

It was known that ribosome biogenesis in eukaryotes requires hybridization between U3 snoRNA and pre-rRNA at three sites. However, these sites are buried in conserved structures. This study shows that the Imp3 protein functions to unwind these structures and make them accessible to U3 binding. The authors hypothesize that Imp3 recruitment could prevent premature or inaccurate rRNA processing.
Keywords: ribosome synthesis, RNA conformational switch, yeast, ribonucleoprotein
Abstract
Eukaryotic ribosome biogenesis requires rapid hybridization between the U3 snoRNA and the pre-rRNA to direct cleavages at the A0, A1, and A2 sites in pre-rRNA that liberate the small subunit precursor. The bases involved in hybridization of one of the three duplexes that U3 makes with pre-rRNA, designated the U3-18S duplex, are buried in conserved structures: box A/A′ stem–loop in U3 snoRNA and helix 1 (H1) in the 18S region of the pre-rRNA. These conserved structures must be unfolded to permit the necessary hybridization. Previously, we reported that Imp3 and Imp4 promote U3-18S hybridization in vitro, but the mechanism by which these proteins facilitate U3-18S duplex formation remained unclear. Here, we directly addressed this question by probing base accessibility with chemical modification and backbone accessibility with ribonuclease activity of U3 and pre-rRNA fragments that mimic the secondary structure observed in vivo. Our results demonstrate that U3-18S hybridization requires only Imp3. Binding to each RNA by Imp3 provides sufficient energy to unfold both the 18S H1 and the U3 box A/A′ stem structures. The Imp3 unfolding activity also increases accessibility at the U3-dependent A0 and A1 sites, perhaps signaling cleavage at these sites to generate the 5′ mature end of 18S. Imp4 destabilizes the U3-18S duplex to aid U3 release, thus differentiating the roles of these proteins. Protein-dependent unfolding of these structures may serve as a switch to block U3-pre-rRNA interactions until recruitment of Imp3, thereby preventing premature and inaccurate U3-dependent pre-rRNA cleavage and folding events in eukaryotic ribosome biogenesis.
INTRODUCTION
Eukaryotic ribosomes are large RNA-protein complexes. They require hundreds of trans-acting proteins and small nucleolar RNAs (snoRNAs) to produce 2000 ribosomes per minute (Warner 1999) in rapidly dividing Saccharomyces cerevisiae cells. With the major outline of ribosome biogenesis established and the trans-acting factors required for this process identified (for review, see Henras et al. 2008; Phipps et al. 2011), attention now focuses on mechanistic investigations of how these factors function. The dynamic nature of this process is expected to involve a large number of RNA–RNA and RNA protein assembly and disassembly steps. Trans-acting factors are expected to help direct this stepwise and dynamic process by recognizing noteworthy structural changes in the precursor ribosomal RNA, pre-rRNA (White et al. 2008; Xu et al. 2008; Lamanna and Karbstein 2009, 2011; Swiatkowska et al. 2012) and in snoRNAs. Previously, we showed that an essential step in ribosome biogenesis requires Imp3 and Imp4: rapid U3 snoRNA-pre-rRNA hybridization (Fig. 1; Gerczei et al. 2009). Herein, we investigate the mechanism and consequences of this activity by probing how protein binding affects the structures of both RNA molecules. As expected, S. cerevisiae is an excellent model system because the pre-rRNA processing pathways and most of the genes identified to play a role in ribosome biogenesis, including Imp3 and Imp4, have counterparts in humans and other higher eukaryotes (for review, see Henras et al. 2008).
FIGURE 1.
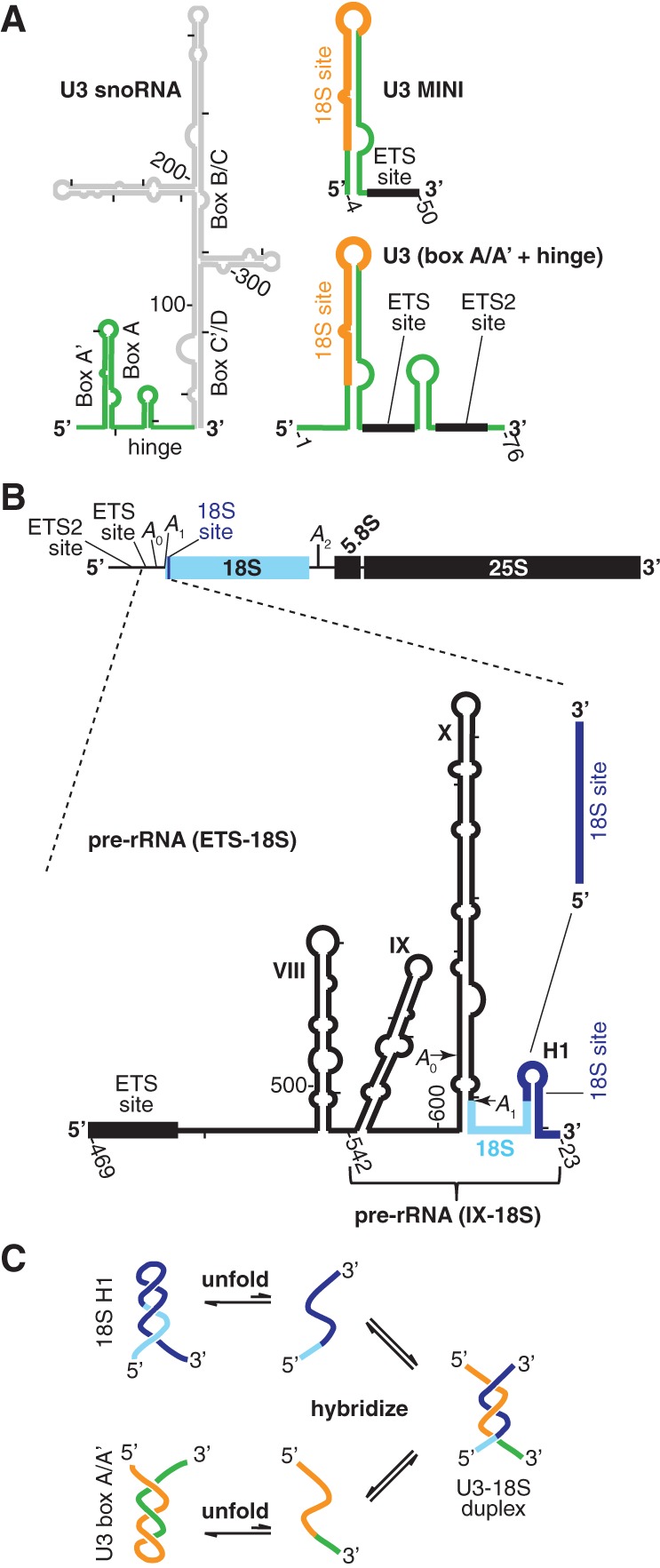
Secondary structure of RNA substrates and the kinetic unfolding barrier to U3-18S duplex formation. (A) U3 snoRNA (left) secondary structure as previously determined (Mereau et al. 1997) with the substrates used herein highlighted (green). The two substrates are U3 MINI, containing two hybridization sites (ETS [black] and 18S [orange]), and U3 (box A/A′ + hinge), containing all three hybridization sites (ETS and ETS2 [black] and 18S [orange]). (B) Schematic overview of the pre-rRNA marked by the U3-dependent cleavage sites (A0, A1, and A2) and the three U3-hybridization sites above. Below is the secondary structure of the pre-rRNA fragments between the ETS and 18S hybridization sites, which is designated pre-rRNA (ETS-18S). The smaller pre-rRNA fragment that begins at stem–loop IX is designated pre-rRNA (IX-18S). The smallest fragment contains just the 18S site. (C) The kinetic unfolding barrier to formation of the U3-18S hybridization involves the unfolding of the U3 box A/A′ and the 18S H1 stem structures.
