

Sub2p/UAP56 is a highly conserved DEAD-box RNA helicase involved in the packaging and nuclear export of mRNA. Here we analyze S. cerevisiae Sub2p functions in vivo and in vitro and find that a conserved N-terminal motif is required for mRNA export without affecting Sub2p helicase activity. Mutants in the helicase domain are trans-dominant negative and cause hyper-recombination.
Keywords: RNA, RNA export, RNA helicase, genome stability
Abstract
Sub2p/UAP56 is a highly conserved DEAD-box RNA helicase involved in the packaging and nuclear export of mRNA/protein particles (mRNPs). In Saccharomyces cerevisiae, Sub2p is recruited to active chromatin by the pentameric THO complex and incorporated into the larger transcription–export (TREX) complex. Sub2p also plays a role in the maintenance of genome integrity as its inactivation causes severe transcription-dependent recombination of DNA. Despite the central role of Sub2p in early mRNP biology, little is known about its function. Here, we report the presence of an N-terminal motif (NTM) conserved specifically in the Sub2p branch of RNA helicases. Mutation of the NTM causes nuclear accumulation of poly(A)+ RNA and impaired growth without affecting core helicase functions. Thus, the NTM functions as an autonomous unit. Moreover, two sub2 mutants, that are deficient in ATP binding, act in a trans-dominant negative fashion for growth and induce high recombination rates in vivo. Although wild-type Sub2p is prevented access to transcribed loci in such a background, this does not mechanistically explain the phenotype.
INTRODUCTION
In eukaryotes, transcription is tightly coupled to mRNA processing and packaging with export factors to produce mature mRNA/protein particles (mRNPs) functional in cytoplasmic translation. In Saccharomyces cerevisiae, the pentameric THO complex (Tho2p, Hpr1p, Mft1p, Thp2p, and Tex1p) functions at the interface between transcription elongation and mRNP formation by recruiting the essential RNA helicase Sub2p, which in turn binds the Yra1p export factor to form the larger TREX (transcription-export) complex (Strasser and Hurt 2001; Jimeno et al. 2002; Strasser et al. 2002; Zenklusen et al. 2002; Peña et al. 2012). This positions Sub2p as a key factor linking the nascent mRNA to its export factors, which allow translocation to, and through, the nuclear pore complex (NPC) (for review, see Tutucci and Stutz 2011).
Sub2p is a highly conserved RNA helicase with orthologs in Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophila melanogaster, mouse, and man (Fan et al. 2001; Jimeno et al. 2002). As such, it contains the typical DEAD-box helicase core in addition to an extended N-terminal region of unknown function. Although motif II of the core helicase domain deviates from the typical DEAD signature (DECD in Sub2p), Sub2p features all characteristic DEAD-box sequence motifs (Fairman-Williams et al. 2010), and the helicase core of the mammalian Sub2p ortholog UAP56 adopts the typical DEAD-box helicase fold (Zhao et al. 2004). Sub2p is involved in multiple stages of mRNA maturation and its inactivation leads to nonproductive spliceosome assembly (Kistler and Guthrie 2001; Libri et al. 2001), decreased polyadenylation efficiency and mRNA instability (Rougemaille et al. 2007; Saguez et al. 2008), as well as nuclear accumulation of poly(A)+ RNA (Jensen et al. 2001a; Strasser and Hurt 2001; Rougemaille et al. 2007; Schmid et al. 2008). In addition, Sub2p is important for chromatin maintenance as its overexpression suppresses the genomic instability caused by deletion of the HPR1 THO component gene (Chavez et al. 2000; Fan et al. 2001), which otherwise causes a recombination rate of >3000 times that of wild-type (wt) cells (Aguilera and Klein 1988; Piruat and Aguilera 1998). The reason for involvement of an RNA helicase in DNA stability is not understood, but it may be related to the unwinding of RNA:DNA hybrid structures (R-loops) formed during transcription (Gomez-Gonzalez et al. 2011).
Here, we report a detailed functional analysis of S. cerevisiae Sub2p, focusing on its two highly conserved domains, a hitherto uncharacterized short N-terminal motif (NTM) as well as the core RNA helicase domain. Mutation of conserved residues in the NTM triggers nuclear accumulation of poly(A)+ RNA without affecting key helicase functions. In contrast, mutation of core helicase residues adversely affects Sub2p ATPase activity, RNA duplex unwinding, and RNA binding. This suggests that the NTM is not modulating the Sub2p core helicase as is often the case for N- or C-terminal extensions in RNA helicases (Mohr et al. 2008). We also report that sub2 variants with mutated ATP-binding sites are trans-dominant negative for genomic stability. Evidence suggests that such trans-dominance may arise from chromatin association of these mutants.
RESULTS AND DISCUSSION
Identification of a conserved and functionally important N-terminal motif in Sub2p
To analyze Sub2p for conserved motifs other than the core helicase domain, we performed a BLAST search of the NCBI database, allowing identification of 186 eukaryotic Sub2p orthologs (data not shown). Alignment of these sequences revealed, in addition to the expected RNA helicase domain (Cordin et al. 2006; Fairman-Williams et al. 2010), a short conserved and separate region contained within the first 50 amino acids of Sub2p orthologs (Fig. 1A). This highly acidic, 11-residue motif, which we term the Sub2p NTM, follows the consensus sequence a(2 − 3)LhaYa(2 − 3), where L = leucine, Y = tyrosine, a = any acidic residue, and h = any aliphatic residue. Within this motif the strictly conserved tyrosine residue is subject to phosphorylation (Albuquerque et al. 2008). Pattern searches of the Swiss-Prot and Uniref-50 nonredundant databases using MyHits (Pagni et al. 2004) and a degenerated NTM consensus (<X(1,50)–[EDNQ]–[EDNQ]–[EDNQ]–[LIVM]–[LIVM]–[ED]–Y–[EDS]–[ED]–[EDSNQ]) as a query produced additional hits within the N termini of two seemingly unrelated proteins, Rmr1p and CCDC47 (data not shown). As these sequence hits are only present in a few distantly related species, we conclude that the NTM most likely is specific to the Sub2p branch of DEAD-box helicases.
