are essential for long-term management of this disease (3).
Oxidative stress, resulting from elevated reactive oxygen species (ROS) is implicated in the initiation and progression of lung cancer (4). The host protective anti-tumor immunity is also often suppressed by several non-malignant leukocyte lineages in the tumor microenvironment (TME) which significantly dampen the long-term efficacy of existing combination therapies for lung cancer (5, 6). Myeloid-derived suppressor cells (MDSC) generate ROS, associated free radicals and immunoregulatory cytokines to suppress host CD4+ and CD8+ T cell responses, thereby promoting tumor progression and metastasis (7–9). In patients with advanced stages of lung cancer, increased numbers of circulating MDSC correlate with immunosuppression and enhanced tumor burden (10). Despite the documented roles of these immunosuppressive cells in lung cancer, few immunomodulatory therapies have been successful at targeting MDSC to enhance long-term immunity. Development of treatment resistance following gemcitabine (Gem) therapy and elevation of ROS in the TME may impact infiltrating immune effector function, induce MDSC proliferation and impair memory response, which is a critical component of protective immunity. We hypothesized, therefore, that a timely uncoupling of the MDSC-induced immunosuppression in the TME and ROS would not only enhance effects of Gem but also reactivate host anti-tumor immunity including both effector and memory functions and significantly enhance survival outcomes.
Herein, we report an innovative therapeutic strategy that both impairs MDSC function and induces effective memory T-cell responses against lung tumors. Using a syngeneic lung cancer mouse model, we demonstrate that Gem, a well-established chemotherapy agent for lung cancer and a known inhibitor of MDSC expansion, along with superoxide dismutase mimetic (SOD mim), reduced tumor growth and resulted in long term immunity and survival. This combination therapy enhanced the quantity and quality of the effector and memory CD8+ T cell responses with an enrichment of self-renewing memory cells. Cellular redox–mediated regulation of STAT-3 activation and metabolism of the central memory and effector memory CD8+ T cells contributed to the long term immunity. Further, adoptive transfer experiments demonstrated that inhibition of MDSC and MDSC-generated ROS enhanced the persistence of memory CD8+ T cells, as well as their vigorous activity in response to re-encountering tumor antigens as in stages of relapse. This study clearly delineates the role of MDSC in lung cancer progression and demonstrates utility of this combination therapy for inhibiting tumor and MDSC expansion, abrogating their key molecular functions/pathways and offers a new strategy for treatment of early lung cancer and prevention of relapse.
MATERIALS AND METHODS
Syngeneic Orthotopic Lung Cancer Model
The murine Lewis Lung Carcinoma (LLC) cell line (ATCC) was propagated in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 10 µg/ml penicillin-streptomycin and 0.1 mM non-essential amino acids (Life Technologies, Carlsbad, CA) LLC cells (106) were injected either intravenously (i.v.), via tail-vein or via an intra-cardiac (i.c.) route in syngeneic, 6–8 weeks old female C57BL/6 mice (Frederick Cancer Research and Development Center, MD). Analyses included assessment of tumor growth and survival. Tumors, lungs and spleen tissues were used for enumeration and characterization of MDSC, regulatory T cells and CD8+ T cell memory subsets.
In vivo treatment regimen
LLC-challenged mice were treated with Gemcitabine (Gem) and a Superoxide dismutase mimetic (SOD mim) either individually or in combination (see the treatment model in Figure 2). Five days post tumor challenge via an i.v. route and 3 days post tumor challenge via an i.c. route, mice were injected intraperitoneally with either PBS or 60 mg/kg Gem (Sigma-Aldrich, St. Louis, MO) in 50 µl/ mouse and 10mg/kg SOD mim (MnTE-2-PyP5+ (manganese (III) mesotetrakis (di-N-diethylimidazole) porphyrin), generously provided by Dr. James Crapo (National Jewish Hospital, Denver, CO), in 100 µl/ mouse, twice in the 1st week and once in the following week. Alternatively, LLC-challenged mice received anti-Gr1 antibody (250 µg/100µl) (11) or IgG control (BioXCell, West Lebanon, NH) intraperitoneally at day 5 either individually or in combination with SOD mim. Catalase deficient mice (a kind gift from Dr. Jaroslaw Zmijewski, UAB) were also challenged with tumor and treated as described above.
Figure 2. Combination therapy targeted MDSC recruitment efficiently and prolonged survival of mice by reducing tumor burden.

(a) Our model depicting the time-line of tumor challenge, therapy and analysis. (b) Kaplan-Meier survival curve displaying percent survival among treatment groups after tumor challenge and treatment (n = 6 mice/group). Statistical significance was determined by Mantel-Cox log-rank test (*p<0.05 for PBS vs SOD mim, *** p<0.001 for PBS vs Gem, ***p<0.001 for PBS vs SOD mim + Gem, **p<0.01 for Gem vs SOD mim+Gem, ***p<0.001 for SOD mim vs SOD mim+ Gem and ***p<0.001 for SOD mim vs Gem) (c) FACS plot showing MDSC infiltration in tumor, lung and spleen of mice from all four treatment groups, **p<0.01 for treatment groups SOD mim + Gem vs Gem, for comparisons of treatment groups Gem vs SOD mim and for all three treatments compared to PBS controls (n= 4 mice/group, 4 replicate experiments) (d) Percent ROS+ MDSC and non-MDSC populations were detected by Dihydroethidium (an indicator of ROS) by flow cytometry within the lung, spleen, and tumor tissues for mice from all the 4 treatment groups. Percent ROS+ MDSC in tumor samples- **p<0.01 for comparisons of treatment groups SOD mim + Gem vs Gem, SOD mim vs PBS and *p<0.05 SOD mim vs Gem, Spleen samples- **p<0.01 SOD mim + Gem vs Gem. Percent Non-MDSC ROS+ cells in tumor samples- **p<0.01 for SOD mim & SOD mim + Gem vs Gem, Lung samples- **p<0.01 SOD mim + Gem compared to SOD mim, Gem and PBS groups. (e) Representative FACS plots showing percent of CD4+FoxP3+ cells in tumor tissues of mice from all the treatment groups, *p<0.05 for comparisons of SOD mim + Gem and SOD mim vs Gem and PBS controls.
