

Experiments are presented which suggest that the binding of the primer tRNA to the primer binding site of the HIV-1 5′ UTR is involved in the dimerization of the genome, as part of the packaging process.
Keywords: tRNALys3, RNA encapsidation, viral assembly, RNA conformation, 5′ untranslated region
Abstract
The 5′ untranslated region (5′ UTR) of HIV-1 genomic RNA (gRNA) includes structural elements that regulate reverse transcription, transcription, translation, tRNALys3 annealing to the gRNA, and gRNA dimerization and packaging into viruses. It has been reported that gRNA dimerization and packaging are regulated by changes in the conformation of the 5′-UTR RNA. In this study, we show that annealing of tRNALys3 or a DNA oligomer complementary to sequences within the primer binding site (PBS) loop of the 5′ UTR enhances its dimerization in vitro. Structural analysis of the 5′-UTR RNA using selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) shows that the annealing promotes a conformational change of the 5′ UTR that has been previously reported to favor gRNA dimerization and packaging into virus. The model predicted by SHAPE analysis is supported by antisense experiments designed to test which annealed sequences will promote or inhibit gRNA dimerization. Based on reports showing that the gRNA dimerization favors its incorporation into viruses, we tested the ability of a mutant gRNA unable to anneal to tRNALys3 to be incorporated into virions. We found a ∼60% decrease in mutant gRNA packaging compared with wild-type gRNA. Together, these data further support a model for viral assembly in which the initial annealing of tRNALys3 to gRNA is cytoplasmic, which in turn aids in the promotion of gRNA dimerization and its incorporation into virions.
INTRODUCTION
Fully assembled, extracellular HIV-1 contains tRNALys3 annealed to sequences in the 5′ region of viral genomic RNA. Upon new HIV-1 infection, this tRNALys3 acts as a primer to initiate the reverse transcription that will produce HIV-1 proviral DNA. After the integration of proviral DNA into the host’s genome, the newly synthesized viral RNA can undergo various splicing events during transcription, resulting in spliced mRNAs coding for proteins such as Tat, Nef, Rev, Vpu, and Env. Full-length, unspliced genomic RNA (gRNA) is also exported into the cytoplasm where it is initially translated into Gag and GagPol, and is later capable of undergoing reactions that appear to be exclusive of translation, such as annealing with tRNALys3, dimerization, and incorporation into the assembling virus. Using inhibitors of cytoplasmic mRNA production, it has been reported that, in HIV-1, this transition from translation to dimerization and packaging uses the same pool of cytoplasmic gRNA (Dorman and Lever 2000).
Based on their discovery of two different in vitro conformations of the 5′ untranslated region (5′ UTR) of gRNA, Berkhout’s group first proposed that the structural changes in the 5′ UTR could act as a molecular switch to regulate the processes carried out by the viral RNA (Huthoff and Berkhout 2001). The 5′ UTR includes the RU5 region, the primer binding site (PBS), the dimerization initiation site (DIS), and the splice donor (SD) and Ψ stem–loops (see Fig. 4 below for a diagram of these regions). The 5′-R region, labeled “R” because it is also repeated at the 3′ end of gRNA, is composed of the TAR and poly(A) stem–loops. TAR is involved in promoting gRNA transcription through binding of transcriptional factors to the TAR stem–loop (Rosen et al. 1985; Muesing et al. 1987), while the poly(A) stem–loop suppresses polyadenylation in the 5′-R region (Klasens et al. 1999). The R region is followed by the U5 region (unique 5′ region), which includes part of the 5′ stem sequence of the PBS stem–loop, and which terminates immediately upstream of the PBS sequence. The PBS sequence is an 18-nucleotide (nt) sequence complementary to the 3′ end of tRNALys3 and is an essential binding site in gRNA for tRNALys3 (Kleiman et al. 2004), although other regions upstream of the PBS also participate in tRNALys3 annealing (Isel et al. 1993; Arts et al. 1996; Beerens et al. 2001; Ooms et al. 2004a; Wilkinson et al. 2008). Downstream from the PBS stem–loop are successive hairpin structures that have been assigned to playing key roles in the various stages of the gRNA