FIGURE 1.

Yeast Sub2p contains a conserved N-terminal motif (NTM) required for normal growth. (A) WebLogo showing conservation of the Sub2p NTM as retrieved from 186 nonredundant protein sequences (Crooks et al. 2004). Numbering denotes the position of amino acids in S. cerevisiae Sub2p. The y-axis indicates the relative frequency of each amino acid. (B–D) Tenfold serial dilutions of indicated S. cerevisiae sub2 NTM mutant strains plated for 3 d on 5-FOA plates at 25°C. Lines and rectangles denoting “NTM,” “linker,” “random sequence,” “core helicase domain,” and deleted residues are explained below image D.
To assess the functional relevance of the NTM, we constructed the Sub2p N-terminal variants sub2Δ[5–49] and sub2Δ[6–17] for which NTM and linker (Δ[5–49]), or only the NTM (Δ[6–17]) sequences had been removed. Although neither of these variants caused as dramatic a growth phenotype as the full SUB2 gene deletion, both gave rise to a significant decrease in growth rate (∼10-fold) (Fig. 1B). In contrast, removal of just the linker sequence between the NTM and the RNA-helicase domain (sub2Δ[25–46]), or replacement of the linker by a random sequence (sub2Δ[25–46]::RanSeq), did not impact growth. Thus, linker length and sequence appear to be of little importance. Within the NTM, mutation of the conserved tyrosine at position 12 (sub2[Y12S]) or of one (or both) of the conserved acidic stretches (sub2[E6Q,E7Q,D8N], sub2[D14N,E16Q], sub2[E6Q,E7Q,D8N,D14N,E16Q], and sub2[E6Q,E7Q,D8N,E11Q,D14N,E16Q]) yielded growth phenotypes comparable to the full NTM deletion (Fig. 1C). Finally, we tested whether the function of the Sub2p NTM depends on its location in relation to the core helicase domain by removing the NTM from the N terminus and instead fusing it to the C terminus of the protein (SUB2–FLIP) (Fig. 1D). Remarkably, Δsub2 cells carrying the SUB2–FLIP locus grew nearly as well as wt cells (Fig. 1D), suggesting that the NTM motif is a separable domain, capable of functioning regardless its location within the protein.
Sub2p NTM mutations affect poly(A)+ RNA export
Having established that Sub2p NTM alterations cause decreased growth, we next focused on how the function of Sub2p might be affected in the sub2Δ[6–17] and sub2[Y12S] variants. We first investigated RNA localization effects by conducting RNA fluorescence in situ hybridization (FISH) analysis using an LNA-spiked oligo-dT20 probe (Thomsen et al. 2003) on fixed cells grown at 25°C or subjected to a 37°C heat induction for 15 min. As previously reported, full deletion of SUB2 caused nuclear accumulation of poly(A)+ RNA (Fig. 2A; Strasser and Hurt 2001). A similar phenotype was detectable in sub2Δ[6–17] and sub2[Y12S] cells at both 25°C and 37°C. Importantly, this was not due to low stability of the mutant proteins, as Western blotting analysis confirmed that both the sub2Δ[6–17]p and sub2[Y12S]p variants were expressed at wt-Sub2p levels (Fig. 2B). Moreover, as demonstrated by microscopic detection of Sub2–GFP fusion constructs, both variants localized to the nuclear compartment in a manner similar to wt Sub2p–GFP (Supplemental Fig. S1A). As a control, both mRNA export (Supplemental Fig. S1B) and growth (Supplemental Fig. S1C) defects were indistinguishable between untagged and GFP-tagged Sub2p mutants. Finally, RNA-FISH analysis of the other growth-impaired NTM mutants shown in Figure 1C yielded comparable levels of nuclear accumulation of poly(A)+ RNA (Supplemental Fig. S1D).
FIGURE 2.