Isolation of immune cells and FACS analysis
Infiltrating leukocytes were isolated from minced tumor and lung tissues from LLC-challenged mice by incubation in DMEM containing collagenase-B (2 mg/ml, Roche) and DNase I (0.02 mg/ml, Sigma Chemical) at 37°C for 30 min. This was followed by the addition of an equal volume of complete DMEM containing 10% FBS (Please see Supplementary Methods for additional details). ROS+ cells in tumor tissues were detected by flow cytometry as described before (12). To quantitate the percentage of apoptotic cells, CD8+ T cells from the tumor tissue were stained by a FITC-Annexin V Apoptosis detection kit (BD Biosciences). Fifty × 103 events were collected for all analyses using the BD LSRII cytometer (BD Biosciences) and the data were analyzed using FlowJo software.
Immunofluorescence Microscopy
Detailed procedures for detection of MDSC and CD8+ T cells in snap frozen tumor tissues are provided in Supplementary Methods.
Cytotoxicity assay
CD8+ T cell memory subsets and effector cells were isolated from LLC-challenged mice with gemcitabine and SOD mimetic treatment as reviewed previously and then used as the effector cells (E). LLC cells were used as the target population (T). The assay was set up with E:T ratios of 5:1, 10:1, 20:1 and 40:1. The cytotoxicity assay was performed using the LIVE/DEAD cell-mediated cytotoxicity kit (Molecular Probes, Eugene, OR). DiOC18+PI+ cells were identified as dead target cells and DiOC18+PI− cells were identified as live target cells by flow cytometry analysis. The percent cytotoxicity was calculated following manufacturer’s guidelines as [(Dead Cells/Live Cells+effectors – (Dead Cells/Live Cells–effectors] × 100.
Detection of thiols
Cell surface thiols on CD8+ T cells from tumor tissues of LLC-challenged mice treated with gemcitabine and SOD mimetic were detected by staining with 5 µM Alexa Fluor 633-coupled maleimeide (ALM-633) (Life Technologies, Grand Island, NY) for 15 minutes on ice, washed with PBS and analyzed by flow cytometry.
Metabolomic Analysis
CD8+ T cell memory cell subsets were immunopurified from tumor tissues of LLC-challenged mice following treatment as described above. Cells (100 mg wet weight) were rinsed with ice-cold, isotonic PBS twice and the cell pellet was resuspended in cold methanol (−45°C in dry ice). The methanol slurry was then transferred to a cooled centrifuged tube and centrifuged at 14,000 rpm at −20°C for 10 min. The methanol extracts were evaporated to dryness, derivatized and reconstituted in 100 ul of water prior to analysis. Metabolites that contain an aldehyde or a ketone in their structure were derivatized using Amplifex (Applied Biosystem, Carlsbad, California). Liquid chromatography Mass Spectrometry-Multiple Reaction Monitoring analysis was then carried out with the mass spectrometer operating in both the positive and negative modes. Addition of amplifex adds an amine group to the carbonyl of the ketone/aldehyde which increases the mass of the compound by 115 amu. The LC system was reverse-phase with the Mobile phase A of 0.1% HCOOH in water and Mobile phase B of Methanol + 0.1% HCOOH. A Synergi Hydro-RP C18 column, 2×250 mm was used for analysis.
Phospho-STAT3 Flow Cytometry and ELISA
Detailed procedures for detection and quantitation of p-STAT-3 activation in CD8+ T cell memory cell subsets are provided in Supplementary Methods.
Isolation of CD8+ T cell memory populations and adoptive transfer
Detailed procedures for characterization, purification and in vitro expansion of CD8+ T cell memory populations are provided in the Supplementary Methods.
Statistical analysis
Data are represented as Mean ± SD. One way ANOVA with Tukey multiple comparison post-test and the Student’s t- test was used for statistical comparisons using Graph Pad Prizm5. Statistical significance was determined at the < 0.05 level (*p<0.05, **p< 0.01, and ***p<0.001). Kaplan-Meier method was used for evaluation of survival patterns in tumor-bearing mice. Results were ranked according to the Mantel-Cox Log-Rank test and p<0.05 was considered statistically significant. Survival was defined as time until death of mice due to excess tumor burden.
RESULTS
MDSC are elevated with a concomitant decrease in CD8+ T cells during progression of lung cancer
For delineating the role of MDSC in progression of lung cancer, a murine lung cancer model was established by intravenous injection (i.v.) of LLC cells (106 cells) in syngeneic C57BL/6 mice. A significant increase in tumor burden was observed by 15 days following in vivo establishment and progression of lung cancer (Fig. 1a-b, p<0.001 compared to early stage tumor burden).
Figure 1. Recruitment of MDSC were increased while the infiltration of CD8+ and CD4+ T cells were decreased with tumor progression.

(a) Tumor weights from mice on days 5, 10, 15, and 19 after i.v. challenge with 106 LLC tumor cells. ***p<0.001 in comparisons to day 5, day 10 compared to day 5, day 15 compared to day 10 and day 19 compared to day 15 (n=5 mice/time point, 3 replicate experiments). (b) H & E staining of lung tissue at indicated time points (c) FACS plots showing percentages of MDSC in tumor on days 10, 15 and 19 post-LLC injection, **p<0.01 day 19 vs day 15 and for day 10 vs day 15. (d) Characterization of MDSC subsets by flow cytometry using additional MDSC markers Ly-6C, Ly-6G and F4/80. (e) FACS plots showing CD8+ and CD4+ T cells in tumor at indicated times, left to right panels *p<0.05 for both CD4+ and CD8+ T cells, day 10 vs day15 vs day19 (n=5 mice/time point, 3 replicate experiments).
We first investigated the progression of tumor growth in the lungs and the significance of infiltrating immunosuppressive cells in the tumor microenvironment. Enumeration of immune cell phenotypes by flow cytometry demonstrated a increase in tumor infiltrating MDSC with increasing tumor growth (Figure 1C). The CD11bintGr-1int MDSC population stained positive for both Ly-6C and F4/80 (markers characteristic of monocytic phenotype of MDSC) whereas the CD11bhiGr-1hi MDSC population expressed both Ly-6G and F4/80 (markers characteristic of granulocytic phenotype of MDSC) (Fig. 1d). These MDSC subsets were also characterized in lung and spleen (Supplementary Fig. 1). As the numbers of MDSC increased with tumor burden, a significant reduction in CD8+ and CD4+ T cells was observed (Fig.1e, same time points as Fig. 1c, p<0.05 with increased tumor growth). Similar enhanced infiltration of MDSC and a steady decline in CD8+ T cells with tumor progression was also noted following intra-cardiac implantation of tumor cells (Supplementary Fig. 1c).