Initiated by pre-rRNA transcription in the nucleolus, the preribosome assembles in a stepwise and complex manner with many trans-acting factors recruited as preformed subparticles (Perez-Fernandez et al. 2011). The U3 snoRNA (Fig. 1A) assembles with five proteins, which contact its 3′ box C′/D region and are common to other box C/D snoRNAs (Phipps et al. 2011). The resulting 20S U3 snoRNA-protein particle is recruited to the pre-rRNA by protein–protein interactions (Perez-Fernandez et al. 2011). After recruitment, formation of three U3-pre-rRNA duplexes is a prerequisite for the U3-dependent pre-rRNA cleavages at A0, A1, and A2 that are essential for cellular growth: the U3-ETS duplex (Beltrame and Tollervey 1995), the U3-ETS2 duplex (Borovjagin and Gerbi 2004; Dutca et al. 2011; Marmier-Gourrier et al. 2011), and the U3-18S duplex (Fig. 1A,B; Hughes et al. 1987; Mereau et al. 1997; Sharma and Tollervey 1999). Cleavages at A0 and A1 liberate the mature 5′ end of 18S, whereas cleavage at A2 produces a 3′ end within ITS1 (Fig. 1B), requiring further processing in the cytoplasm at a later time.
The initial U3-pre-rRNA duplex to form arises from the first pre-rRNA hybridization site to be transcribed: the U3-ETS2 duplex, same as 3H (Dutca et al. 2011), and as helix VI (Marmier-Gourrier et al. 2011). In vivo, formation of the U3-ETS2 duplex was recently shown to be a prerequisite for forming the other two U3-pre-rRNA duplexes (Dutca et al. 2011; Marmier-Gourrier et al. 2011). Because the relevant U3 and pre-rRNA bases are exposed, the U3-ETS2 duplex is expected to form spontaneously, but in vitro formation of this duplex has not yet been tested. As transcription continues, the U3-ETS duplex is the next to form, followed by U3-18S hybridization.
A fourth U3-pre-rRNA base-pairing (Hughes et al. 1987; Hughes 1996; Sharma and Tollervey 1999; Kudla et al. 2011; Swiatkowska et al. 2012) may chaperone formation of a universal and central structure of small ribosomal subunits: the central pseudoknot. This pseudoknot involves base pairs between bases in the loop of helix 1 (H1) of 18S and a complementary site >1000 nt downstream: 1139 to 1143. Juxtaposing these two distal elements is expected to favor pseudoknot formation upon release of U3 snoRNA from the U3-pre-rRNA base pair. Formation of this U3-pre-rRNA interaction is mutually exclusive with formation of the central pseudoknot because both structures involve the same bases in the loop of 18S H1.
Previously, we showed in vitro that Imp3 and Imp4 stabilize the U3-ETS duplex, which is unstable due to its short length and AU-rich sequence (Gerczei et al. 2009). The increased stability results in a duplex yield high enough to satisfy the in vivo demands of rapidly growing cells. Unlike the duplexes involving the 5′-ETS nucleotides, formation of the U3-18S duplex is not observed in vitro due to two barriers (Fig. 1C). First, bases involved in this duplex are buried in two stem–loop structures: the U3 box A/A′ stem and 18S H1, which forms at the 5′ end of 18S. Second, once unfolded, each stem–loop structure is predicted to refold faster than the competing hybridization reaction. Using minimal substrates, the U3 fragment was sufficient to form the box A/A′ structure, but the pre-rRNA fragment was too short to form the 18S H1; both proteins (Imp3 and Imp4) are required for U3-18S duplex formation (Gerczei et al. 2009). Addition of these proteins accelerates hybridization from an undetectable rate (≤400 M−1 s−1) to one indistinguishable from the spontaneous rate of hybridization between two short duplexes (∼106 M−1 s−1). Time-resolved FRET data indicated protein-dependent changes to the distance between two fluorophore-labeled U3 nucleotides at the bottom of the U3 box A/A′ stem structure. Unfortunately, these fluorophore probes were up to 15 nt away from bases involved in U3-18S hybridization. Furthermore, no data were available for how Imp3 and Imp4 affect the pre-rRNA structure. Thus, how protein binding affects accessibility of the U3 box A/A′ and the 18S H1 nucleotides involved in U3-18S hybridization remained unknown, and the fundamental mechanistic question of how proteins remove the barriers to U3-18S hybridization remained unanswered.
Here, we addressed this mechanistic question by probing how protein binding affects the base and backbone accessibility of the pre-rRNA and the U3 structures. The data indicate that Imp3 and Imp4 unfold the U3 box A/A′ stem structure and promote U3-18S duplex formation using minimal substrates. In contrast, only Imp3 is able to promote formation of this duplex using larger pre-rRNA fragments, whose conformations closely resemble the pre-rRNA secondary structure observed in vivo. Imp3 binds to these pre-rRNA fragments, unfolding the 18S H1 and exposing nucleotides adjacent to the A0 and A1 cleavage site, perhaps to promote endonucleolytic cleavage. These pre-rRNA and U3 conformational changes needed for hybridization may not only play a crucial role in preventing premature or incorrect cleavage at A0, A1, and A2 but also avert premature formation of the universal central pseudoknot of 18S.
RESULTS
Stability of the U3 box A/A′ stem structure
Conserved among eukaryotes, the U3 box A/A′ stem structure buries the bases involved in U3-18S hybridization, thereby blocking formation of this duplex. Estimates of the upper limits of the energy needed to unfold the box A/A′ structure were derived from UV melting data of a minimal U3 snoRNA substrate (U3 MINI) (Fig. 1A), with a melting temperature of 54°C (Gerczei et al. 2009). Thus, at the growth temperature of vertebrates (37–42°C) it is expected to remain folded. The unfolding free energy, extracted from the melting data and fit with Meltwin3 V3.5 (Petersheim and Turner 1983; Longfellow et al. 1990), is 5.4 kcal/mol at 30°C, corresponding to a folded-to-unfolded Keq of ∼1 × 10−4. This free energy barrier requires helix destabilization activity to ensure rapid U3-18S duplex formation. Previously, we showed that Imp3 and Imp4 bind to nucleotides 1–76 of U3 snoRNA (Gerczei and Correll 2004), which contain the box A/A′ stem structure and the hinge region and are designated U3 (box A/A′ + hinge) (Fig. 1A). Moreover, the presence of both proteins enables U3-18S hybridization (Gerczei et al. 2009). Importantly, it was unknown whether protein binding unfolds the box A/A′ stem structure.
Imp3 binding opens up the U3 box A/A′ stem
To directly test whether Imp3 or Imp4 binding unfolds the box A/A′ stem structure of U3 snoRNA, we probed base accessibility throughout this stem structure with 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate (CMCT). CMCT modifies the Watson-Crick face of two bases (U at N3 and G at N1) with sites of enhanced or protected base accessibility identified by comparing the primer extension pausing of a CMCT-modified U3 (box A/A′ + hinge) template in the absence and presence of protein. Protein-dependent enhancement arises from conformational changes that expose bases. Conversely, protein-dependent protection arises from either direct protein binding that sterically blocks base accessibility or indirect effects that trigger RNA conformational rearrangements, resulting in reduced base accessibility. To ensure that pausing in primer extension monitors base accessibility rather than template degradation or some other trivial cause, we compared primer extension pausing of RNA and RNA/protein complexes in the presence and absence of a modifying agent, where each sample was subjected to the same procedure (Fig. 2A, cf. lane 5 with lanes 1–4).
FIGURE 2.
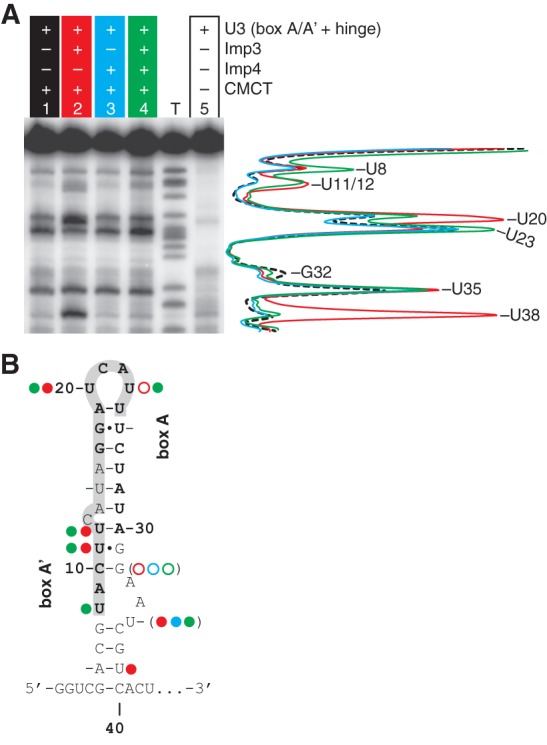
Protein binding increases base accessibility in the A/A′ region of U3 snoRNA as probed by reactivity of CMCT modification. (A) Representative data for CMCT modification for 80 nM U3 (box A/A′ + hinge) incubated in the absence or presence of 1.5 μM Imp3 for 45 min and then treated with 20 mg/mL CMCT for 30 min at RT. Sites modified by CMCT were detected by pauses in primer extension and mapped using a sequencing ladder. Lane intensity quantification is shown on the right for lane 1 (dashed black), lane 2 (red), lane 3 (cyan), and lane 4 (green); each lane was normalized based on counts from a band near the primer (band not shown) that remained unchanged in each lane. Lane T, here and in Figures 5A and 6A, is the dideoxy TTP sequencing ladder. (B) Bases that are protected (open circles) or enhanced (filled circles) upon addition of Imp3 (red), Imp4 (cyan), and both proteins (green) as compared to lane 1 (A) are mapped on the secondary structure of the U3 box A/A′ structure. Parentheses indicate small effects at 32 and 35.