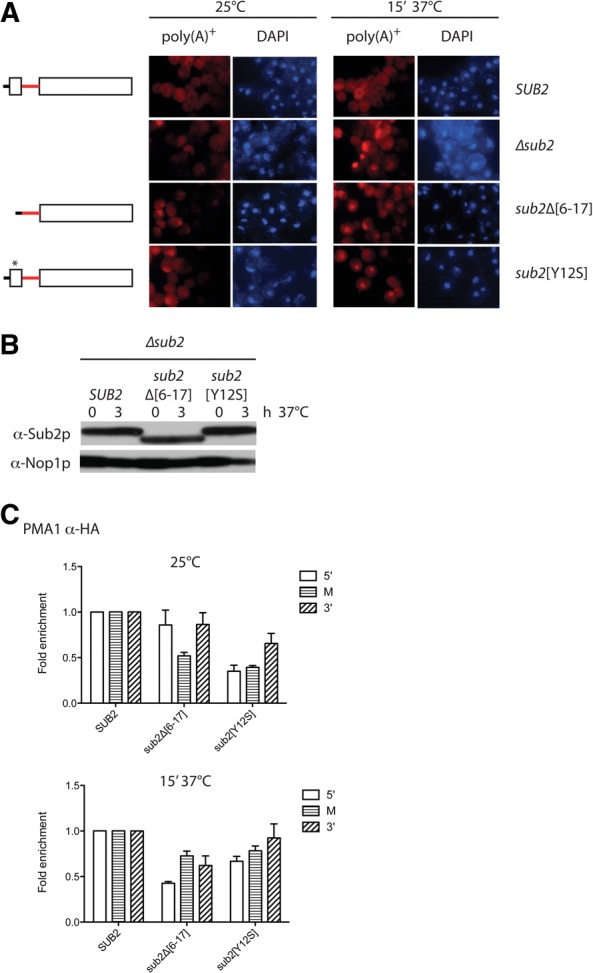
Mutation of the Sub2p NTM causes nuclear accumulation of poly(A)+ RNA. (A) Poly(A)+ RNA-FISH analysis of the indicated yeast strains grown at 25°C or heat-induced at 37°C for 15 min before fixation and processing. A Cy3-labeled and LNA-spiked dT20 probe was used for poly(A)+ targeting, and DAPI staining visualized the chromatin-rich part of the nuclei. (B) Western blotting analysis of proteins expressed in A. Cells were harvested after 0 or 3 h of growth at 37°C as indicated. Membranes were sequentially probed with α-Sub2p and α-Nop1p (loading control) antibodies. (C) Recruitment of HA-tagged Sub2p wt or indicated NTM mutants to the PMA1 gene at 25°C (top) or 37°C 15′ (bottom) analyzed by ChIP with amplicons directed toward the 5′ end (5′), middle (M), or 3′end (3′) of the gene. HA-tagged fusion proteins were expressed in a Δsub2 background and α-HA ChIP levels were normalized to Rpb3p ChIP values obtained from the same extracts and to Sub2p-HA wt levels from the same experiment. Average signals and standard deviations reflect ChIP results from three different chromatin extracts.
Since Sub2p is recruited to the nascent RNA during transcription, the observed poly(A)+ RNA export phenotypes of the sub2Δ[6–17] and sub2[Y12S] strains could in principle be due to altered co-transcriptional recruitment of the variant proteins (Strasser et al. 2002; Zenklusen et al. 2002; Abruzzi et al. 2004; Johnson et al. 2009). To resolve this question, we analyzed cells grown in conditions similar to those subjected to RNA-FISH by Sub2p chromatin immuno-precipitation (ChIP) analysis. Sub2p ChIP efficiencies of the constitutively expressed PMA1 gene were monitored using HA-antibody against HA-tagged variant proteins. ChIP levels were normalized to those of RNAPII as measured by the anti-POLR2C antibody targeting the Rpb3p subunit and to the levels of wt Sub2p-HA. For both samples treated at 25°C and 37°C, ChIP amplicons directed toward three positions along the PMA1 gene revealed only slightly decreased signals of co-transcriptional recruitment of the sub2Δ[7–19]p and sub2Δ[Y12S]p variants as compared with HA-tagged wt Sub2p (Fig. 2C). Thus, the function of the Sub2p NTM in mRNA export is not likely to be due to an inability to target active chromatin.
Mutation of the NTM does not affect Sub2p RNA helicase activity in vitro
Next, we asked if the NTM might be able to modulate the classical helicase functions, i.e., RNA binding, ATP hydrolysis, and RNA duplex unwinding. To approach this, we compared activities of the Y12S NTM mutant with Sub2p mutants for which conserved helicase residues with known functions had been altered. For RNA binding, we thus introduced mutations in highly conserved arginines in the RNA-binding motifs Ia (R140M) and Ic (R194M) (Fig. 3A; Andersen et al. 2006; Sengoku et al. 2006). To inhibit ATP binding and hydrolysis, we mutated D215 and E216 in motif II (Fig. 3A), noting that mutation of E216 in the human homolog of Sub2p, UAP56, abolishes both ATPase activity and RNA unwinding (Shen et al. 2007). Finally, to impair the coupling between ATP and RNA binding we mutated D218, the second aspartate in motif II, which has been shown to uncouple ATP hydrolysis and RNA unwinding in UAP56 (Fig. 3A; Shen et al. 2007; Henn et al. 2012). Crystal structures of several DEAD-box proteins with bound RNA and ATP analogs have revealed an interaction network involving this aspartate and residues in motif III, motif Va, and the conserved histidine (H) in motif VI, which could be responsible for the observed functional effects.
FIGURE 3.

Mutation of the NTM does not affect Sub2p helicase activity. (A) Schematic representation of mutations in the helicase core of Sub2p analyzed in this study. Location of the characteristic helicase motifs in DEAD-box proteins (red, motifs involved in ATP binding and hydrolysis; blue, motifs involved in RNA binding; green, motifs involved primarily in coupling between ATP and RNA binding). Boxes show the DEAD-box family consensus in motifs Ia, Ic, and II (Fairman-Williams et al. 2010); corresponding sequences of Sub2p are given underneath. The label for E216 is omitted for clarity. (B) In vitro RNA-binding gel-shift assay using a single-stranded 5′-radiolabeled 13-mer RNA. The migration of free and protein-bound RNA is shown. Sub2 was present at a concentration of 2 µM and the RNA at 0.6 nM. (C) ATPase activity measured by a modified Baginski-type assay (Cariani et al. 2004) using wt Sub2p or mutant variants as indicated and poly(U). The assay measures the relative production of Pi in nmol during a fixed time interval. (D) RNA-unwinding assay using a 13-bp duplex substrate with 12-bp overhang and wt or mutant Sub2p protein without (−) or with (+) ATP/Mg, as indicated. The positions of duplex substrate and unwound product are indicated at the left, where the stars show the position of the radiolabel. (E) RNA:DNA hybrid unwinding assay as in D but using a duplex of a 13-nt DNA annealed to a 38-nt RNA. (F) Tenfold serial dilution of yeast Δsub2 cells complemented by plasmids expressing wild type (SUB2), mutant Sub2p variants, or an empty vector control as indicated. Cells were plated on 5-FOA plates for 3 days at 25°C.