Treatment of tumor-bearing mice with gemcitabine and a SOD mimetic targets MDSC and reduces tumor progression
MDSC are negative regulators of protective anti-tumor immune responses in cancer (7, 8) and use ROS as their primary mechanism for immunosuppression. Therefore, we used Gemcitabine (Gem), a current frontline chemotherapy for lung cancer, to preferentially target and deplete proliferating MDSC (13–15) in combination with a superoxide dismutase mimetic (SOD mim) (16, 17) a metalloporphyrin catalytic anti-oxidant which scavenges ROS in the TME (see treatment model in Fig. 2a). As shown in Fig. 2b, combination therapy of SOD mim+Gem, significantly prolonged the survival of tumor-bearing mice compared to control and individual treatment groups (p<0.01 for Gem vs SOD mim+Gem, p<0.001 for PBS vs SOD mim + Gem, p<0.001 for SOD mim vs SOD mim+ Gem). Additionally, reduced tumor burden correlated with increased survival (Supplementary Fig. 2b).
A significant reduction in tumor infiltrating MDSC numbers was noted following combination therapy compared to all other treatment groups (Fig. 2c, p<0.01), with similar observations in lung and spleen tissues (Supplementary Fig. 2a). Further, ROS levels associated with MDSC and other ROS contributing immune cell types including tumor associated macrophages (TAM) and tumor associated neutrophils (TAN) were significantly reduced in the combination therapy group as compared to the PBS control group (Fig. 2d, p<0.01 for ROS+ MDSC and non-MDSC cells), reflecting an overall reduction of total ROS in TME. Although we observed a significant reduction in tumor infiltrating neutrophils (Supplementary Fig. 3a), macrophage infiltration was not modulated by combination therapy.
MDSC may also induce development and migration of regulatory T cells (Treg) to the TME, which can then inhibit anti-tumor responses and contribute to immunological tolerance in cancer (18). As shown in (Fig. 2e), combination therapy of SOD mim+Gem also significantly reduced the infiltration of Treg p<0.05).
Combination therapy with a SOD mimetic and Gemcitabine enhances the CD8+ T cell response
ROS mediated inhibition of CD8+ T cell response is the primary immunosuppressive mechanism of MDSC (19, 20). Since increased MDSC infiltration was associated with a reduction of CD8+T cells during tumor progression, we investigated whether depleting MDSC with combination therapy would modulate the CD8+ T cell response. The total percentages and absolute numbers of CD8+ T cells increased in tumor, lung and spleen tissues of mice treated with SOD mim+Gem (Fig. 3a & b) Immunohistochemical analysis further showed a significant increase in the infiltration of CD8+ T cells and a decrease in infiltration of Gr-1+ cells in the periphery as well as center of tumor tissue in mice treated with combination therapy as compared to controls (Fig. 3c). Furthermore, the combination therapy significantly reduced the percentages of apoptotic Annexin+ CD8+ T cells (Fig. 3d, p<0.001 in comparison to all other treatments and control groups).
Figure 3. SOD mim + Gem treatment results in enhanced CD8+ T cell recruitment and diminished presence of MDSC.

(a) The percentage of CD8+ T cells was determined by flow cytometric analyses of cells harvested from lung, spleen and tumor tissues. Live CD3+ cells in the lymphocyte gate were analyzed for percentage of CD8+ cells. ***p<0.001 when compared to all treatment groups as derived by ANOVA. (b) Total numbers of CD8+ T cells in lung, spleen and tumor tissues from each treatment group are presented. (c) Localization of Gr-1+ and CD8+ cells as determined by immunofluorescent analyses of OCT frozen tumor tissue stained with anti-Gr-1 and CD8 antibodies. (d) Percent Annexin+ CD8+ T cells as determined by flow cytometry of cells harvested from lung, spleen and tumor tissues. ***p<0.001 in comparison to all other treatment and control groups.
Combination therapy enhances the memory CD8+ T cell response
The effectiveness of CD8+ T cells to tumor challenge is dependent on state of their differentiation. We investigated whether the increased CD8+ T cell viability and cell numbers following targeted depletion of MDSC and ROS pathways reflected an increased differentiation into various effector and memory subsets. As shown in Fig. 4a, a significant and rapid increase in the percentages of effector (TEM), central (TCM), and stem cell (TSCM) memory CD8+T cells was observed with tumor progression (p< 0.001 for SOD mim+Gem vs Gem for TEM and TSCM). At 14 days after tumor establishment and 6–7 days after therapy, TEM and TSCM subsets were observed in increased numbers. However, the pool of CD8+ memory subsets changed 72 hours later, with significantly higher percentages of all three subsets (Fig. 4b, p<0.01 for SOD mim+Gem vs Gem for TEM and TSCM, p<0.05 for the same comparison for TCM). Increased TEM levels were also observed in the group treated with only gemcitabine. Further characterization of CD8+ T cells indicated that they expressed stem cell antigen- 1 (Sca-1) (Fig. 4c, p<0.001 for SOD mim+Gem compared to Gem and PBS, p<0.01 for SOD mim vs Gem) (21), a marker found on self-renewing CD8+ T cells capable of generating central and effector memory populations. Co-expression of high CD62L expression defined these cells as TSCM as shown in (Fig. 4d). The combination therapy, however, did not directly affect the viability of CD4+ or CD8+ T cells ex vivo (data not shown). Enhanced CD8+ memory T cell response was also noted following anti-Gr-1 mAb-mediated depletion of MDSC and targeting MDSC function with SOD mimetic (Supplementary Fig. 3b). We did not observe any enhanced targeting effects of SOD mim on LLC tumor cells in vitro as pretreatment of LLC tumor cells with increasing concentrations of SOD mim did not sensitize the cells further to treatment with Gem (Supplementary Fig. 3c). Combination treatment also reduced MDSC infiltration and ROS levels in the TME of catalase deficient mice, which normally have elevated ROS levels. In addition, it reduced the tumor burden (Supplemental Fig. 4a & d) and enhanced the memory response (increased percentages of TEMTCM and TSCM to levels noted in the WT mice (Supplemental Fig. 4b & c).