Addition of Imp3 to U3 (box A/A′ + hinge) increases base accessibility throughout the box A/A′ stem structure: in box A′ at U11 and U12, in box A at U20, and in the base of the stem at U35 (slightly) and U38 (strongly) (Fig. 2A, cf. lanes 1 and 2). Imp3 binding also led to a decrease in base accessibility: in box A at U23 and the stem structure at G32. Larger changes were observed when Imp4 was added to a preformed Imp3-RNA complex. Accessibility of U8 increased and two reversals in accessibility were observed: U38 was now inaccessible, as observed in the absence of protein; and U23, with decreased accessibility in the presence of Imp3 alone, now exhibited increased accessibility relative to either the unbound or Imp3-bound RNA. In contrast, addition of Imp4 alone produced small changes: slightly increasing base accessibility in the stem structure at U35 and slightly decreasing accessibility in the box A′ stem at U32 (Fig. 2A, cf. lanes 1 and 3). Evidently, addition of Imp3, either in the presence or absence of Imp4, increases base accessibility throughout the box A/A′ stem structure. These accessibility data concur with previous time resolved FRET data, indicating that Imp3 binding increased the distance between nucleotides at the base of the box A/A′ stem structure (Gerczei et al. 2009). These data provide evidence that Imp3 binding unfolds the box A/A′ structure, raising the possibility that addition of this protein alone is sufficient to mediate U3-18S hybridization.
Imp3 mediated U3-18S duplex formation with a minimal 18S substrate
To test whether Imp3 has annealing activity, we used our previously developed fluorescence-based assays and substrates (Gerczei et al. 2009). The U3-18S duplex association and dissociation rates, kon and koff, respectively (Figs. 1, 3; Table 1), were determined by monitoring the time-dependent quenching of fluorescein labeled at the 5′ end of U3 MINI (Fl-U3 MINI) after addition of 18S labeled with tetramethylrhodamine at either its 5′ or 3′ end, Rh-18S and 18S-Rh, respectively.
FIGURE 3.

Protein dependent U3-18S association (kon) and dissociation (koff) rate constants (Table 2). (A) Representative trace (left y-axis, gray squares) is shown for Fl quenching upon addition of Rh-18S to the Fl-U3 MINI/Imp3 complex to a final concentration of 10 nM of each RNA. The [AB]apparent values were calculated, plotted on the right y-axis (black circles) and fit (gray line) as previously described (Gerczei et al. 2009) to give a kon of 1.3 × 106 M−1 s−1. (B) Representative dissociation reaction traces were initiated by addition of a 100-fold excess chase of unlabeled 18S to a preformed complex with equimolar concentrations of Fl-U3 MINI and Rh-18S (10 or 15 nM) in the presence of either Imp3 (gray) or Imp4 (black). The time-dependent recovery of the quenched Fl fluorescence was fit to a single exponential as described previously (Gerczei et al. 2009) to yield koff.
TABLE 1.
U3-18S duplex kinetic and thermodynamic parameters
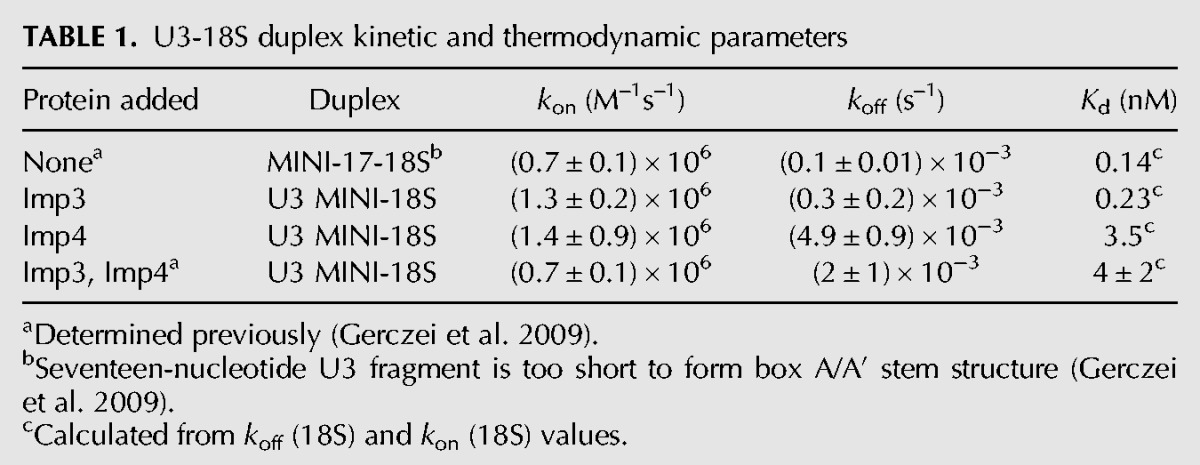
In the presence of Imp3, Fl-U3 MINI associates with Rh-18S to form the U3-18S duplex with a kon of (1.3 ± 0.2) × 106 M−1 s−1 and a koff of (0.3 ± 0.2) × 10−3 M−1s−1 (Table 1). Whereas the kon is only slightly more than the value previously reported in the presence of both Imp3 and Imp4 (Table 1; Gerczei et al. 2009), koff differs. This koff is similar to that determined for a U3 substrate that retains only the 18S binding nucleotides, designated MINI-17 (Gerczei et al. 2009). MINI-17 is unable to form a hairpin because the 3′ half of this stem structure is deleted. In the absence of a kinetic unfolding barrier, duplex formation occurs rapidly, as expected. U3-18S hybridization was verified using nondenaturing electrophoretic mobility shift assay (EMSA) (data not shown; Gerczei and Correll 2004). These data demonstrate that Imp3 alone is sufficient to remove the kinetic unfolding barrier to enable rapid U3-18S hybridization. Differences between the observed chaperone activity of Imp3 presented herein and our previous studies (Gerczei and Correll 2004) may arise from removal of detergent from the purification protocol. In contrast, removal of detergent did not show any effect on the activity of Imp4 (Gerczei et al. 2009). The kon in the presence of Imp4 shows a similar value to that observed for Imp3 (Table 1). In sharp contrast, the koff of (4.9 ± 0.9) × 10−3 M−1 s−1 is more than 16-fold faster than the koff in the presence of Imp3 (Fig. 3B, cf. the blue and the red traces) and is only somewhat more than the rate observed in the presence of both proteins (Gerczei et al. 2009). These kinetic dissociation constants provide evidence that each protein individually promotes U3-18S hybridization. In contrast, Imp4, either alone or when bound with Imp3, destabilizes the U3-18S duplex but Imp3 alone does not. To assess the role of this destabilization activity in promoting U3-18S release after the U3-dependent cleavage events, further investigation is needed but is beyond the scope of this study.
The RNA binding energy of either Imp3 or Imp4 is sufficient to open up the box A/A′ stem structure, thereby stabilizing the unfolded conformation. Chemical modification data (Fig. 2) indicate that the protein-bound state of the RNA is unfolded by Imp3 alone, not by Imp4 alone. Thus, the observed binding energy is the sum of energies from RNA-protein docking and unfolding. Imp3 binds to the U3 (box A/A′ + hinge) substrate with a Kd of 218 ± 62 nM (Gerczei and Correll 2004), indicating that the unfolded U3 stem-structure is stabilized when bound to protein. This dissociation constant corresponds to a folded-to-unfolded Keq of 4.6, given a standard state defined by the estimated nucleolar U3 snoRNA concentration of ∼1 μM (Gerczei et al. 2009). Thus, protein binding shifts this Keq by greater than 36,000-fold relative to the value in the absence of protein. This large and protein-dependent shift in Keq may create a protein-dependent switch.