Sub2p and all mutant variants were expressed in Escherichia coli and purified for functional analysis in vitro (Supplemental Fig. S2A). Firstly, we probed the ability of the recombinant proteins to bind a 13-nt single-stranded RNA (5′-AGCACCGUAAAGC-3′) in the presence of the non-hydrolysable ATP analog ADPNP using a gel-shift assay. Sub2p and the Y12S mutant bound RNA with similar efficiencies, indicating that the NTM does not contribute to the RNA-binding ability of the helicase core (Fig. 3B). In contrast, none of the other mutants shifted the RNA. This outcome was not unexpected, as complex formation requires both binding of the ATP analog and the RNA, as well as their coupling. The only exception was the D218A mutant, which appeared to bind RNA weakly as indicated by a smearing of the signal, suggesting that coupling between RNA and ATP binding might not be completely interrupted by this mutation.
Secondly, we analyzed the capacity of Sub2p and its variants to hydrolyze ATP in an RNA-dependent fashion as measured by the release of inorganic Pi. In this assay, the Y12S variant exhibited ATPase activity comparable to that of wt Sub2 (Fig. 3C). As expected, none of the helicase core domain mutants yielded significant activity, again with the exception of D218A, which showed about half the activity of the wt enzyme. This observation supports the notion that the D218A mutation maintains residual coupling between RNA and ATP binding.
Thirdly, we tested the RNA-unwinding activities of the enzymes using a substrate consisting of a 13-bp RNA duplex with a 25-nt single-stranded 3′ overhang. Both wt Sub2p and the Y12S mutant displayed very similar unwinding activities, as evidenced by the similar extent of strand separation in the single time-point experiment (Fig. 3D), as well as in measurements of unwinding rate constants as a function of enzyme concentration (Supplemental Fig. S2B). In contrast, any mutation in the helicase core virtually eliminated unwinding activity (Fig. 3D). The D218A protein again showed weak activity as revealed by a measurable unwinding rate constant (Supplemental Fig. S2C). The unwinding activity of wt Sub2p is not restricted to RNA:RNA hybrids, as RNA:DNA hybrids were also efficiently resolved (Fig. 3E).
Finally, we introduced the analyzed mutants into a Δsub2 background and measured growth. The D215A and E216Q mutations acted strongly inhibitory while the D218A strain grew with almost wt kinetics at 25°C and showed a minor growth defect at 37°C (Fig. 3F; Supplemental Fig. S2D). Cells harboring the R194M RNA-binding mutation showed a partial growth defect at 25°C, whereas mutating two RNA-binding residues (R140M/R194M) strongly impeded growth. In addition, and consistent with the growth defects observed at elevated temperatures, all tested mutants displayed mRNA export defects at 37°C (Supplemental Fig. S2E).
Collectively, our data show that the critical mutation (Y12S) of the Sub2p NTM does not affect known helicase functions of the enzyme. Thus, the NTM does not appear to be directly involved in Sub2p helicase activity but might instead mediate Sub2p function via binding of other factors in vivo that are important for mRNA processing.
Mutations in conserved helicase domains are dominant negative
In a final set of experiments, we asked whether transformation of plasmid-borne sub2 helicase-deficient mutants (R194M, R140M/R194M, D215A, and E216Q) into yeast cells harboring an endogenous wt copy of SUB2 would have any growth phenotypic consequences. Indeed, introduction of the D215A or E216Q mutants reduced growth at 37°C, while expression of the D218Ap, R194Mp, R140M/R194Mp, Y12Sp, or Δ[6–16]p mutants showed no, or only a marginal, effect (Fig. 4A; Supplemental Fig. S3A). Such growth decrease was also previously observed (Kistler and Guthrie 2001; Libri et al. 2001). Besides its critical role in mRNP maturation (Jensen et al. 2001a; Libri et al. 2001; Strasser and Hurt 2001), over-expression of Sub2p also suppresses the genomic instability phenotype of Δhpr1 cells (Fan et al. 2001; Jimeno et al. 2002). Moreover, conditional alleles of SUB2 cause an increased recombination frequency between direct DNA repeats (Fan et al. 2001; Jimeno et al. 2002). To analyze whether genomic stability would be affected by co-expression of the sub2 core helicase mutants, we took advantage of a yeast strain carrying the duplication system leu2-k::ADE2-URA3::leu2-k, previously used to detect hyper-recombination in Δhpr1 cells (Fig. 4B, bottom; Aguilera and Klein 1990). Remarkably, cells harboring the D215A and E216Q mutants on top of SUB2 displayed a dramatically increased recombination frequency (Fig. 4B, note the Δhpr1 hyper-recombinogenic background control). In contrast, and consistent with the normal growth of R140M/R194M-containing cells, no major recombination phenotype was detected.