Figure 4. Combination therapy modulates the quantity of the memory CD8+ T cell response.

Stacked histogram plots display percentages of subsets within total CD8+ populations in the tumor tissue at days 14 (a) (p< 0.001 for SODmim+Gem vs Gem for TEM and TSCM) and 17 (b) p<0.01 for SODmim+Gem vs Gem for TEM and TSCM, p<0.05 for the same comparison for TCM after establishment of tumor. (c) Percent CD8+Sca-1+ T cells in different treatment groups are shown, p<0.001 for SODmim+Gem compared to Gem and PBS, p<0.01 for SOD mim vs Gem. (d) Representative FACS plots showing percent CD8+ T cells which are Sca-1+CD62L+ identified as TSCM.
Thiol dependent STAT-3 activation is enhanced in memory cells following combination therapy
Reduction of ROS levels is associated with reduced thiol groups on T cell surfaces which contribute to persistence of memory cells and protection from apoptotic cell death (22). TCM are reported to have more reduced thiols as compared to TEM and can withstand oxidative stress (22, 23). Therefore, we hypothesized that CD8+ T cells purified from the combination therapy treated mice may express high levels of thiols and therefore, are more effective in anti-tumor response. Based on maleimide reactivity, an increase in reduced thiols was detected in CD8+ T cells obtained from tumor, spleen and lungs of mice treated with combination therapy (Fig. 5a, p<0.01 for SOD mim+Gem vs Gem). Furthermore, phosphorylation of a thiol dependent transcription factor STAT-3 (24, 25), critical for memory CD8+ T cell function (26), was in memory cells following combination therapy. As demonstrated by phospho-flow analysis of memory subsets (Fig. 5b), increased levels of phosphorylated STAT-3 were detected in tumor-derived TEM and TCM obtained from mice given combination therapy when compared to Gem therapy alone. Additionally, the ratio of p-STAT-3 to total STAT-3 changed significantly in the memory cells as compared to effector cells and the percentage of phosphorylation was enhanced in the combination therapy group as compared to all other individual treatments (Fig. 5c, p<0.05 for SOD mim+Gem vs Gem for TEM and TCM). We then investigated whether the combination therapy triggers switching of the metabolic pathways which fuel the energy production required for function of the memory T cell subsets. LC-MS-MRM analysis of metabolites of these CD8+ T cell memory subsets (Fig.5d) indicated that both TEM and TCM subsets from combination therapy had a higher dependency on glycolysis compared to those from individual therapies alone. Thus the reduction of MDSC infiltration and MDSC-associated ROS altered the metabolic status of these tumor-specific CD8+ T cell memory subsets.
Figure 5. Combination therapy modulates redox mediated STAT-3 signaling and metabolic switching of memory CD8+T cell subsets.

(a) FACS plots of memory subsets stained with maleimide (detects cell surface thiols) and anti-CD8 antibody, **p<0.01 for comparison of treatment groups SOD mim+Gem vs Gem for lung, spleen and tumor tissues (n=3 mice/group, 4 replicate experiments) (b) Overlaid histograms of Phospho-flow cytometric analyses of TEM and TCM cells from Gem and SOD mim + Gem treatment groups stained with phospho-STAT-3 antibody. Gray tracings displaying the memory subsets from the SOD mim group. (c) Stacked plot graphs showing an increased percent of TCM with p-STAT3 as compared to total STAT-3. (d) Concentrations of metabolites of glycolysis and Krebs cycle detected in tumor-specific memory CD8+ T cell subsets analyzed by LC-MS-MRM in SOD mim, Gem and SOD mim + Gem treatment groups, **p<0.01 for treatment group S+G (SOD mim+Gem) compared to Gem alone (G) or SOD mim alone (S) for all metabolites (e) Cytotoxic activity was significant in CD8+ T cells from mice treated with CD8+ T Central Memory cells from SOD mim + Gem and from SOD mim alone. The cytotoxicity assay was carried out using a Live/Dead Cell-mediated cytotoxicity Kit (Molecular probes). LLC cells were labeled with DiOC18 and incubated for 1 hour with the effector memory CD8+ T cell subsets at the Effector:Target ratios described in the text. Percent cytotoxicity was determined by flow cytometric analysis as described in methods, p<0.05 SOD mim+Gem TCM compared to SOD mim+Gem TEM, p<0.05 SOD mim + Gem memory T cell subsets compared to Gem or SOD mim alone groups (n=3 mice/group, n=3 replicates/group).
Targeting MDSC and the ROS pathway enhances poly-functional activity and the cytotoxic potential of memory CD8+ T cells
We then investigated whether the quality of the CD8+ T cell response is modulated by the combination therapy. We first compared the cytotoxic potential of these tumor specific memory CD8+ T cells subsets. As shown in Fig. 5e, the cytolytic activity of both TEM and TCM derived from tumor tissues of SOD mim+Gem therapy mice was more potent against target lung cancer cells when compared to those obtained from mice receiving individual therapies (p<0.05). The TCM purified from the SOD mim therapy group were also cytolytic. Importantly, the CD8+ T cells purified from mice treated with combination therapy showed an enhancement of the polyfunctional response with increased percentages of IFN-γ, cytoplasmic perforin and IL-2+ CD8+ T cells (Fig. 6a-c, p<0.01 for IFN-γ expression, p<0.001 for perforin+ CD8+T cells, and IL-2+ CD8+ T cells from tumor). Thus, it is likely that the central role of establishing memory response following combination therapy was not only for long-term anti-tumor immune senescence but also against relapse following long-term remission.
Figure 6. Combination therapy increased the percentage of multi-functional CD8+ T cells producing IFN-γ, IL-2 and perforin.