Stability of the 18S H1 in pre-rRNA
To explore whether there is a corresponding switch occurring in the pre-rRNA, larger substrates must be investigated. The pre-rRNA nucleotides involved in the U3-18S duplex form a stable and universal stem–loop structure designated H1. In previous assays (Fig. 1B; Gerczei et al. 2009), the pre-rRNA substrate was truncated to its 18S hybridization site, preventing formation of the universal H1 stem–loop structure. H1 is stabilized by 5.3 kcal/mol at 30°C based on Mfold v2.3 calculations (Zuker 2003). To unfold 18S H1 in the pre-rRNA and the U3 box A/A′ stem structures, chaperone activity is needed to satisfy the in vivo demand for rapid U3-pre-rRNA hybridization (Kos and Tollervey 2010).
The larger pre-rRNA fragments
To test whether Imp3 binding unfolds the 18S H1 stem–loop structure, we probed base accessibility throughout this stem structure with CMCT using two larger pre-rRNA fragments: the pre-rRNA (ETS-18S) and pre-rRNA (IX-18S) (Fig. 1B). The pre-rRNA (ETS-18S) consists of nucleotides 469–700 of the 5′ ETS and H1 of 18S (nucleotides 1–23) to provide the bases involved in the U3-18S and U3-ETS duplexes. This second substrate is shorter, spans the region between stem–loop IX of the ETS and H1 of 18S and is designated pre-rRNA (IX-18S) (Fig. 1B).
To ensure that the conformations of the pre-rRNA fragments are in agreement with the secondary structure previously mapped in vivo (Yeh and Lee 1992), we probed pre-rRNA (ETS-18S) reactivity to CMCT modification and to T1 ribonuclease cleavage (Supplemental Figs. S1, S2). These probing results are also in agreement with the pre-rRNA (ETS-18S) SHAPE reactivity (Supplemental Fig. S3). The determined secondary structure concurs with the earlier work (Yeh and Lee 1992) except at the base of stem X, where our data suggest that this stem extends further than previously modeled (Yeh and Lee 1992). The RNA in this region is, however, expected to be dynamic due to its limited base reactivity with CMCT in this region. Others predicted the extension to stem X based on sequence complementarity but lacked experimental evidence (Elela et al. 1996; Venema and Tollervey 1999). It remains to be established whether this extended stem structure exists in vivo. Shape, CMCT, and T1 data show that both pre-rRNA fragments fold into the secondary structure that was observed in vivo.
Imp3 binds to pre-rRNA fragments
To determine whether protein binds to the pre-rRNA fragments, we used EMSAs. In agreement with earlier experiments (Gerczei and Correll 2004), Imp4 does not bind specifically to pre-rRNA (ETS-18S). On nondenaturing PAGE, the bound species are a smear at high protein concentrations, indicating a rapid equilibrium between the bound and the free RNA species within the time frame of the assay (data not shown). In contrast to our previous studies using filter binding assays (Gerczei and Correll 2004), Imp3 bound to pre-rRNA (ETS-18S) in the presence of the nonspecific RNA competitor tRNA with a Kd of 1.4 ± 0.3 μM (Fig. 4; Table 2). The absence of stem–loop VIII and the ETS site in pre-rRNA (IX-18S) had no effect on binding affinity (Table 2), consistent with the protein binding to the 3′ end of the ETS region and the 5′ end of the 18S.
FIGURE 4.

Imp3 binds to pre-rRNA (ETS-18S). Representative EMSA data for binding of Imp3 to 32P-labeled pre-rRNA (ETS-18S) in the presence of a nonspecific competitor (0.1 mg/mL tRNA). Fraction bound is defined as the fraction of labeled RNA shifted and plotted against increasing protein concentration. Binding constant (Kd) is calculated by fitting to a hyperbolic Hill binding curve (Table 2; Materials and Methods).
TABLE 2.
Apparent dissociation constants (Kd)

Imp3 binding unfolds 18S H1 and an adjacent stem structure of the pre-rRNA
To investigate whether Imp3 or Imp4 binding to pre-rRNA alters its structure, we probed the base accessibility of relevant pre-rRNA fragments by CMCT in the absence and presence of each protein. Addition of Imp3 to pre-rRNA (ETS-18S) enhances base accessibility to CMCT at the 3′ end of ETS and the adjacent A1 site and at the 5′ end of 18S (Fig. 5A, cf. lanes 1 and 2), in accord with protein binding opening up the stem structures of this region. The largest enhancements occur at U700 in the ETS and at U1, U3, and U9 in the 18S region. Intermediate levels of enhancements occur in H1 of 18S at U5, U8, and U12. The remaining changes are small and flank the A0 and A1 cleavage sites at the base of the X stem structure. The strength of enhancement is time-dependent, indicating that the signal of the modifying reagent is not saturated, consistent with modification serving as a proxy for accessibility. Interestingly, the sites strongly enhanced by Imp3 are either around the A1 cleavage site or in H1, containing the 18S base-pairing site (Fig. 5). We obtained an equivalent result when using the shorter pre-rRNA (IX-18S), in which nucleotides from ETS to stem VIII are deleted (Fig. 1B; Supplemental Fig. S4). In contrast to Imp3 binding, Imp4 binding does not show this chaperone activity (data not shown), and when Imp4 is added to a preformed complex between Imp3 and a pre-rRNA fragment, the activity is indistinguishable from that observed upon addition of Imp3 alone to the pre-rRNA (ETS-18S) (data not shown). Apparently, Imp3 binding exposes the 18S hybridization site by unfolding H1. The Imp3-dependent unfolding activity raises the possibility that this protein promotes U3-18S hybridization. To verify that the observed changes in base accessibility arise from a specific Imp3-pre-rRNA interaction, we repeated these modification reactions using the basic protein U1A added at high enough concentrations to promote nonspecific binding. Under these conditions, U1A did not cause the Im3p specific increases in base accessibility (data not shown). Moreover, these Imp3 changes in base accessibility were unchanged upon addition of an RNA competitor (0.1 mg/mL tRNA) (data not shown).
FIGURE 5.

Primer extension analysis of base accessibility of the pre-rRNA (ETS-18S) probed by CMCT modification. (A) The CMCT reactions were carried out with 40 nM of pre-rRNA (ETS-18S) incubated in the absence or presence of 3 μM Imp3 for 45 min and then treated with 20 mg/mL CMCT for either 7 or 25 min at RT. Lane intensity was quantified, normalized to total counts in the lane, and is shown on the right for lane 1 (blue), lane 2 (red), and lane 4 (gray). (B) Modification sites enhanced by CMCT upon addition of Imp3 were divided into three levels (strong, medium, and weak) based on normalized intensities and are marked with corresponding circle sizes on the pre-rRNA secondary structure.
Imp3 mediates U3-18S hybridization with larger pre-rRNA fragments
To test whether the Imp3 binding promotes annealing between U3 (box A/A′ + hinge) and a pre-rRNA fragment, we probed the hybridization with CMCT. CMCT modification can monitor base pair formation because hybridization buries the Watson-Crick face of nucleotides to block CMCT modification. To minimize CMCT reactivity arising from duplex dissociation, we reduced the CMCT reaction time. As observed above (Fig. 5), Imp3 enhanced base accessibility of pre-rRNA (ETS-18S) at the 3′ end of the ETS and the adjacent 5′ end of the 18S (Fig. 6A, cf. lanes 1 and 2). Importantly, the Imp3-dependent enhancement was protected by subsequent addition of U3 (box A/A′ + hinge), which contains the U3-18S hybridization sites (Fig. 6A, cf. lanes 2 and 3). The protection localized to a region inside the U3-18S hybridization site (U8, U9, and U21) and adjacent nucleotides, including the A1 site, U1, U3, and U5. To verify that protection arises from hybridization and not from conformational changes, modifications were carried out with an antisense U3 analog. This analog, designated U3 (AS box A/A′ + hinge), contains noncomplementary RNA sequences at the U3-18S hybridization site and other mutations to maintain a box A/A′-like stem structure. As expected, the antisense analog does not show protection (Fig. 6A, cf. lanes 2, 3, and 4). We obtained an equivalent result when using the shorter pre-rRNA (IX-18S) incubated with either the sense or the antisense U3 analog (Supplemental Fig. S4). The CMCT results were unable to address whether U3-18S hybridization occurs in the absence of protein because under these conditions there is effectively no CMCT signal to monitor.