cycle. For example, the DIS stem–loop has a central role in promoting gRNA dimerization. Based on in vitro studies, dimerization is thought to occur in two steps, first by a kissing–loop interaction between two DIS loops, followed by a further extension of the RNA duplex to form a more stable dimer (Laughrea and Jetté 1994; Skripkin et al. 1994). The role of the kissing–loop interaction has been confirmed by in vivo studies, in which the loop sequence has been shown to play a central role in recruiting the dimerization partners (Moore et al. 2007). Further downstream, the SD stem–loop regulates the donation of the 5′ terminus of gRNA during splicing events (Abbink and Berkhout 2008), and the Ψ stem–loop promotes the packaging of the gRNA into viruses through its binding to Gag (Lever et al. 1989; Aldovini and Young 1990; Clavel and Orenstein 1990). However, although separate functions have been assigned to the structural elements of the 5′ UTR, these elements contribute to the overall structure of the 5′ UTR, and mutations in one element could thereby affect functions associated with the other structural elements (Vrolijk et al. 2008).
FIGURE 4.

Secondary structure models of the HIV-1 5′ UTR derived from SHAPE analysis. The figure shows the predominant RNA structure in the absence (A) or presence (B) of cPBS(182–199). The structures were determined using RNAstructure software and were drawn using Adobe Illustrator. The palindromic sequences involved in the kissing–loop interaction between two DIS loops are boxed. The AUG start codon of Gag is encircled. Nucleotides are color-coded according to the key to reactivites shown in the figure. The 3′ end terminal 50 nt of the 5′-UTR RNA were not shown in the drawing as described in the Materials and Methods section.
Studies in vitro have shown that dimerization of the 5′ UTR is controlled by two mutually exclusive RNA conformations, i.e., the long distance interaction (LDI) and the branched multiple-hairpin (BMH) forms, in which the DIS stem–loop is, or is not, respectively, bound to other regions in the same RNA molecule, thereby affecting its availability for dimerization (Huthoff and Berkhout 2001; Lu et al. 2011). The structure of the LDI and BMH conformations, first determined by chemical and enzymatic probes (Huthoff and Berkhout 2001), was also confirmed by the same group using microarray scanning (Ooms et al. 2004b). Mutations in the 5′ UTR that promote the BMH conformation also result in an enhancement of gRNA incorporation into virus (Ooms et al. 2004a). The effect on gRNA packaging could be the direct result of the changes in the Ψ stem structure (Abbink and Berkhout 2003), which is sequestered in the LDI model and/or by the requirement for gRNA dimerization for packaging into the virion (Moore et al. 2009). The ratio of the LDI to the BMH forms can be shifted toward one form or the other using different salt concentrations, introducing specific mutations, or incubating with nucleocapsid protein, NCp7 (Marquet et al. 1991; Huthoff and Berkhout 2001; Ooms et al. 2004a; Lu et al. 2011).
The role of the PBS stem–loop in dimerization has been studied by several groups. Based on in vitro experiments, it has been reported that annealing oligomers to the PBS loop induces a small increase in dimerization (Berkhout et al. 2002; Reyes-Darias et al. 2012), but tRNA annealing failed to increase dimerization when annealed to wild-type 5′ UTR. A small increase in dimerization could be observed when tRNALys3 is annealed with a mutant 5′ UTR that favors the BMH conformation (Berkhout et al. 2002). A later study has also shown that deleting the PBS upper stem–loop had only a minor effect on dimerization in vitro and no effect in vivo (Heng et al. 2012).
In this report, we show that the annealing of either tRNALys3 or DNA oligomers complementary to sequences in the PBS loop does promote a BMH-like RNA conformation that facilitates wild-type 5′-UTR dimerization in vitro, and possibly in vivo as well, since preventing tRNALys3 annealing through deletion of the PBS sequence causes a 60% reduction in the packaging of gRNA into virus. Based on SHAPE and antisense experiments, we propose a model that explains how tRNALys3 annealing aids in promoting viral RNA dimerization and packaging, and discuss how this model can explain the different conclusions mentioned above.