FIGURE 4.

Sub2p helicase mutants display dominant negative growth and recombination phenotypes. (A) Tenfold serial dilutions of cells plated for 3 d at 25°C or 37°C and transformed with plasmids expressing either wt or mutant Sub2p as indicated. (B) Cells carrying the integrated leu2k–ADE–URA–leu2k repeat system (bottom) were used as vehicles to examine the impact of ectopic expression of Sub2p mutants on genomic stability. The frequency of recombination was measured as previously described (Prado and Aguilera 1995) with the hyper-recombinant Δhpr1 strain as a positive control. (C) Recruitment as analyzed by ChIP of HA-tagged (top) or total (bottom) Sub2p to PMA1 (left) or HSP104 (right) genes after a 37°C heat induction for 15 min. Cells were transfected with centromeric plasmids expressing HA-tagged wt SUB2 or its mutant variants, as indicated. An α-HA antibody was used to specifically precipitate exogenous HA-tagged proteins, and α-Sub2p antibody was used to precipitate total Sub2p (exogenous and endogenous proteins). ChIP levels as measured to the 5′, middle, or 3′ regions of the genes were normalized to Rpb3p values and to levels of wt Sub2p cells from the same samples. Average signals and standard deviations reflect ChIP results from three different chromatin extracts.
To understand what caused the decreased growth at 37°C and high recombination frequencies observed upon D215Ap and E216Qp expression, we analyzed the recruitment of core helicase mutants to the PMA1 and HSP104 genes by ChIP. To this end, Sub2p and the D215Ap, E216Qp and R140M/R194Mp mutants were tagged with a triple HA epitope and expressed in cells containing untagged, endogenous Sub2p (see Supplemental Fig. S3B for expression levels of HA-tagged and endogenous proteins). Exponentially growing cells were collected after 15-min incubation at 37°C, subjected to ChIP using α-HA beads (for HA-tagged proteins), α-Sub2p beads (for tagged as well as untagged Sub2p proteins), and α-POLR2C beads to normalize for levels of RNAPII. Subsequently, protein occupancies at three positions along the PMA1 and HSP104 loci were measured by qPCR. Of these, the α-HA IPs revealed a considerably decreased recruitment of all three helicase mutants compared with that of wt Sub2p (Fig. 4C, top panels). Interestingly, while the D215Ap and E216Qp proteins could be detected over background, the R140M/R194Mp variant was barely present. Expression of the three mutants also led to the detection of less total Sub2p at the reporter genes compared with the wt Sub2p-HA expressing strain (Fig. 4C, bottom panels). Between the three mutants the total Sub2p levels only varied approximately twofold and were virtually identical between the D215Ap and R140M/R194Mp expressing strains (Supplemental Fig. S3B). Furthermore, we also conducted ChIP of the nondominant-negative mutants D218Ap and R140M/R194Mp in a Δsub2 background. In this setting the co-transcriptional recruitment of R140M/R194Mp was also severely impaired, whereas D218Ap ChIP signals were ∼50% of wt Sub2p (Supplemental Fig. S3C), despite only minor changes in protein expression from these constructs (Supplemental Fig. S3D). Although this amount is roughly similar to that of wt Sub2p in the presence of the dominant-negative mutant E216Qp (Fig. 4C), the D218A strain only modestly increased recombination rates, even in a sub2 “single copy” setting (Fig. 4B; Supplemental Fig. S3E). Similarly, levels of total Sub2p recruited upon co-expression of the R140M/R194Mp construct were also decreased without significantly increased recombination rates (Fig. 4B,C). We therefore conclude that even if the low-chromatin recruitment of Sub2p could account for the growth deficiency of the various mutant-expressing strains, it does not explain the dominant-negative growth and severe hyper-recombination phenotypes specifically observed in D215Ap- and E216Qp-expressing cells. Rather the presence of low levels of these ATPase mutants at transcribed loci may directly trigger hyper-recombination, for example by preventing access of wt Sub2p to R-loops. Alternatively, these mutants may sequester other factors away from chromatin and thereby indirectly elicit the dominant-negative effect. Such sequestration could also underlie the low recruitment of wt Sub2p in the presence of mutant protein (Fig. 4C). However, since this is also observed upon co-expression of the R140M/R194M double mutant, it does not explain hyper-recombination. Instead it may perturb mRNA export, consistent with an observable mRNA export defect in wt cells co-expressing the dominant-negative mutants (Supplemental Fig. S3F).
Interestingly, there is precedence for dominant-negative effects of mutant RNA helicases. Previous work demonstrated that eIF4A-driven translation in rabbit reticulocyte lysates is inhibited when defective eIF4A is added (Pause et al. 1994). Moreover, introduction of ATPase-deficient UPF1 helicase abrogates nonsense-mediated decay (NMD) in human cells (Sun et al. 1998). Thus, inhibition of ATPase activity may generally yield dominant-negative effects across cellular functions. ATP is required for the interaction of UAP56/Sub2p with other factors involved in mRNA export (Dufu et al. 2010; Folco et al. 2012) and with RNA (Taniguchi and Ohno 2008). Since these activities predictably occur co-transcriptionally, failure to bind, or release, key interaction partners may be the trigger of transcription-dependent hyper-recombination, either via an inability to resolve DNA::RNA hybrids or by increasing R-loop formation (Huertas and Aguilera 2003). Consistent with its apparent detachment from core helicase function, expression of NTM mutants does not lead to hyper-recombination (data not shown). We suggest that the NTM represents an extension to Sub2p potentially involved in the binding of an, as yet, unidentified co-factor(s) or in subnuclear localization of the protein. These possibilities remain to be investigated.