FIGURE 6.

Imp3 is required for U3-18S hybridization. (A) Primer extension analysis of the signal of U3-18S hybridization detected by CMCT modification of the pre-rRNA (ETS-18S). Lane intensity quantification, normalized as in Figure 5A, is shown on the right for lanes containing Imp3 alone (red) or in the presence of either U3 (box A/A′ + hinge) (orange) or U3 (AS box A/A′ + hinge) (cyan). The CMCT reactions were performed as in Figure 5A except that samples were treated with 40 mg/mL CMCT for 1 min at 30°C. Ignore primer extension pausing at C4 (asterisk), which is due to a break in the template. (B) T1 cleavages were detected by monitoring breaks in 32P-labeled pre-rRNA (IX-18S) in the absence or presence of 0.3 μM Imp3. Lane intensity quantification, normalized to counts in the full-length substrate, is shown on the right for lanes containing pre-rRNA (IX-18S) alone (dashed black) or in the presence of Imp3 incubated with either U3 (box A/A′ + hinge) (orange), U3 (AS box A/A′ + hinge) (cyan), or buffer (red). RNA-protein complexes were first incubated for 45 min, then treated with 1.6 units of T1 in a 20-μL reaction for 15 min at RT. (C) Pre-rRNA (IX-18S) sites whose modification changes upon addition of U3 (box A/A′ + hinge) in the presence of only Imp3 are indicated on the secondary structure of the pre-rRNA: CMCT (open circle) and T1 (bulls eye).
To address whether Imp3 is needed to promote U3-18S hybridization and verify Imp3-dependent formation of the U3-18S duplex, we performed T1 ribonuclease protection assays. This enzyme monitors backbone accessibility by cleaving single-stranded regions of RNA on the 3′ side of G nucleotides. The 3′ end of the substrate was easier to detect using the pre-rRNA (IX-18S) (Fig. 1B); however, similar results were observed using the larger fragment pre-rRNA (ETS-18S) (Supplemental Fig. S4B,C). Consistent with CMCT modification data (Fig. 5), we observed that Imp3 binding enhances the backbone accessibility of pre-rRNA (IX-18S) at G6 and G7 of 18S H1, providing evidence that this stem–loop unfolds (Fig. 6B, cf. lanes 3 and 6). Formation of the U3-18S duplex is expected to block T1 digestion at G6, G7, G10, G16, G20, and G23 of the 18S portion of the pre-rRNA substrates. As expected, the backbone accessibility of these sites was protected by subsequent addition of U3 (box A/A′ + hinge) but not of U3 (AS box A/A′ + hinge) (Fig. 6B, cf. differences between lanes 5 and 6 with differences between lanes 4 and 6). Upon hybridization, the largest change occurred at G10, consistent with the only G transitioning from an exposed single-stranded loop region to double-stranded RNA. The other sites of hybridization-dependent protection are in H1 at G6, G7, G16, G20, and G23 and around the A1 site at G695 and G699 (Fig. 6B, cf. lanes 5 and 6) but not in other regions of RNA (Supplemental Fig. S5). The CMCT and T1 data provide evidence that Imp3 is needed to mediate U3-18S hybridization with the larger pre-rRNA substrates. The extra sites of protection at U1, U3, and U5 that are adjacent to the U3-18S duplex may arise from additional U3-pre-rRNA base pair formation (Supplemental Fig. S6).
U3-ETS and U3-ETS2 duplex formation
Lastly, we investigated the role of protein in mediating the other two U3-pre-rRNA interactions. In addition to the U3-18S duplex, U3 (box A/A′ + hinge) also contains the bases involved in the U3-ETS2 and U3-ETS duplexes. The ETS2 site binds to full-length U3 snoRNA with a Kd of 2.2 nM in the absence of protein (Supplemental Fig. S7), demonstrating that protein is not required to form this duplex. Previously, we demonstrated that Imp3 and Imp4 mediate the formation of the U3-ETS duplex by increasing duplex stability and thus yield (Gerczei et al. 2009). Here, we investigated, using T1 protection assays, whether Imp3 alone mediates formation of the U3-ETS duplex with larger RNA substrates: pre-rRNA (ETS-18S) and U3 (box A/A′ + hinge). The protection of nucleotides inside the ETS site was only detected in the presence of Imp3 and U3 (box A/A′ + hinge) but not in the absence of protein or U3 substrate (Supplemental Fig. S8). Moreover, the sites of protection were localized to inside the ETS site. Apparently, Imp3 is needed to mediate both the U3-ETS and the U3-18S duplex for larger RNAs that more closely mimic the full-length substrates in vivo.
Protein-dependent stem–loop unfolding
The reaction profile in Figure 7 illustrates the two barriers that prevent duplex formation: unfolding and hybridization. Both RNAs of the duplex must unfold—the U3 box A/A′ and the 18S H1 stem–loop structures. Due to the >5 kcal/mol stability of each structure, only a small percentage of either RNA exists in the unfolded form in the absence of protein. Duplex formation is not observed for this small percent of unfolded RNA because the barrier to hybridization is higher than the barrier to reform U3 and 18S stem–loop structures, under estimated physiological conditions (Hughes et al. 1987; French et al. 2003; Berger et al. 2008; Gerczei et al. 2009). In sharp contrast, protein binding stabilizes the unfolded form of both structures and may also lower the activation barrier to unfolding. As a result, the barrier to hybridization is lower than the barrier to refold the U3 and pre-rRNA stem structures, such that duplex formation is observed in the presence of protein but not in its absence. These structural changes can function as protein-dependent switches to regulate higher-order events of ribosome biogenesis, such as the U3-dependent cleavage events.
FIGURE 7.
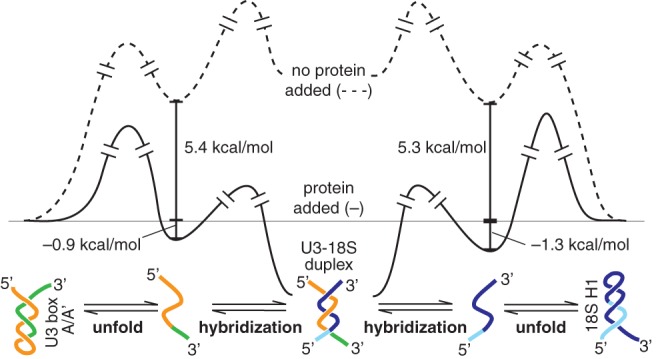
Reaction profiles illustrate a mechanism for protein binding enabling U3-18S duplex formation by unfolding of both the 18S H1 and the U3 box A/A′ stem structures (first step) rather than directly accelerating hybridization (second step). In the absence of protein (dashed gray line), the >5 kcal/mol needed to unfold either of these stem structures limits the amount of unfolded RNAs, which, in turn, reduces the subsequent product duplex to an undetectable level. In contrast, protein binding (black line) unfolds both U3 and 18S stem structures to form protein-stabilized open structures that can proceed to spontaneous hybridization. The standard state is defined as 30°C in reaction buffer, with estimated in vivo RNA concentrations (Gerczei et al. 2009): 1 μM U3 snoRNA and 13 μM pre-rRNA.
DISCUSSION
This report determines important insights into the mechanism by which Imp3 and Imp4 promote U3-pre-rRNA duplex formation that is essential for small subunit processing. Imp3 alone unfolds stem–loop structures in both the pre-rRNA and U3 snoRNA to expose the bases involved in the U3-18S duplex (Figs. 2, 5), thereby promoting hybridization (Fig. 6). Moreover, this unfolding activity increases accessibility of bases surrounding the A0 and A1 cleavage sites, perhaps signaling cleavage at these sites to generate the 5′ mature end of 18S. Though Imp4 does not interact with the pre-rRNA fragments, it aids in unfolding the U3 stem structure (Fig. 2). Imp4 may also facilitate U3 release because its binding destabilizes the U3-18S duplex (Fig. 3).