RESULTS
Annealing to the PBS loop promotes dimerization of the 5′ UTR in vitro
We have examined the ability of an 18-nt DNA oligomer, cPBS(182–199), complementary to the viral PBS sequence (nt 182–199), to promote dimerization of a 406-nt RNA representing the 5′ UTR. The tested 5′-UTR RNA contains sequences from R (+1) through the initial coding sequence of Gag, i.e., in addition to the actual sequence of the 5′ UTR, this RNA contains 71 nt of translated RNA. For ease of reference, this RNA will be referred to herein as the 5′-UTR RNA. Unless stated otherwise, a concentration of 100 nM of 5′-UTR RNA was used in the following experiments, and annealing was done with a fourfold excess of DNA oligomers. Monomeric and dimeric RNAs were separated by 1.7% agarose electrophoresis.
As shown in Figure 1A, annealing of the cPBS(182–199) to the 5′-UTR RNA increased the appearance of a high molecular weight band identified as dimer. To confirm the nature of that band, we used oligomers complementary to the DIS stem–loop, cDIS(257–280) and cDIS(236–254), that should inhibit RNA dimerization. As expected, both cDIS(257–280) and cDIS(236–254) decreased the high molecular weight band, confirming that this RNA is an extended dimer (Fig. 1A). Annealing of an oligomer complementary to nt 369–386 within the coding sequence of Gag (cGag[369–386]) had no effect upon dimerization. Because the 5′-UTR structure is sensitive to salt concentration, we tested the effect of cPBS(182–199) annealing on dimerization in KCl concentrations ranging from 0 to 200 mM. We show in Figure 1B that cPBS(182–199) annealing promotes dimerization at all the KCl concentrations tested. Considering these results, we chose to perform later experiments in 10 mM HEPES pH 8.0, 150 mM KCl, 1 mM MgCl2, 10 mM NaCl, a condition that is close to physiological osmolarity.
FIGURE 1.

Dimerization of the 5′ UTR in the presence of DNA oligomers annealed to different regions of this RNA. (A) The wild-type 5′-UTR RNA (the first 406 nt of gRNA) was annealed with different DNA oligomers: cPBS(182–199), complementary to the PBS; cGAG(369–386), complementary to a coding sequence of Gag; cDIS(257–280) and cDIS(236–254), complementary to two different regions of the DIS hairpin. Monomeric and dimeric 5′-UTR RNA were resolved by electrophoresis in agarose gels, and detected by EtBr staining. (B) 5′-UTR RNA dimerization in presence or absence of cPBS(182–199) in dimerization buffer using varying concentrations of KCl.
It can be noticed in Figure 1A,B (and later, in Fig. 2A,C) that the dimer induced by cPBS is brighter than would be expected based on the corresponding loss of monomer. Ethidium bromide binds better to double-stranded regions of nucleic acid than to single-stranded regions, and while the agarose gels demonstrate a qualitative shift from monomer to dimer upon annealing with cPBS, we have not attempted to quantitate this shift.
FIGURE 2.

Annealing to the PBS loop increases the dimerization of the 5′ UTR in a DIS dependent mechanism. (A) Resolution by agarose gel electrophoresis of the monomer and dimer of the 5′-UTR RNA, either wild-type or mutant RNA, ΔDIS, with nt 248–270 deleted (i.e., deletion of the DIS loop and part of the DIS stem). Gels are stained with ethidium bromide. The assays were performed as described in Materials and Methods in presence or absence of cPBS(182–199). (B) Dimerization of the 5′ UTR missing the PBS sequence, ΔPBS, in the presence or absence of cPBS(182–199). (C) Dimerization assay using wild-type 5′-UTR RNA that has been annealed with either a PBS oligomer, which has a sequence identical to the PBS sequence, or cPBS(182–199). This assay was performed in 200 nM 5′-UTR RNA. (D) Dimerization of the 5′ UTR annealed with either cPBS(182–199) or cPBS(199–216), oligomers complementary to two different regions within the PBS loop.