Finally, our work shows that the physiological function of Sub2p correlates with classical RNA helicase activities, ATP/RNA binding, ATP hydrolysis, and RNA duplex unwinding. Curiously, both mutants R194Mp and D218Ap, which show no, or weak, RNA-binding and unwinding activities in vitro, rescue growth of the sub2 deletion in vivo. Since these biochemical activities of Sub2p are probably essential, these mutants may support residual activity in the presence of cellular protein interaction partners. In contrast, mutant strains D215A, E216Q, and R140M/R194M may be fully compromised. This observation suggests that the cell can cope with a decreased RNA affinity of Sub2p until a critical threshold is reached. In a potentially similar fashion, defects in the coupling between ATP and RNA binding are tolerated without drastic growth effects.
MATERIALS AND METHODS
Strains, plasmids, and primers
All strains, plasmids, and primers used in this study are listed in Supplemental Tables S1, S2, and S3, respectively. A 2-kb DNA fragment containing the coding region of SUB2 was PCR-amplified from genomic DNA and cloned into the pRS415 plasmid (Sikorski and Hieter 1989) using SacI and ApaI restriction sites. All sub2 alleles were generated by site-directed mutagenesis using standard PCR-driven overlap extension (Heckman and Pease 2007) and sub-cloned into pRS415–SUB2, pRS415-3HA–SUB2, or pRS415–SUB2–GFP using unique restriction sites surrounding the mutation. Plasmids carrying the different mutant alleles of SUB2 were introduced into the THJ2235 strain, carrying a disrupted chromosomal copy of SUB2 and the wt SUB2 gene on plasmid pCM188–SUB2 (URA3). Transformants were selected on medium lacking leucine, and then cured for pCM188–SUB2 as previously described (Boeke et al. 1984). sub2Δ cells containing empty vector or D215A, E216Q, and R140M/R194M mutant plasmids grew only poorly on 5-FOA plates. Even so, viable colonies cured for the URA plasmid could be obtained and cultivated on YPAD medium at 25°C. For production of recombinant proteins, the SUB2 ORF and its mutant variants were cloned into pET30 and transformed into the E. coli BL21 (DE3) Rosetta strain.
Recombination assays
The FM-1 strain used for recombination assays was constructed by back-crossing the A3Y1-T6 strain (Aguilera and Klein 1990) three times into the BY4741 background. Recombination frequencies were determined as previously described (Prado and Aguilera 1995). Dominant effects on recombination efficiencies were analyzed after transformation of centromeric plasmids expressing various sub2 mutants in the FM-1 strain. To measure recombination rates of sub2 mutants alone, FM-1 was crossed to various sub2 mutants in the BY background. sub2Δ, sub2–D215A, sub2–E216Q, and sub2–R140M/R194M were found not to be viable in the BY background, consistent with information at SGD.
RNA-FISH and Sub2–GFP localization analysis
Poly(A)+-RNA FISH assays and image preparation were conducted as previously described (Jensen et al. 2001b; Thomsen et al. 2003).
Western blotting analysis
Western blotting analysis was performed using standard techniques. Relevant proteins were detected using a 1/3000 dilution of polyclonal α-Rabbit Sub2p antibody, a 1/10000 dilution of monoclonal α-Mouse Nop1p antibody (Abcam), and a 1/5000 dilution of polyclonal α-Rabbit HA-tag antibody (Abcam).
ChIP analysis
Overnight cultures were diluted to OD600 0.2, grown in synthetic complete media lacking leucine at 25°C for 4 h, and temperature-shifted to 37°C for 15 min before fixation with 1% formaldehyde for 20 min at room temperature. IP experiments were carried out as previously described (Jimeno-Gonzalez et al. 2010) using the following reagents: α-HA, Dynabeads M-280 sheep α-Rabbit IgG (Invitrogen) with rabbit polyclonal α-HA tag antibody (Abcam ab9110); α-Sub2p, Dynabeads M-280 sheep α-Rabbit IgG with rabbit polyclonal Sub2p antibody; α-RNAPII (Rpb3p), Dynabeads M-280 sheep α-Mouse IgG with mouse monoclonal α-POLR2C antibody (Abcam); α-RNAPII (CTD); Dynabeads M-280 sheep α-Mouse IgG (Invitrogen) with mouse monoclonal 8WG16 antibody (Abcam).
De-crosslinked DNA was analyzed by quantitative PCR (Mx 3005 Stratagene) using Platinum SyBRGreen qPCR SuperMix UDG (Invitrogen). Standard PCR conditions: 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. Primers targeting 5′, middle, and 3′ regions of PMA1 and HSP104 were as described (Zenklusen et al. 2002; Jensen et al. 2008). All primers were used at a final concentration of 100 nM. IP efficiencies were calculated after subtraction of background values (no antibody control). α-HA and α-Sub2p IP efficiencies were normalized to the background-subtracted values for α-Sub2p and shown relative to levels for wt Sub2p.