Our studies differentiate the roles of Imp3 and Imp4 in mediating each of the U3-pre-rRNA duplexes. Of the three essential duplexes formed between the U3 snoRNA and the pre-rRNA, the U3-ETS2 forms first (Hughes et al. 1987; Beltrame and Tollervey 1995; Sharma and Tollervey 1999; Dutca et al. 2011; Marmier-Gourrier et al. 2011). Our results indicate that no protein is needed to form this duplex (Supplemental Fig. S7). Formation of the U3-ETS duplex is next. With the minimal substrate, Imp3 and Imp4 are needed to increase the duplex yield enough to satisfy in vivo demands (Gerczei et al. 2009), whereas with larger pre-rRNA substrates that mimic the secondary structure found in vivo, Imp3 alone is sufficient for U3-ETS duplex formation (Supplemental Fig. S8).
For the third duplex, U3-18S duplex formation is promoted by either Imp3 or Imp4 using minimal substrates (Fig. 3), evidently by unfolding the box A/A′ stem structure (Fig. 2). However, Imp3 is more efficient at unfolding this U3 stem structure. Protein-dependent structural changes to the pre-rRNA were not investigated with the minimal substrate because it is too small to form H1. Unlike the U3-18S duplex association rates, which are the same in the presence of either protein, the dissociation rate in the presence of Imp4 is 16-fold faster than the koff in the presence of Imp3 (Fig. 3B). Rapid U3-pre-rRNA release is important to ensure release of the pre-18S subunit for transport to the cytoplasm, where final processing and assembly occur. This release is also needed to recycle the U3 snoRNA and its associated proteins and thus expose the bases in the loop of H1 to permit formation of the 18S central pseudoknot. The helix destabilizing activity of Imp4 is too slow to support in vivo demands for rapid U3 release (Kos and Tollervey 2010), suggesting that a helicase is needed. Of the 19 helicases that are required for ribosome biogenesis in yeast (for review, see Rodriguez-Galan et al. 2013), Ecm16 (Dhr1) (Colley et al. 2000) and Has1 (Liang and Fournier 2006) are candidates to dislodge U3 from the pre-ribosome. Perhaps Imp4 aids this helicase activity.
With larger pre-rRNA fragments (>250 nt), formation of the U3-18S duplex has different requirements. Imp3, but not Imp4, binds to larger pre-rRNA fragments (Fig. 4; Table 2). Protein binding unfolds both the U3 box A/A′ and the 18S H1 stem–loop structures (Fig. 5) and is necessary and sufficient to form the U3-18S duplex (Fig. 6). The binding energy of Imp3 is sufficient to form stable structures where the bases in the U3 snoRNA and pre-rRNA stem structures are accessible and thus available for hybridization. Moreover, protein binding may lower the activation barrier for unfolding these structures (Fig. 7). Protein binding not only removes the first barrier to hybridization by unfolding the U3 and pre-rRNA stem structures, it also removes the second barrier. By stabilizing the unfolded form of the U3 and pre-rRNA stem–loop structures, the RNAs bound to Imp3 are expected to hybridize faster than the competing reaction of stem–loop refolding. In contrast, hybridization is blocked in the absence of protein because unfolded stem–loop structures that are present are predicted to refold faster than the subsequent hybridization step. A matchmaker mechanism (Sancar and Hearst 1993) is possible because Imp3 binds to either substrate: the U3 snoRNA or pre-rRNA. Even though simultaneous binding of both RNA molecules to one Imp3 is unlikely, it may be possible for a dimer of Imp3 (data not shown) to have one monomer bind to one RNA and the other bind to the complementary strand. To unfold the U3 box A/A′ or the 18S H1 stem structure, Imp3 binding pushes and/or pulls the strands apart, thereby exposing the bases involved in base pair formation (Fig. 5).
We envisage that the protein-dependent formation of the U3-18S duplex acts as a switch to direct the U3-dependent cleavage events. Imp3 binding controls whether the U3 and 18S stem structures are folded or unfolded and thus serves as a switch for U3-18S hybridization, which, in turn, is a prerequisite for the U3-dependent cleavage reactions. The off state, associated with folded U3 and 18S stem structures in the absence of protein, blocks the U3-dependent cleavages by preventing formation of the prerequisite U3-18S duplex. The on state, associated with protein binding that unfolds the U3 and 18S stem structures, allows rapid formation of the U3-18S duplex to permit U3-dependent cleavage events and thus initiates subsequent steps in small subunit (SSU) biogenesis. Perhaps this switch ensures that premature and inaccurate pre-rRNA cleavages are prevented during the stepwise assembly of the pre-ribosome. The bases in 18S H1 are also involved in the universal pseudoknot by pairing with nucleotides >1000 nt downstream. Upon release of U3 snoRNA, these juxtaposed distal elements can pair to form the pseudoknot. Thus, the on state may also block premature and incorrect formation of the 18S central pseudoknot.
A central question in understanding the molecular bases of ribosome biogenesis is how trans-acting factors direct progression from one step to the next in this stepwise process. To mark a transition from one step to the next, investigations by others illustrated the role of protein binding to specific pre-rRNA structures. The KsgA/Dim1 family of methyltransferases binds to part of the decoding center (helix 45) of a precursor of the small subunit to prevent immature SSU from participating in translation, whereas, methylation and release transition the subunit to the functional form (Lafontaine et al. 1995; Connolly et al. 2008; Xu et al. 2008). Bud23, another methyltransferase, recognizes a fold in the P-site of the small subunit; in this case, methylation and enzyme release may transition the ribosome to the next step in biogenesis (White et al. 2008). There is also an example of how pre-rRNA structural rearrangements regulate the order of pre-rRNA cleavage. In this case, structural rearrangements in the pre-rRNA triggered by cleavage at A2 may promote 3′ end formation of 18S by recruiting Nob1 to its correct pre-rRNA cleavage at site D (Lamanna and Karbstein 2011).
Here, we illustrate how protein recognizes a structure in the pre-rRNA to rearrange it and thus signal the next step of ribosome biogenesis. In our model, Imp3 is recruited to the preribosome after protein-independent formation of the U3-ETS2 duplex. Imp3 then stabilizes the U3-ETS duplex and unfolds the U3 box A/A′ and 18S H1 stem structures to permit formation of the U3-18S duplex. Formation of these three U3-pre-rRNA duplexes signals pre-rRNA cleavage at A0, A1, and A2, which may be aided by increased accessibility of nucleotides surrounding the A0 and A1 cleavage sites arising from Imp3 binding. Future challenges will address how Mpp10 binding to Imp3 and Imp4 (Lee and Baserga 1999; Granneman et al. 2003) affects their activity, how helicase activity dislodges the U3 complex from the pre-ribosome, and how U3-release promotes formation of the central pseudoknot.
MATERIALS AND METHODS
Sequences for proteins (Imp3 and Imp4) and RNA molecules are from S. cerevisiae. All reactions mediated by Imp3 or Imp4 were performed in reaction buffer (100 mM Tris pH 8.0, 100 mM KCl, 2 mM MgCl2 [Sigma-Aldrich, 255777]) at room temperature (RT) unless otherwise noted. Reactions catalyzed by purchased enzymes and purifications using purchased kits followed the recommendations of the manufacturer, unless otherwise noted.
Protein purification
Purification of Imp3 and Imp4 was carried out as described before (Gerczei and Correll 2004), except that no detergent was used and proteins were concentrated to ∼30 μM before storage in protein buffer (50 mM MES pH 6.5, 50 mM MgCl2, 1 M urea and 5 mM 2-mercaptoethanol).