To confirm that cPBS(182–199) oligomer is enhancing dimerization through a DIS-dependent mechanism, and through binding to the PBS sequence, we tested the ability of cPBS(182–199) oligomer to enhance dimerization of a DIS mutant (ΔDIS) missing part of the DIS stem–loop (del 248–270, Fig. 2A), and a mutant where the PBS sequence was deleted (ΔPBS[del 183–200], Fig. 2B). The ΔPBS mutant has been shown to be unable to anneal in vitro with either tRNALys3 or cPBS(182–199) (Xing et al. 2011). As expected, both of these mutant RNAs showed no enhancement of dimerization in the presence of cPBS(182–199). In the ΔPBS mutant (deletion of nt 183–200), a small amount of dimer is formed, independent of the presence or absence of cPBS(182–199), resembling the monomer/dimer pattern seen for wild-type RNA in the absence of cPBS(182–199) (Fig. 2B). This argues against an RNA structural change resulting from deletion of the PBS sequence that would affect dimerization. In addition, mfold thermodynamic calculations estimate only a minor (2 kcal) free energy change in the upper PBS stem–loop for wild-type and ΔPBS RNAs (Zuker 2003).
The increase in dimerization induced by the cPBS(182–199) oligomer annealing to the PBS loop could be due to either preventing the PBS sequence from interacting with complementary sequences elsewhere in the 5′ UTR, or to a thermodynamic stabilization of the PBS internal loop (nt 179–216). If the PBS sequence is involved in an interaction that inhibits dimerization, we would expect deletion of the PBS sequence to alter the dimer/monomer ratio, but as shown in Figure 2B, this does not occur. We also tested the ability of an eightfold excess of a PBS DNA oligomer, with sequence identical to the PBS sequence, to alter the efficiency of dimerization at two different RNA concentrations, 100 nM and 200 nM, but only results for 200 nM are shown (Fig. 2C). Excess PBS oligomer had no effect upon dimerization (Fig. 2C), although increasing the 5′-UTR concentration does increase the efficiency of dimerization (cf. lanes 1 in Fig. 2A,C,D).
These results do not support the hypothesis that the observed increase in dimerization is caused by the inhibition of an interaction between the PBS sequence and a region elsewhere in the RNA.
On the other hand, as shown in Figure 2D, dimerization is also promoted by annealing to the lower 3′-half of the PBS loop using the oligomer cPBS(199–216). This observation favors the hypothesis that the increase in dimerization is due to the thermodynamic stabilization of the PBS loop. This is supported by estimations of the free energy of annealing of cPBS(182–199) to its RNA target, as calculated using the DINAMelt webserver (Markham and Zuker 2005). Estimated free energy values for annealing to the PBS for a DNA:DNA or DNA/RNA hybrid are −22.5 and −36.2 kcal/mol, respectively, and we therefore estimate the free energy of the cPBS(182–199) DNA oligomer annealing to the PBS loop to be between −22.5 and −36.2 kcal/mol. The free energy of the region that includes the two stems surrounding the PBS loop, starting at position 125 and ending at position 223, was calculated using mfold (Zuker 2003) to be −27.7 kcal/mol, with the appropriate structural constraints to mimic the structure of that region as described in Wilkinson et al. (2008). Since the decrease in the free energy due to cPBS(182–199) annealing (between −22.5 and −36.2 kcal/mol) is significant compared with that of the upper PBS stem–loop (−27.7 kcal/mol), we propose that this annealing stabilizes the PBS internal loop and the helices surrounding it, and that this effect may translate into a change in RNA conformation compared with the unannealed form.