(a) FACS plots showing IFN-g expression in CD8+ Perforin+ T cells in tumor tissue, *p<0.05 for comparisons of SOD mim+Gem vs Gem, **p<0.01 for comparisons of SOD mim+Gem and Gem vs SOD mim, and for all treatment groups compared to PBS controls (n=4 mice/group, 3 replicate experiments) (b) FACS plots showing the percentage of IL-2 secreting CD8+ Perforin+ T cells in tumor tissue, ***p#x0003C;0.001 for SOD mim+Gem compared to Gem or SOD mim alone (n=4 mice/group, 3 replicate experiments) (c) FACS plots of gated CD8+ T cells stained for intracellular expression of perforin in lung, spleen and tumor tissues of all treatment groups, lung samples: **p<0.01 for SOD mim+Gem compared to Gem, SOD mim alone or PBS controls, spleen samples: *p<0.05 for SOD mim+ Gem compared to all other treatment groups, tumor samples: ***p<0.001 for SOD mim+Gem compared to all other treatment groups and PBS controls (n=4 mice/group, 3 replicate experiments).
Adoptive transfer of memory CD8+T cells from therapy groups significantly improved long-term survival of mice bearing lung cancer
Since the increased infiltration of the memory CD8+ T cells following combination therapy lowered lung cancer progression, we investigated whether adoptive transfer of these memory cells would improve survival of mice with established lung tumors. For these studies, effector, TCM and TEM CD8+ T cells were purified from Gem only and SOD mim+Gem therapy groups and were adoptively transferred into tumor-bearing mice seven days post-lung cancer challenge (Fig. 7a). As shown in Fig. 7b, 80% of mice which received either TCM or TEM from the SOD mim+Gem group survived for up-to 80 days after lung cancer challenge as compared to 25 day mean survival of treated mice in control groups (p<0.001 for G+S- E CD8, G+S- TCM CD8, G+S- TEM CD8 and Gem- TEM CD8 compared to PBS group, p<0.05 for PBS group when compared to Gem- E CD8).
Figure 7. Central and effector memory CD8+ subsets from mice treated with combination therapy efficiently targets tumor cells and prolongs survival following adoptive therapy.
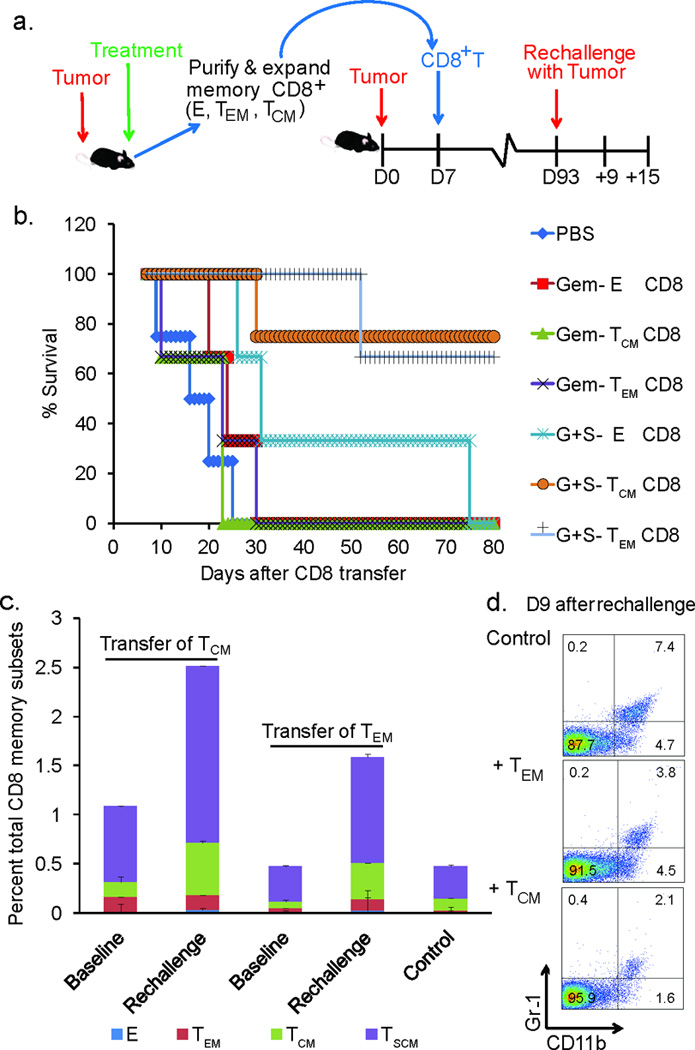
(a) Schematic showing the time line of adoptive transfer and tumor re-challenge. (b) A Kaplan-Meier survival curve showing an increase in percent survival of mice with established lung cancer after i.v. adoptive transfer with 105 indicated memory CD8+ T cell subsets (n=4–6 mice/group). Data shown are cumulative results from two independent experiments with five mice/group and analyzed by Mantel-Cox log-rank test (*p<0.05 for PBS vs Gem- E CD8, *p< 0.05 for PBS vs Gem- TEM CD8, ***p<0.001 for PBS vs G+S- E CD8, ***p<0.001 for PBS vs G+S- TCM CD8, and ***p<0.001 for PBS vs G+S- TEM CD8) (c) Stacked histogram depicting the percent of total CD8+ memory subsets at baseline and at Day 9 after re-challenge with tumor in adoptive transfer recipient mice with TCM or TEM transfer (n=3 mice/group, 2 replicate experiments, ***p<0.001 for TSCM and TCM in rechallenged groups compared to baseline and controls. (d) FACS plots of circulating Gr-1+ CD11b+ MDSC at day 9 after re-challenge in adoptive transfer recipients compared to tumor challenged controls, *p<0.05 TCM compared TEM and **p<0.01 TCM compared to controls (n=3 mice/group, 2 replicate experiments).