RNA substrates and radiolabeling
Three of the four RNA substrates <50 nt have been described before (Gerczei and Correll 2004): ETS, 18S, and U3 MINI (Fig. 1). The fourth, ETS2, represents nucleotides 282–292 of the pre-rRNA: 5′-GGAUUUGGUGG-3′. Of the four larger RNA substrates, two were previously described (Gerczei and Correll 2004): U3 (box A/A′ + hinge) and U3 snoRNA. The third, designated pre-rRNA (ETS-18S), represents nucleotides 469–723, stretching from the ETS hybridization site to the first 23 nt of the 18S sequence to include the first 18S H1. This RNA also contains two 5′-terminal G nucleotides to enable T7 transcription and a 3′ addition to enable primer extension readout of the 3′ end of the pre-rRNA (5′-GGGGCGGGGGAUCCUCUAGAGUCGA-3′). The fourth, designated pre-rRNA (IX-18S), represents nucleotides 542–723, including nucleotides from stem IX through to 18S H1. To resolve 3′ T1 cleavage product bands on denaturing PAGE, the sequence 5′-GCGGGCCUUCGGGCCAA-3′ was added to the 5′ end of this RNA. To enable primer extension readout of the 3′ end, the following sequence was added to the 3′ end: 5′-UCGAUCCGGUUCGCCGGAUCCAAAUCGGGCUUCGGUCCGGUUCAGUCGA-3′. The 5′- and 3′-flanking sequences are also designed to form stable hairpin structures in order that these nucleotides do not interfere with the proper folding of the pre-rRNA. Hereafter, these two RNAs will be designated the pre-rRNA fragments because they are treated in the same manner. The four small RNA substrates were made by solid-phase oligonucleotide synthesis (Integrated DNA Technologies, Inc.). The four larger RNAs were made by T7 run-off transcription using linearized plasmid DNA templates. The full- length U3 snoRNA, the U3 (box A/A′ + hinge) were prepared as previously described (Gerczei and Correll 2004). The pre-rRNA (ETS-18S) and the pre-rRNA (IX-18S) were produced by run-off transcription using plasmid DNA templates linearized with Sal1 (New England Biolabs).
All RNAs analyzed by primer extension were purified by gel electrophoresis, precipitated by ethanol, resuspended in water, and stored in aliquots at −80°C. To avoid degradation, the two pre-rRNA fragments and the U3 snoRNA samples were thawed only twice.
Before use, each RNA substrate was refolded, except for ETS, ETS2, and 18S. Two of these RNAs (U3 [box A/A′ + hinge] and U3 MINI) were refolded using a quick heat and cool protocol (Gerczei and Correll 2004; Gerczei et al. 2009). The remaining three RNAs (the two pre-rRNA fragments and U3 snoRNA) were refolded by heating at 42°C for 20 min, followed by incubation on ice for at least 10 min. The previous refolding method (Gerczei and Correll 2004) gave the same results but was avoided due to increased RNA degradation.
To minimize degradation of large RNA substrates, standard 5′-32P radiolabeling procedures (Sambrook et al. 1989) were modified by adding 40 units of RNasin (Promega) to each step and by avoiding heat inactivation. After antarctic phosphatase (New England Biolabs) treatment, the reaction was quenched and protein was removed by phenol/chloroform extraction, followed by ethanol precipitation. To label the 5′ end of each RNA, T4 Polynucleotide Kinase (New England Biolabs) was used in the presence of γ-[32P]-ATP (PerkinElmer, 150 μCi/μL). Radiolabeled RNAs were purified by gel electrophoresis, precipitated by ethanol, resuspended in water, and stored at −20°C. Degradation was also minimized by storing RNA in low-volume aliquots to avoid repeated freeze-thaw cycles.
DNA primers and radiolabeling
For pre-rRNA templates, primer extension reactions were primed with SalI_SLP, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCGACT-3′. For U3 (box A/A′ + hinge) templates, primer extension reactions were primed with Hinge_SLP, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTTGG-3′. Both of these stem–loop primers were made synthetically (Integrated DNA Technologies, Inc.), radiolabeled at the 5′ end using standard protocols (Sambrook et al. 1989), purified by gel electrophoresis, concentrated to ∼1 μM, and stored at −20°C.
Chemical modification and primer extension
Before use, each RNA substrate was refolded, as described above, and kept on ice until use. N-methylisatoic anhydride (NMIA, Invitrogen, M-25) was dissolved in DMSO and added to pre-rRNA (ETS-18S) at a final concentration of 6.5 mM for the SHAPE reagent and 40 nM for the RNA. A mock reaction was also carried out in the absence of NMIA but in the presence of 3 μM Imp3 and an equivalent amount of DMSO, to ensure that the modification as detected by primer extension arose from NMIA modification and not from a degraded RNA template or from DMSO. After reacting for 210 min at room temperature, the reaction was quenched by phenol/chloroform extraction, followed by an overnight ethanol precipitation using a carrier (2 μg glycogen) to increase yield.
To probe base accessibility of both pre-rRNA fragments, the RNA was reacted with the CMCT modifying reagent (Sigma-Aldrich, 29469). CMCT was dissolved in reaction buffer at 100 mg/mL, and the reaction was initiated by adding CMCT to a preformed RNA-protein complex (40 nM of pre-rRNA fragment and 3 μM of Imp3) to a final volume of 150 μL and concentration of 20 mg/mL CMCT. A mock reaction was performed by adding an equivalent volume of reaction buffer. After reacting for 25 min at room temperature, the reaction was quenched and precipitated as described above. In the hybridization reactions (Fig. 6A), the CMCT final concentration was increased to 40 mg/mL and incubated at 30°C for 1 min with 400 nM of U3 (box A/A′ + hinge). For the data in Figure 2, the base modifications were carried out using 80 nM of U3 (box A/A′ + hinge), 1.5 μM of Imp3 in 50 mM potassium borate pH 8.0, 100 mM ammonium chloride, and 10 mM MgCl2 instead of the reaction buffer.
To probe sites of NMIA and CMCT modifications, we used a standard primer extension assay (Tijerina et al. 2007) modified as follows. The precipitated pre-rRNA fragment from each modification assay was resuspended in 3 μL of water, of which 2 μL was used for primer extension with a stem–loop primer (SalI_SLP). Before annealing the SalI_SLP with each pre-rRNA fragment, the template was heated to 42°C for 10 min, and its primer was heated to 75°C for 2 min and cooled slowly to 42°C. Next, 1 μL of primer was hybridized to its template at room temperature for 15 min in 1 μL of 5× First-Strand Buffer (Invitrogen). Primer extension with SalI_SLP used 40 units Superscript II Reverse Transcriptase (Invitrogen) in 50 mM Tris pH 8.3, 10 mM DTT, 75 mM KCl, 3 mM MgCl2, and 0.15 mM dNTP.
Precipitated U3 (box A/A′ + hinge) from modification assays was suspended in 2 μL of water. Next, 1 μL of a stem–loop primer (Hinge_SLP) was hybridized by heating for 1 min at 90°C, followed by a slow cooling to <42°C in hybridization buffer. The cDNA was synthesized at 42°C for 40 min using 2 units of AMV Reverse Transcriptase (Life Sciences Advanced Technologies, Inc., LME704) in 25 mM Tris pH 8.3, 50 mM KCl, 5 mM MgCl2, 2 mM DTT, and 0.125 mM dNTP. Prior to use, AMV RT was diluted in 25 mM Tris pH 8.0, 50 mM KCl, and 2 mM DTT.
After primer extension, each RNA template was degraded, and the cDNA was precipitated (Tijerina et al. 2007) and resuspended in 9 M urea, 0.5× TBE, 0.02% (w/v) bromophenol blue, and 0.02% (w/v) xylene cyanol (made fresh). Nucleotide resolution was obtained on 12% (w/v) denaturing sequencing PAGE (29:1 acrylamide:bis acrylamide, 0.5× TBE, and 8 M urea). Dried gels were exposed on a Fuji imaging plate (BAS 2024), scanned by a Typhoon 9400 (Amersham Biosciences, GE), and quantified using Image Quant TL 7.0 (GE Healthcase Life Sciences). Normalized lane intensity quantification was displayed using Prism 6 (GraphPad, Inc.). When primer extension pausing was observed in control reactions in the absence of modifying agent, these sites were not used when investigating protein-dependent conformational changes and hybridization.
T1 ribonuclease protection assays
Up to 10 nM of 5′-32P labeled pre-rRNA (ETS-18S) was incubated for 45 min in the presence of either 0.7 μM Imp3 or protein buffer in a 20-μL reaction followed by digestion with 2 units of T1 ribonuclease (Life Technologies, AM2280) for 15 min at RT. The same reaction was used with the 5′-32P labeled pre-rRNA (IX-18S), except that Imp3 was 0.3 μM. To probe cleavage at each G on the RNA substrate, a denatured sample was also digested by 0.4 units of T1 in 7 M Urea, 20 mM sodium citrate pH 5.0, and 1 mM EDTA for 15 min. All reactions were quenched by phenol/chloroform extraction, and 10 μL of supernatant was mixed with an equal volume of loading dye (9 M Urea, 1× TBE) and resolved on denaturing PAGE (29:1 acrylamide:bis acrylamide, 0.5× TBE, and 8 M urea). Prior to use, T1 ribonuclease was diluted to 0.5 units/μL in 20 mM sodium Hepes pH 7.25, 1 mM EDTA, 0.1% (w/v) Triton, and 30% (v/v) glycerol. The 0.5× TBE is 45 mM Tris, 40 mM Borate, and 1.25 mM ETDA.