Protein purification
E. coli BL21(DE3) Rosetta cells containing the expression plasmid were grown to mid-log phase at 37°C, induced with 0.5 mM of IPTG, grown overnight at 20°C, and lysed in lysis buffer (300 mM KCl, 50 mM HEPES pH 7.5, 10% glycerol, 15 mM imidazole, and 5 mM BME). After clearing the lysate by centrifugation, it was passed over a Ni affinity column (Qiagen) and washed in high salt buffer (1 M KCl, 50 mM HEPES pH 7.5, 10% glycerol, 15 mM imidazole, and 5 mM BME). Bound protein was eluted in lysis buffer with 500 mM imidazole using a gradient and was dialyzed overnight in the presence of TEV protease in 50 mM Bis-Tris pH 6.5, 50 mM KCl, and 5 mM BME. The cleaved protein was passed a second time over the Ni column to remove impurities, then loaded onto a Source Q column equilibrated in buffer A (100 mM KCl, 50 mM Bis-Tris pH 6.5, and 5 mM BME) and eluted using a gradient into buffer B (1 M KCl, 50 mM Bis-Tris pH 6.5, and 5 mM BME). Peak fractions were concentrated and run on a Superdex 75 gel filtration column in 100 mM KCl, 10 mM HEPES pH 6.5, and 5 mM BME, and finally concentrated and stored in 30% glycerol at −80°C.
RNA-binding assays
A 13-mer single-stranded RNA (5′-AGCACCGUAAAGC-3′, Dharmacon) was 5′ end-labeled with 32P-radiolabeled γATP. Protein (2 μM) and RNA (0.6 nM) were incubated in binding buffer (10 mM MES pH 6.5, 5% glycerol, 2.5 mM DTT, 3 mM MgCl2, 2 mM ADPNP, and RNase inhibitor) at room temperature for 30 min. Samples were loaded onto a pre-chilled 10% native (1× TBE) gel and run at 300 Volts for 1 h in the cold. Gels were then dried and exposed to a phosphor screen overnight and scanned using a Typhoon Trio (GE Healthcare).
Unwinding reactions
Reaction solutions (30 μL) contained 40 mM MES pH 6.5, 0.01% IGEPAL, 0.6 U/µL RNasin, 2 mM DTT, 5% Glycerol, 25 mM NaCl, and 0.5 nM 32P-labeled 13-nt RNA or 13-nt DNA of identical sequence annealed with an unlabeled 38-nt RNA leaving a 25-nt 3′ overhang. Sub2p or the respective mutant protein was incubated with the RNA substrate at 30°C for 5 min prior to the reaction start. Unwinding reactions were initiated with an equimolar mixture of 2 mM ATP and MgCl2. Aliquots (3 µL) were removed from the reactions and stopped by addition to 2× Helicase Stop Buffer (50 mM EDTA, 1% SDS, 20% Glycerol, 0.1% bromphenol blue, xylene cylanol). Samples were loaded onto a 15% non-denaturing polyacrylamide gel and products were separated at 10 V/cm at 4°C. Gels were dried and visualized using a phosphor imager (STORM), and fraction of unwound substrate was quantified using the ImageQuant 5.2 software (Molecular Dynamics). Unwinding rate constants were calculated as described (Yang and Jankowsky 2005).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Danish National Research Foundation (T.H.J. and D.E.B.), the Novo Nordisk Foundation (T.H.J. and D.E.B.), and the NIH (E.J.). We thank Gregers Rom Andersen for help with identification of structurally critical residues in Sub2p. Polyclonal α-Rabbit Sub2p antibodies were generous gifts from Elaine Lahue and Christine Guthrie.
REFERENCES
- Abruzzi KC, Lacadie S, Rosbash M 2004. Biochemical analysis of TREX complex recruitment to intronless and intron-containing yeast genes. EMBO J 23: 2620–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Klein HL 1988. Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I. Isolation and genetic characterization of hyper-recombination mutations. Genetics 119: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Klein HL 1990. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol Cell Biol 10: 1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H 2008. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 7: 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR 2006. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 313: 1968–1972 [DOI] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197: 345–346 [DOI] [PubMed] [Google Scholar]
- Cariani L, Thomas L, Brito J, del Castillo JR 2004. Bismuth citrate in the quantification of inorganic phosphate and its utility in the determination of membrane-bound phosphatases. Anal Biochem 324: 79–83 [DOI] [PubMed] [Google Scholar]
- Chavez S, Beilharz T, Rondon AG, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Lithgow T, Aguilera A 2000. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J 19: 5824–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordin O, Banroques J, Tanner NK, Linder P 2006. The DEAD-box protein family of RNA helicases. Gene 367: 17–37 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE 2004. WebLogo: A sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufu K, Livingstone MJ, Seebacher J, Gygi SP, Wilson SA, Reed R 2010. ATP is required for interactions between UAP56 and two conserved mRNA export proteins, Aly and CIP29, to assemble the TREX complex. Genes Dev 24: 2043–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams ME, Guenther UP, Jankowsky E 2010. SF1 and SF2 helicases: Family matters. Curr Opin Struct Biol 20: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HY, Merker RJ, Klein HL 2001. High-copy-number expression of Sub2p, a member of the RNA helicase superfamily, suppresses hpr1-mediated genomic instability. Mol Cell Biol 21: 5459–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Lee CS, Dufu K, Yamazaki T, Reed R 2012. The proteins PDIP3 and ZC11A associate with the human TREX complex in an ATP-dependent manner and function in mRNA export. PloS one 7: e43804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalez B, Garcia-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marin A, Foiani M, Aguilera A 2011. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J 30: 3106–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman KL, Pease LR 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2: 924–932 [DOI] [PubMed] [Google Scholar]