Mice which survived for up-to 80 days post-adoptive transfer were re-challenged with LLC cells to investigate the efficacy of these adoptively transferred memory subsets and their potential to be reactivated in the presence of tumor antigens. Enumeration of circulating memory CD8+ T memory subsets showed a significant increase in the TSCM population in re-challenged mice as compared to the baseline levels (levels of memory cells in adoptive transferred mice prior to lung cancer re-challenge) and in comparison to tumor challenged control mice (tumor bearing mice without CD8+ T cell adoptive transfer) (Fig. 7c and Supplemental Fig. 5, p<0.001 for TSCM and TCM in re-challenged groups compared to baseline and controls). These persistent memory CD8+ T cell subsets were efficient in maintaining reduced tumor burden. A reduction in infiltration of MDSC levels was also noted in the adoptive transfer recipients (Fig. 7d and Supplemental Fig. 6, p<0.05 and p<0.01 for TCM compared to TEM and control, respectively). In addition to the increased memory subsets in circulation, congenic adoptive transfer of memory subsets further confirmed the expansion of adoptively transferred CD8+ T cell memory subsets specifically in the tumor tissue (Supplemental Fig. 7).
Collectively, this study convincingly demonstrates that targeting the proliferation and immunosuppressive functions of MDSC promote anti-tumor immunity by enhancing the quantity and quality of the CD8+ T cell responses and promoting persistent immunological memory, thereby reducing tumor burden and prolonging survival of mice with lung cancer.
DISCUSSION
Recent evidence suggests that immune regulation contributed by MDSC in the TME dampens the long-term efficacy of existing combination chemotherapies for lung cancer (10). ROS produced by MDSC contributes to immunosuppression in the TME (7, 8). MDSC also replenish the tumor stroma with precursors of both TAM and TAN which contribute to oxidative stress in the TME (27). The multifaceted impact of oxidative stress in the TME is reflected not only in the impairment of T cell and NK cell activity, but also on redox mediated regulation of T cell signaling and T cell survival (22, 28). The differentiation state of the lymphocytes determines their function and persistence (29, 30). Mechanisms that modulate survival and function of T cell subsets, particularly in regard to their differential sensitivity to oxidative stress in the TME, have yet to be completely elucidated. This is of great relevance in the context of memory CD8+ T cells, which are a critical component of protective immunity against cancer. Differential sensitivity of memory cell phenotypes to oxidative stress may skew the memory repertoire to a particular subset leading to its persistence in TME. Therefore, strategies which eliminate MDSC-mediated immunosuppression in combination with anti-oxidant therapeutics to enhance the CD8+ T cell effector and memory response may be efficacious as novel anti-lung cancer regimens.
In this study, we provide evidence that gemcitabine when used in combination with an SOD mimetic, result in successful inhibition of MDSC, Treg and tumor associated neutrophil infiltration and a significant reduction of ROS in the TME (Figure 2). Inhibition of MDSC observed with this therapy is supported by recent studies which demonstrate efficacy of chemotherapeutic strategies in targeting MDSC (31–33). The observed reduction of infiltration of Treg is consistent with depletion of MDSC, as MDSC induce development (34) and promote migration and recruitment of Treg to sites of chronic inflammation (35). The reduction in neutrophils observed in the tumor tissue following combination therapy is consistent with the reduction in MDSC, since MDSC infiltration have been associated with increased TAN in the TME. The enhanced percentages and absolute numbers of CD8+ T cells and reduction in their apoptotic death following combination therapy (Figure 3) suggest a potential utility of this approach for reducing tumor burden and stimulating anti-tumor immunity by providing an ideal microenvironment for optimal T cell function.
Our novel observation that the combination therapy enhances both the quantity and quality of the memory response with polyfunctional and cytolytic activity, all support the potential of this strategy to promote T cell survival and function (Figure 4 & 5). In an MDSC and ROS depleted environment, the memory response to tumor challenge were skewed towards TCM, TEM and more interestingly, a subset TSCM with a stem cell memory phenotype (Figure 4). Similar observations noted with MDSC depletion using anti-Gr-1 Ab and SOD mim further validates these findings. TSCM CD8+ T cells have extensive replicative potential in vivo and maintain the naïve CD44−CD62L+ phenotype (30). These cells display the glycosylphosphatidyl inositol-linked molecule Sca-1, a marker for self-renewing cells (22, 29). Our studies showed a significant early increase in TEM and TSCM phenotypes followed by a shift in the expansion of both TCM and TSCM (Figure 4) as a late response to combination therapy. It is possible that redox regulation by MDSC may cause an intrinsic defect in the ability of TEM and TCM to proliferate in response to tumor challenge and this defect could be reversed or rectified by depletion of MDSC and associated ROS in the TME. Oxidative stress in the TME may also affect the differentiation pathways of these memory subsets and skew the memory repertoire toward subsets that are less sensitive to oxidative stress such as the TCM and TEM 22, 28). Depletion of ROS and MDSC from TME could then trigger the expansion of these resistant memory cell subsets, as well as TSCM (stem cell phenotype with high proliferative capacity). Consistent with these possibilities, our data suggest that combination therapy-based depletion of the excess ROS and MDSC enhanced the CD8+ T cell memory response, consistently skewing the memory repertoire to TCM and TEM cells, and increased numbers of TSCM (Figure 4). These observations were also noted in catalase deficient mice (Supplementary Figure 4). Our studies do not, however, delineate whether alterations in differentiation pathways of these T cell subsets account for the observed cellular redistribution in the memory compartment.
SOD mimetics are effective free radical scavengers in many disease models including those of stroke (36), diabetes (37) and sickle cell anemia (38). They have been shown to protect normal tissues against radiation-induced damage presumably by alleviating oxidative stress (39). In addition, there is mounting evidence that SOD mimetics may enhance tumor radioresponsiveness (40, 41) Although the SOD mim which we have used in our combination therapy does not enhance tumor responsiveness to chemotherapy, it offers significant advantages for adoptive cell therapy strategies as it enhances persistent anti-tumor immunity.
Our data suggest that modulation of redox regulation and signaling by gemcitabine and combination therapy may impact long lived memory populations. We show that lowered oxidative stress in the TME, resulting from depletion of MDSC and ROS, is associated with an increased number of cell surface thiols on TCM Figure 5). This is consistent with earlier reports that memory subsets resistant to activation induced cell death or apoptosis have increased thiols contributing to their persistence (22, 29). We do not anticipate any direct effects of SOD mim on thiols, but rather the combination therapy modulates upstream events resulting in a change in the redox status of the memory cells. The modulation of reduced thiols on T cell surface may reduce the threshold for T cell activation and enhanced their proliferative capacity in vivo. The observed increase in phosphorylation of thiol-dependent STAT-3 in TCM as compared to TEM (Figure 5b & c), may allow them to persist as memory cells, as STAT-3 activation is critical for maintenance of long-term memory (26).