cPBS(182–199) annealing alters the 5′-UTR secondary structure, as determined by SHAPE analysis
Using selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE), we have determined the secondary structure of the 5′-UTR RNA with and without annealing of cPBS(182–199). We compared the difference in SHAPE reactivities between unannealed and cPBS(182–199)-annealed data sets that are available as Supplemental Material. The differences in nucleotide reactivities occur mainly in the annealed PBS sequence, the DIS stem–loop, and G224 (summarized in Fig. 3), and are compatible with the secondary structure models shown in Figure 4. More detailed changes in reactivities of these regions are shown in Supplemental Figure S1. Without annealing (Fig. 4A), part of the DIS loop (boxed nucleotides) and the 3′ part of the DIS stem are sequestered to the lower part of the PBS stem. The sequences that form the SD and Ψ stem–loops, as well as the AUG start codon for Gag (nt 336–338, encircled), are part of one long stem–loop. After annealing with cPBS(182–199) (Fig. 4B), the DIS stem–loop is fully formed and the DIS loop is unsequestered. The SD and Ψ stem–loops are also formed separately, while the AUG start codon is part of the lower PBS stem. These experiments were done using 10 nM 5′ UTR, a concentration at which the 5′ UTR does not form a dimer even when annealed with the cPBS(182–199). This is shown in Figure 5A, which represents an experiment in which unlabeled 5′ UTR at concentrations of either 10 or 100 nM was annealed with 32P-cPBS, and then resolved by 1D PAGE. At a 5′-UTR concentration of 10 nM, the annealed RNA remains predominantly a monomer in presence or absence of 1 mM Mg++ in the gel, the loading dye, and the gel buffer (Fig. 5A). When the concentration is increased to 100 nM, the annealed 5′ UTR forms dimers. Thus, the structure of the annealed 5′ UTR shown in Figure 4B is that of monomeric 5′-UTR RNA in a dimer-susceptible conformation.
FIGURE 3.

The difference in SHAPE reactivities of annealed 5′ UTR relative to the reactivities of unannealed 5′ UTR. The difference in reactivities between annealed and unannealed 5′ UTR was calculated by subtracting the sum of the standard deviations at each nucleotide position from the absolute value of the difference of reactivites at every position for each data set ([{reactivityannealed} − {reactivityunannealed}] − [Stdannealed + Stdunannealed]). A zero value was assigned to positions where the result was negative, implying that the error is higher than the difference in reactivities. Negative values were assigned to the differences when the unannealed positions reactivities were higher. The calculations and the graph plot were done using Excel.
FIGURE 5.

5′ UTR annealed with different radioactive probes, and resolved into monomers and dimers in native 1D PAGE. (A) Annealing of radiolabeled cPBS(182–199) to either 100 nM or 10 nM 5′-UTR RNA, in the absence (TB) or presence (TBM) of Mg++ in the loading dye, electrophoretic buffer, and gel. The 10 nM 5′-UTR concentration is that used for SHAPE experiments. (B) Annealing with either radiolabeled c(115–124), an oligomer with locked nucleotides, or cGag(369–386). (C) Control for oligomer c(115–124) binding. The extended product and the ladder were prepared as described in Materials and Methods. One-fourth of the radiolabeled product was loaded next to a radiolabeled DNA size marker covering 100 nt (Invitrogen). The size was estimated by comparing with the ladder and corresponds to the expected product. (D) Annealing of 3′-32P-tRNALys3 or radiolabeled cGag(369–386) to unlabeled 5′-UTR RNA. Radiolabeled unannealed tRNALys3 and oligomers run out of the gel in long runs that separate the monomeric and dimeric RNAs.