Fluorophore labeling of RNA and kon and koff assays
Fluorescein-labeled U3 MINI, tetramethylrhodamine-labeled 18S, and fluorescence-based kon and koff kinetic assays are as described before (Gerczei et al. 2009).
Binding affinity (Kd determination)
Kd values were determined by fitting the fraction of 32P-labeled RNA bound as a function of protein concentration, [protein], using
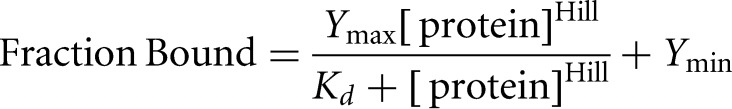 |
where Ymax and Ymin are the fraction-bound values at saturating and limiting [protein], respectively, and Hill is the Hill coefficient. Binding data were fit to the equation above using nonlinear regression (Prism 6, Graphpad, Inc.).
Experimental errors
Reported values represent the average and standard deviation of at least three measurements. Modification results were reproduced at least three times, and the results were independent of RNA and protein batches.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The work was supported by a grant from the National Institutes of Health to C.C.C. (GM070491). We thank Drs. M.J. Glucksman, A.W. Johnson, D.M. Mueller, and K.E. Neet for valuable advice and discussion.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Beltrame M, Tollervey D 1995. Base pairing between U3 and the pre-ribosomal RNA is required for 18S rRNA synthesis. EMBO J 14: 4350–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AB, Cabal GG, Fabre E, Duong T, Buc H, Nehrbass U, Olivo-Marin JC, Gadal O, Zimmer C 2008. High-resolution statistical mapping reveals gene territories in live yeast. Nat Methods 5: 1031–1037 [DOI] [PubMed] [Google Scholar]
- Borovjagin AV, Gerbi SA 2004. Xenopus U3 snoRNA docks on pre-rRNA through a novel base-pairing interaction. RNA 10: 942–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley A, Beggs JD, Tollervey D, Lafontaine DL 2000. Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNP U3. Mol Cell Biol 20: 7238–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Rife JP, Culver G 2008. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol Microbiol 70: 1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutca LM, Gallagher JE, Baserga SJ 2011. The initial U3 snoRNA:pre-rRNA base pairing interaction required for pre-18S rRNA folding revealed by in vivo chemical probing. Nucleic Acids Res 39: 5164–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elela SA, Igel H, Ares M Jr 1996. RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell 85: 115–124 [DOI] [PubMed] [Google Scholar]
- French SL, Osheim YN, Cioci F, Nomura M, Beyer AL 2003. In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol Cell Biol 23: 1558–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerczei T, Correll CC 2004. Imp3p and Imp4p mediate formation of essential U3–precursor rRNA (pre-rRNA) duplexes, possibly to recruit the small subunit processome to the pre-rRNA. Proc Natl Acad Sci 101: 15301–15306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerczei T, Shah BN, Manzo AJ, Walter NG, Correll CC 2009. RNA chaperones stimulate formation and yield of the U3 snoRNA–Pre-rRNA duplexes needed for eukaryotic ribosome biogenesis. J Mol Biol 390: 991–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Gallagher JE, Vogelzangs J, Horstman W, van Venrooij WJ, Baserga SJ, Pruijn GJ 2003. The human Imp3 and Imp4 proteins form a ternary complex with hMpp10, which only interacts with the U3 snoRNA in 60–80S ribonucleoprotein complexes. Nucleic Acids Res 31: 1877–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65: 2334–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JM 1996. Functional base-pairing interaction between highly conserved elements of U3 small nucleolar RNA and the small ribosomal subunit RNA. J Mol Biol 259: 645–654 [DOI] [PubMed] [Google Scholar]
- Hughes JM, Konings DA, Cesareni G 1987. The yeast homologue of U3 snRNA. EMBO J 6: 2145–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos M, Tollervey D 2010. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37: 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D 2011. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc Natl Acad Sci 108: 10010–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D, Vandenhaute J, Tollervey D 1995. The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev 9: 2470–2481 [DOI] [PubMed] [Google Scholar]
- Lamanna AC, Karbstein K 2009. Nob1 binds the single-stranded cleavage site D at the 3′-end of 18S rRNA with its PIN domain. Proc Natl Acad Sci 106: 14259–14264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanna AC, Karbstein K 2011. An RNA conformational switch regulates pre-18S rRNA cleavage. J Mol Biol 405: 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Baserga SJ 1999. Imp3p and Imp4p, two specific components of the U3 small nucleolar ribonucleoprotein that are essential for pre-18S rRNA processing. Mol Cell Biol 19: 5441–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Fournier MJ 2006. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol Cell Biol 26: 7437–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longfellow CE, Kierzek R, Turner DH 1990. Thermodynamic and spectroscopic study of bulge loops in oligoribonucleotides. Biochemistry 29: 278–285 [DOI] [PubMed] [Google Scholar]
- Marmier-Gourrier N, Clery A, Schlotter F, Senty-Segault V, Branlant C 2011. A second base pair interaction between U3 small nucleolar RNA and the 5′-ETS region is required for early cleavage of the yeast pre-ribosomal RNA. Nucleic Acids Res 39: 9731–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereau A, Fournier R, Gregoire A, Mougin A, Fabrizio P, Luhrmann R, Branlant C 1997. An in vivo and in vitro structure-function analysis of the Saccharomyces cerevisiae U3A snoRNP: Protein-RNA contacts and base-pair interaction with the pre-ribosomal RNA. J Mol Biol 273: 552–571 [DOI] [PubMed] [Google Scholar]
- Perez-Fernandez J, Martin-Marcos P, Dosil M 2011. Elucidation of the assembly events required for the recruitment of Utp20, Imp4 and Bms1 onto nascent pre-ribosomes. Nucleic Acids Res 39: 8105–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersheim M, Turner DH 1983. Base-stacking and base-pairing contributions to helix stability: Thermodynamics of double-helix formation with CCGG, CCGGp, CCGGAp, ACCGGp, CCGGUp, and ACCGGUp. Biochemistry 22: 256–263 [DOI] [PubMed] [Google Scholar]
- Phipps KR, Charette J, Baserga SJ 2011. The small subunit processome in ribosome biogenesis-progress and prospects. Wiley Interdiscip Rev RNA 2: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Galan O, Garcia-Gomez JJ, de la Cruz J 2013. Yeast and human RNA helicases involved in ribosome biogenesis: Current status and perspectives. Biochim Biophys Acta 1829: 775–790 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Sancar A, Hearst JE 1993. Molecular matchmakers. Science 259: 1415–1420 [DOI] [PubMed] [Google Scholar]
- Sharma K, Tollervey D 1999. Base pairing between U3 small nucleolar RNA and the 5′ end of 18S rRNA is required for pre-rRNA processing. Mol Cell Biol 19: 6012–6019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatkowska A, Wlotzka W, Tuck A, Barrass JD, Beggs JD, Tollervey D 2012. Kinetic analysis of pre-ribosome structure in vivo. RNA 18: 2187–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijerina P, Mohr S, Russell R 2007. DMS footprinting of structured RNAs and RNA–protein complexes. Nat Protoc 2: 2608–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Tollervey D 1999. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 33: 261–311 [DOI] [PubMed] [Google Scholar]
- Warner JR 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24: 437–440 [DOI] [PubMed] [Google Scholar]
- White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW 2008. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol Cell Biol 28: 3151–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, O’Farrell HC, Rife JP, Culver GM 2008. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol 15: 534–536 [DOI] [PubMed] [Google Scholar]
- Yeh LC, Lee JC 1992. Structure analysis of the 5′ external transcribed spacer of the precursor ribosomal RNA from Saccharomyces cerevisiae. J Mol Biol 228: 827–839 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]