Since metabolism underlies the functional capacity of T cells, their maintenance and their persistence during an immune response, therapeutic strategies that manipulate the T cell metabolism may alter the outcome of the anti-tumor response (42). This is particularly relevant in our observations that the combination therapy not only enhanced the CD8+ memory T cell response, but also modulated the metabolism of TCM and TEM subsets. These memory cells were glycolysis-dependent and relied on a more readily available ATP source than those resulting from individual therapies (Figure 5d). These observations are also consistent with the association of STAT-3 activation and persistence of TCM and TEM subsets in adoptive transfers and rechallenges with tumor cells. TCM and TEM subsets from combination therapy responded quickly and vigorously to the tumor rechallenge compared to controls. Adoptive transfer recipients of TCM purified from combination therapy treated mice were more efficient than TEM in eradicating tumor burden and prolonging survival (Figure 7). Additionally, rechallenging the surviving animals with a second round of tumor cells produced an increased expansion of TCM as compared to TEM, successful eradication of tumor burden and a reduced infiltration of MDSC (Figure 7). We have not delineated, however, whether the changes in the metabolic program of CD8+ memory T cell subsets triggered by combination therapy is to either increase energy production to cope with the increased demand to proliferate, to provide biosynthetic precursors or to generate reducing equivalents to balance the change in redox status in the TME.
We have not investigated the efficacy of these memory cells after repeated exposure to the antigen or their efficiency in targeting tumor cells following repeated cycles of adoptive transfer. Additionally, we have not addressed the potential of this strategy for small cell lung cancer, cancers other than lung cancers and/or other cancers that metastasize to the lung. Nevertheless, our investigations certainly suggest that depletion of MDSC and ROS may provide a preferential advantage for TCM to be more functionally active, proliferative and persistent in order to provide long-term immunity against lung cancer.
This is particularly relevant for adoptive cell transfer therapies that are being tested in early stage clinical trials for advanced cancer. There is also an impending need for novel effective approaches to generate persistent immunity in patients in whom the potential for complete elimination of tumor antigens are highly unlikely. Therefore, defining determinants for successful CD8+ T cell adoptive immunotherapy are essential. Our combination therapy offers an attractive strategy for adoptive T cell therapy in that it has potential to generate long-lived populations of TCM equipped with the dual potential of both immune surveillance, as well as tumor eradication. Additionally, this therapeutic strategy enhances memory cells with a self-renewing phenotype which may increase their long-term efficacy and persistence. We believe there is a potential role for this combination therapy as an adjuvant therapy in treatment strategies to eradicate early lung cancer and has implications for prevention of lung cancer relapse or recurrence.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Doyle Ray Moore and Dr. Stephen Barnes of the Targeted Metabolomics and Proteomics Laboratory, UAB for the metabolomic analyses included in this study. We thank Marion Spell (Center for AIDS Research Flow Facility Core for cell sorting) and Enid Keyser (Rheumatic Diseases Analytic and Preparative Cytometry Facility) for their technical support for Fluorescent Activated Cell Sorting.
GRANT SUPPORT: This study was supported by the Collaborative Development Grant for Immunology and Cancer Immunotherapeutics funded by the National Cancer Institute (NCI CA13148-39 to J.S.D. and S.P.), R01CA132077 and R01CA133737 to S.P. Funds for the operation of the Targeted Metabolomics and Proteomics Laboratory come in part from the UAB Center for Nutrient-Gene Interaction (U54 CA 100949), the Purdue-UAB Botanicals Center for Age-Related Disease (P50 AT00477), the UAB O'Brien Acute Kidney Injury Center (P30 DK079337), the UAB Skin Disease Research Center (P30 AR50948), and the UAB Lung Health Center.
Footnotes
Conflict of interest: There is no financial conflict of interest for any of the authors listed
REFERENCES
- 1.Society AC. Cancer Facts and Figures. Atlanta:American Cancer Society 2012. 2012 [Google Scholar]
- 2.Howlader N NA, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review. U.S. National Institutes of Health, National Cancer Institute. 1973–2008 [Google Scholar]
- 3.William WN, Jr, Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol. 2011;8:611–619. doi: 10.1038/nrclinonc.2011.90. [DOI] [PubMed] [Google Scholar]
- 4.Lawless MW, O'Byrne KJ, Gray SG. Targeting oxidative stress in cancer. Expert Opin Ther Targets. 2010;14:1225–1245. doi: 10.1517/14728222.2010.526933. [DOI] [PubMed] [Google Scholar]
- 5.Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, Dranoff G. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest. 2011;121:2436–2446. doi: 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vacchelli E, Galluzzi L, Rousseau V, Rigoni A, Tesniere A, Delahaye N, Schlemmer FD, Menger L, Sukkurwala AQ, Adjemian S, et al. Loss-of-function alleles of P2RX7 and TLR4 fail to affect the response to chemotherapy in non-small cell lung cancer. Oncoimmunology. 2012;1:271–278. doi: 10.4161/onci.18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava MK, Andersson A, Zhu L, Harris-White M, Lee JM, Dubinett S, Sharma S. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy. 2012;4:291–304. doi: 10.2217/imt.11.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula SV, Chaudhury AR, La Perle KM, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101–5110. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deshane J, Zmijewski JW, Luther R, Gaggar A, Deshane R, Lai JF, Xu X, Spell M, Estell K, Weaver CT, et al. Free radical-producing myeloid-derived regulatory cells: potent activators and suppressors of lung inflammation and airway hyperresponsiveness. Mucosal Immunol. 