Several groups have studied the structure of HIV-1 5′ UTR. The annealed model we have determined is very similar to the structure reported using SHAPE analysis (Wilkinson et al. 2008), with a few minor deviations, such as our observations of a Watson-Crick interaction between nt 115 and 335, and interactions between nt 121, 122 and nt 226, 227. It is also similar to the BMH model determined through chemical and enzymatic probing (Abbink et al. 2005) and by that of Lu et al. (2011) using NMR, with variations in the loop between the DIS, SD, and Ψ stem–loops. The unannealed form drawn in Figure 4A is similar in structure to the LDI model reported by Abbink et al. (2005), in regard to the TAR hairpin, the upper part of PBS stem–loop, the SD, and the Ψ stem–loops, and the area surrounding the AUG start codon of Gag. However, they found that the sequestered DIS stem–loop was bound to the poly(A) stem, rather than to U5 sequences in the PBS stem as shown in Figure 4. An NMR model of the LDI-like structure suggests that the DIS loop interacts with the U5:AUG stem. Although our model predicts an interaction between the U5 and the DIS stem–loop, our SHAPE reactivities do not support the model proposed by the NMR study. Reactivities at positions A255 and A256 are zero in the unannealed data set and high (0.6 and 0.7, respectively) in the annealed data set (as shown in Figs. 3, 4, and Supplemental Fig. S1). The changes at these positions do not agree with the LDI-like model proposed by the NMR study (Lu et al. 2011), where these two nucleotides are unpaired in both LDI- and BMH-like models. The difference between our model and the NMR model could be attributed to the difference in the buffer composition. The buffer used in our study contains 1 mM MgCl2 while the buffer used in the NMR study to determine the LDI-like structure contains no Mg++ ions.
We tested the site of interaction of the DIS loop in the LDI-like model by determining whether annealing of an oligomer complementary to nt 115–124 in the PBS stem, c(115–124), could shift the unannealed RNA from monomer to dimer, by competing with the DIS stem–loop for nt 115–124. The short length of this 10-nt oligomer might reduce the efficiency of hybridization. Consequently, we used a DNA oligomer containing locked nucleic acids (LNA), and labeled the oligomer instead of the 5′ UTR to monitor the effect upon the dimerization of 5′-UTR molecules that did bind to the 32P-oligomer. The annealed RNAs were resolved in 1D PAGE. The results, shown in Figure 5B, indicate that annealing of c(115–124) does promote dimerization of the 5′ UTR, compared with cGag(369–386), whose annealing has been shown to have no effect on dimerization (Fig. 2A). This supports the model shown in Figure 4A, where part of the DIS stem–loop is annealed with nt 115–124. To confirm that the oligomer c(115–124) did bind specifically to its target, the oligomer c(115–124) was 5′-labeled with γ–32P-ATP, annealed to the 5′ UTR, and, using reverse transcriptase (RT), we extended the oligomer and resolved the RT product(s), along with a radiolabeled 100-bp DNA ladder, using denaturing 1D PAGE. We detected only one cDNA product that had the expected size corresponding to a transcript initiating at nt 124 and ending at the beginning of the transcript (Fig. 5C).
In vitro annealing of tRNALys3 also promotes dimerization of the 5′ UTR
The ability of DNA oligomers complementary to the PBS loop to promote dimerization of the 5′ UTR in vitro raises the question whether tRNALys3 annealing would show the same effect. Because we found that full-length synthetic tRNALys3 does not anneal to the 5′ UTR with the same efficiency as cPBS(182–199) under the annealing conditions used (heat), we radiolabeled the tRNALys3 instead of the 5′ UTR so as to see the effect upon the dimerization of 5′-UTR molecules that did bind to the 32P-tRNALys3. The results shown in Figure 5D indicate that tRNALys3 is able to anneal to both monomeric and dimeric RNA, which is consistent with tRNALys3 annealing occurring prior to dimer formation. Annealing of tRNALys3 to the 5′ UTR does promote dimerization when compared with annealing with cGag(369–386) oligomer, but not as efficiently as does cPBS(182–199).