- Henn A, Bradley MJ, De La Cruz EM 2012. ATP utilization and RNA conformational rearrangement by DEAD-box proteins. Annu Rev Biophys 41: 247–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Aguilera A 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12: 711–721 [DOI] [PubMed] [Google Scholar]
- Jensen TH, Boulay J, Rosbash M, Libri D 2001a. The DECD box putative ATPase Sub2p is an early mRNA export factor. Curr Biol 11: 1711–1715 [DOI] [PubMed] [Google Scholar]
- Jensen TH, Patricio K, McCarthy T, Rosbash M 2001b. A block to mRNA nuclear export in S. cerevisiae leads to hyperadenylation of transcripts that accumulate at the site of transcription. Mol Cell 7: 887–898 [DOI] [PubMed] [Google Scholar]
- Jensen MM, Christensen MS, Bonven B, Jensen TH 2008. Requirements for chromatin reassembly during transcriptional downregulation of a heat shock gene in Saccharomyces cerevisiae. FEBS J 275: 2956–2964 [DOI] [PubMed] [Google Scholar]
- Jimeno S, Rondon AG, Luna R, Aguilera A 2002. The yeast THO complex and mRNA export factors link RNA metabolism with transcription and genome instability. EMBO J 21: 3526–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno-Gonzalez S, Haaning LL, Malagon F, Jensen TH 2010. The yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol Cell 37: 580–587 [DOI] [PubMed] [Google Scholar]
- Johnson SA, Cubberley G, Bentley DL 2009. Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3′ end processing factor Pcf11. Mol Cell 33: 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler AL, Guthrie C 2001. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes Dev 15: 42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri D, Graziani N, Saguez C, Boulay J 2001. Multiple roles for the yeast SUB2/yUAP56 gene in splicing. Genes Dev 15: 36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr G, Del Campo M, Mohr S, Yang Q, Jia H, Jankowsky E, Lambowitz AM 2008. Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro. J Mol Biol 375: 1344–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Falquet L 2004. MyHits: A new interactive resource for protein annotation and domain identification. Nucleic Acids Res 32: W332–W335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Methot N, Svitkin Y, Merrick WC, Sonenberg N 1994. Dominant negative mutants of mammalian translation initiation factor eIF-4A define a critical role for eIF-4F in cap-dependent and cap-independent initiation of translation. EMBO J 13: 1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña A, Gewartowski K, Mroczek S, Cuéllar J, Szykowska A, Prokop A, Czarnocki-Cieciura M, Piwowarski J, Tous C, Aguilera A, et al. 2012. Architecture and nucleic acids recognition mechanism of the THO complex, an mRNP assembly factor. EMBO J 31: 1605–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piruat JI, Aguilera A 1998. A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J 17: 4859–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A 1995. Role of reciprocal exchange, one-ended invasion crossover and single-strand annealing on inverted and direct repeat recombination in yeast: Different requirements for the RAD1, RAD10, and RAD52 genes. Genetics 139: 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemaille M, Gudipati RK, Olesen JR, Thomsen R, Seraphin B, Libri D, Jensen TH 2007. Dissecting mechanisms of nuclear mRNA surveillance in THO/sub2 complex mutants. EMBO J 26: 2317–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saguez C, Schmid M, Olesen JR, Ghazy MA, Qu X, Poulsen MB, Nasser T, Moore C, Jensen TH 2008. Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol Cell 31: 91–103 [DOI] [PubMed] [Google Scholar]
- Schmid M, Rougemaille M, Libri D, Jensen TH 2008. Chapter 10. Estimating nuclear mRNA decay in Saccharomyces cerevisiae. Methods Enzymol 449: 205–219 [DOI] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S 2006. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125: 287–300 [DOI] [PubMed] [Google Scholar]
- Shen J, Zhang L, Zhao R 2007. Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. J Biol Chem 282: 22544–22550 [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser K, Hurt E 2001. Splicing factor Sub2p is required for nuclear mRNA export through its interaction with Yra1p. Nature 413: 648–652 [DOI] [PubMed] [Google Scholar]
- Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, et al. 2002. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417: 304–308 [DOI] [PubMed] [Google Scholar]
- Sun X, Perlick HA, Dietz HC, Maquat LE 1998. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsense-containing mRNAs in mammalian cells. Proc Natl Acad Sci 95: 10009–10014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi I, Ohno M 2008. ATP-dependent recruitment of export factor Aly/REF onto intronless mRNAs by RNA helicase UAP56. Mol Cell Biol 28: 601–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R, Libri D, Boulay J, Rosbash M, Jensen TH 2003. Localization of nuclear retained mRNAs in Saccharomyces cerevisiae. RNA 9: 1049–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutucci E, Stutz F 2011. Keeping mRNPs in check during assembly and nuclear export. Nat Rev Mol Cell Biol 12: 377–384 [DOI] [PubMed] [Google Scholar]
- Yang Q, Jankowsky E 2005. ATP- and ADP-dependent modulation of RNA unwinding and strand annealing activities by the DEAD-box protein DED1. Biochemistry 44: 13591–13601 [DOI] [PubMed] [Google Scholar]
- Zenklusen D, Vinciguerra P, Wyss JC, Stutz F 2002. Stable mRNP formation and export require cotranscriptional recruitment of the mRNA export factors Yra1p and Sub2p by Hpr1p. Mol Cell Biol 22: 8241–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Shen J, Green MR, MacMorris M, Blumenthal T 2004. Crystal structure of UAP56, a DExD/H-box protein involved in pre-mRNA splicing and mRNA export. Structure 8: 1373–1381 [DOI] [PubMed] [Google Scholar]