2011;4:503–518. doi: 10.1038/mi.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun SH, Lee JE, Park MH, Kang JH, Kim YK, Wang YP, Park JK, Kim HK. Gemcitabine Plus Platinum Combination Chemotherapy for Elderly Patients with Advanced Non-small Cell Lung Cancer: A Retrospective Analysis. Cancer Res Treat. 2011;43:217–224. doi: 10.4143/crt.2011.43.4.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 15.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Batinic-Haberle I, Spasojevic I, Tse HM, Tovmasyan A, Rajic Z, St Clair DK, Vujaskovic Z, Dewhirst MW, Piganelli JD. Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids. 2012;42:95–113. doi: 10.1007/s00726-010-0603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batinic-Haberle I, Reboucas JS, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal. 2010;13:877–918. doi: 10.1089/ars.2009.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162–2171. doi: 10.1158/0008-5472.CAN-11-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy BC, Shimato S, Anderson RC, Bruce JN. Defining the mechanisms of CD8 T-cell tumor tolerance. Immunotherapy. 2011;3:23–26. doi: 10.2217/imt.10.84. [DOI] [PubMed] [Google Scholar]
- 20.Apetoh L, Vegran F, Ladoire S, Ghiringhelli F. Restoration of antitumor immunity through selective inhibition of myeloid derived suppressor cells by anticancer therapies. Curr Mol Med. 2011;11:365–372. doi: 10.2174/156652411795976574. [DOI] [PubMed] [Google Scholar]
- 21.Yu X-ZaAC. Memory stem cells sustain disease. Nat Med. 2005;11:1282–1283. doi: 10.1038/nm1205-1282. [DOI] [PubMed] [Google Scholar]
- 22.Mehrotra S, Mougiakakos D, Johansson CC, Voelkel-Johnson C, Kiessling R. Oxidative stress and lymphocyte persistence: implications in immunotherapy. Adv Cancer Res. 2009;102:197–227. doi: 10.1016/S0065-230X(09)02006-5. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi A, Hanson MG, Norell HR, Havelka AM, Kono K, Malmberg KJ, Kiessling RV. Preferential cell death of CD8+ effector memory (CCR7-CD45RA-) T cells by hydrogen peroxide-induced oxidative stress. J Immunol. 2005;174:6080–6087. doi: 10.4049/jimmunol.174.10.6080. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Ma X, Yan S, Shen S, Zhu H, Gu Y, Wang H, Qin G, Yu Q. 17-hydroxy-jolkinolide B inhibits signal transducers and activators of transcription 3 signaling by covalently cross-linking Janus kinases and induces apoptosis of human cancer cells. Cancer Res. 2009;69:7302–7310. doi: 10.1158/0008-5472.CAN-09-0462. [DOI] [PubMed] [Google Scholar]
- 25.Xie Y, Kole S, Precht P, Pazin MJ, Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150:1122–1131. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, Douek DC, Fahle GH, Cohen JI, Holland SM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35:806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peden DB. The role of oxidative stress and innate immunity in O(3) and endotoxin-induced human allergic airway disease. Immunol Rev. 2011;242:91–105. doi: 10.1111/j.1600-065X.2011.01035.x. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura K, Yube K, Miyatake A, Cambier JC, Hirashima M. Involvement of CD4 D3-D4 membrane proximal extracellular domain for the inhibitory effect of oxidative stress on activation-induced CD4 down-regulation and its possible role for T cell activation. Mol Immunol. 2003;39:909–921. doi: 10.1016/s0161-5890(03)00030-0. [DOI] [PubMed] [Google Scholar]
- 29.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 30.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121:4015–4029. doi: 10.1172/JCI45862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tongu M, Harashima N, Monma H, Inao T, Yamada T, Kawauchi H, Harada M. Metronomic chemotherapy with low-dose cyclophosphamide plus gemcitabine can induce anti-tumor T cell immunity in vivo. Cancer Immunol Immunother. 2012 doi: 10.1007/s00262-012-1343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sim SH, Ahn YO, Yoon J, Kim TM, Lee SH, Kim DW, Heo DS. Influence of chemotherapy on nitric oxide synthase, indole-amine-2,3-dioxygenase and CD124 expression in granulocytes and monocytes of non-small cell lung cancer. Cancer Sci. 2012;103:155–160. doi: 10.1111/j.1349-7006.2011.02158.x. [DOI] [PubMed] [Google Scholar]
- 34.Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu K, Rajapakse N, Horiguchi T, Payne RM, Busija DW. Protective effect of a new nonpeptidyl mimetic of SOD, M40401, against focal cerebral ischemia in the rat. Brain Res. 2003;963:8–14. doi: 10.1016/s0006-8993(02)03796-4. [DOI] [PubMed] [Google Scholar]
- 37.Haskins K, Bradley B, Powers K, Fadok V, Flores S, Ling X, Pugazhenthi S, Reusch J, Kench J. Oxidative stress in type 1 diabetes. Ann N Y Acad Sci. 2003;1005:43–54. doi: 10.1196/annals.1288.006. [DOI] [PubMed] [Google Scholar]
- 38.Kaul DK, Liu XD, Zhang X, Ma L, Hsia CJ, Nagel RL. Inhibition of sickle red cell adhesion and vasoocclusion in the microcirculation by antioxidants. Am J Physiol Heart Circ Physiol. 2006;291:H167–H175. doi: 10.1152/ajpheart.01096.2005. [DOI] [PubMed] [Google Scholar]
- 39.Vujaskovic Z, Batinic-Haberle I, Rabbani ZN, Feng QF, Kang SK, Spasojevic I, Samulski TV, Fridovich I, Dewhirst MW, Anscher MS. A small molecular weight catalytic metalloporphyrin antioxidant with superoxide dismutase (SOD) mimetic properties protects lungs from radiation-induced injury. Free Radic Biol Med. 2002;33:857–863. doi: 10.1016/s0891-5849(02)00980-2. [DOI] [PubMed] [Google Scholar]
- 40.Moeller BJ, Batinic-Haberle I, Spasojevic I, Rabbani ZN, Anscher MS, Vujaskovic Z, Dewhirst MW. A manganese porphyrin superoxide dismutase mimetic enhances tumor radioresponsiveness. Int J Radiat Oncol Biol Phys. 2005;63:545–552. doi: 10.1016/j.ijrobp.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 41.Gridley DS, Makinde AY, Luo X, Rizvi A, Crapo JD, Dewhirst MW, Moeller BJ, Pearlstein RD, Slater JM. Radiation and a metalloporphyrin radioprotectant in a mouse prostate tumor model. Anticancer Res. 2007;27:3101–3109. [PubMed] [Google Scholar]
- 42.van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev. 2012;249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.