The PBS sequence is required for optimal packaging of gRNA into HIV-1
Since the BMH-like conformation of the 5′ UTR has been reported to enhance the incorporation of genomic RNA into HIV-1 (Ooms et al. 2004a; Heng et al. 2012), we next investigated whether deletion of the PBS sequence might affect gRNA packaging into HIV-1 in vivo. It has been previously shown that deleting 8 nt at the 3′ terminus of the PBS sequence is sufficient to reduce annealing in vivo to <4% (Liang et al. 1997). 293T cells were transfected with wild-type viral DNA or viral DNA with the 18-nt PBS deleted. Forty-eight hours post-transfection, viruses were isolated, and a Western blot of lysates of viruses taken from equal volumes of cell culture supernatant was probed with anti-CAp24, as shown in Figure 6A. RNA was then extracted from viruses taken from equal volumes of cell culture supernatant, blotted onto membranes, and hybridized with 32P-DNA oligomers complementary to HIV-1 genomic RNA, tRNALys3, and 7SL RNA. This experiment was repeated five times. The dot blots shown in Figure 6B,C represent membranes probed for HIV-1 gRNA and tRNALys3 (panel B) or HIV-1 gRNA and 7SL RNA (panel C). In each experiment, a phosphorimager was used to measure the amount of gRNA present relative to either tRNALys3 or 7SL RNA. Viral packaging of tRNALys3 and 7SL RNA occurs independently of gRNA packaging (Mak et al. 1994; Onafuwa-Nuga et al. 2006). In PBS-negative virions, the amount of gRNA packaged, relative to tRNALys3 or 7SL RNA, is reduced 60%, which is consistent with a role for tRNALys3 annealing to the PBS aiding in the promotion of gRNA dimerization and incorporation in viruses.
FIGURE 6.
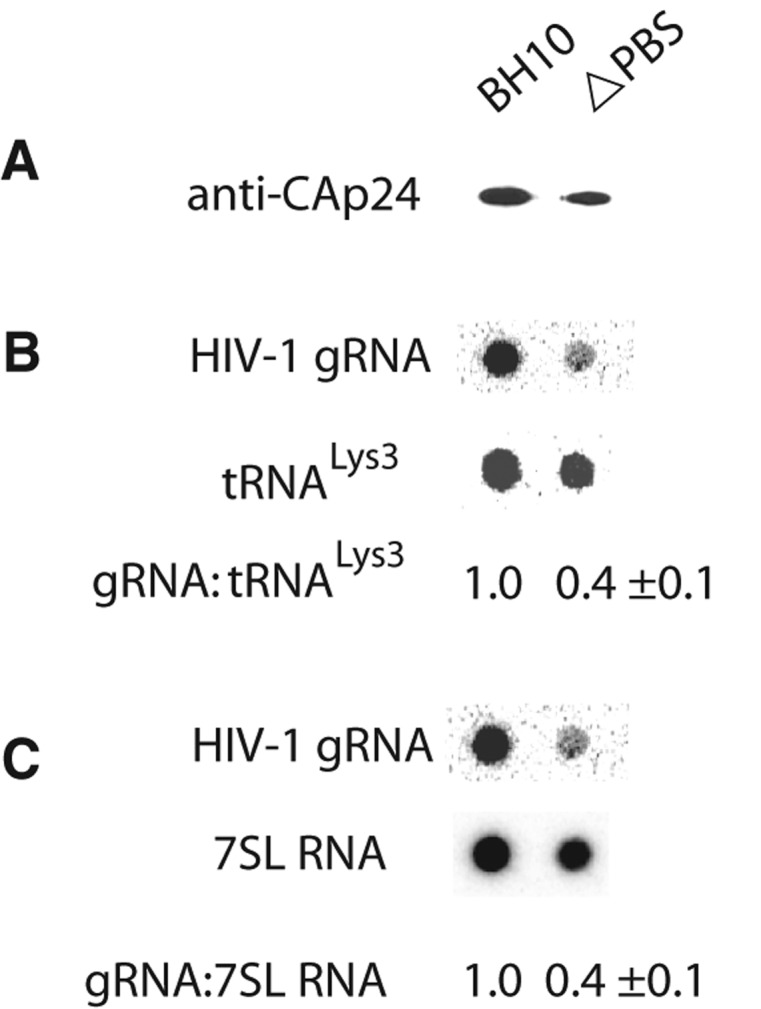
Effect of deletion of the PBS sequence upon gRNA incorporation into viruses. 293T cells were transfected with wild type or ΔPBS BH10. Forty-eight hours post-transfection, HIV-1 were purified from the culture supernatant. (A) Western blot of equal volumes of viral lysates probed with anti-CAp24. (B,C) Dot blots of total viral RNA isolated from equal volumes of viral lysates, and probed with DNA oligomers complementary to either gRNA, tRNALys3, or 7SL RNA. The bands were quantitated by phosphorimager, and the calculated ratios of gRNA:tRNALys3 (B) and gRNA:7SL RNA (C) are listed. The experiment was repeated five times, and the values shown correspond to the average and standard deviation calculated from all five experiments.
DISCUSSION
In this work, we found that annealing of oligonucleotides complementary to the PBS loop promotes dimerization of the 5′ UTR in vitro. The increase in dimerization by these DNA oligomers is associated with a shift in the equilibrium from the LDI-like to the BMH-like conformation, which exposes the DIS loop (Fig. 4) and makes it available for intermolecular interactions favoring dimerization. We propose that this shift is caused by an increased thermodynamic stability of the upper region of the PBS stem–loop induced by annealing to the PBS loop.
Several earlier reports have reached conclusions differing from the above. For example, the effect of annealing either a DNA oligomer complementary to the PBS (nt 182–202) or a full-length synthetic tRNALys3 to a 290-nt HIV-1 5′ UTR was examined in vitro (Berkhout et al. 2002). Under their initial annealing conditions used, the main conformation of the unannealed 290-nt 5′ UTR is the LDI conformation, which has a distinct electrophoretic mobility from both the BMH and dimer conformations. Using this electrophoretic assay to determine the effects of annealing upon RNA conformation, they found that annealing to the 5′ UTR of an oligomer complementary to the PBS (182–202) produced an RNA band whose electrophoretic mobility was neither that of the LDI or BMH forms, and resulted in a barely detectable dimer band. Annealing with tRNALys3 did not cause any change in electrophoretic mobility that could not be accounted for by the added molecular weight of the tRNALys3, and no dimer formation was seen. This inability of annealing to the PBS to move the 5′ UTR to a BMH conformation may be related to their use of a shorter 290-nt 5′ UTR that does not contain the SD, Ψ, or AUG stems, and would be missing the U5:AUG base-pairing, which has been shown to have an essential role in the LDI/BMH equilibrium (Abbink and Berkhout 2003; Lu et al. 2011). The lack of dimer formation may also be related to the low concentration of 5′ UTR used in their report (8 nM), which even under our annealing conditions, will not promote dimer formation (see Fig. 5A).
Two other groups have also reported that a deletion of the upper portion of the PBS stem–loop, that included the PBS loop itself, did not affect either 5′-UTR dimerization in vitro or viral RNA packaging (Sakuragi et al. 2007; Heng et al. 2012; Sakuragi et al. 2012). However, internal loops are known to introduce thermodynamic instability in the stems, and deleting the internal PBS loop could have a stabilizing effect on the lower region of the PBS stem, which has been reported to be important for dimerization and packaging of RNA (Sakuragi et al. 2003, 2012). We therefore postulate that annealing to the PBS sequence, or the removal of the less stable upper region of the PBS stem–loop, may have the same effect of stabilizing the lower stem region and promoting the BMH-like conformation.
The BMH-like model shown in Figure 4 has minor deviations from the model also determined using SHAPE analysis (Wilkinson et al. 2008). The LDI-like model in Figure 4 also shares similarities with the one previously determined by Berkhout’s group (Abbink et al. 2005). For example, the AUG start codon for Gag and the Ψ stem are in a similar conformation to what has been proposed (Abbink et al. 2005). Our SHAPE data did not detect any significant differences in reactivity of the AUG start codon of Gag within the LDI-like and BMH-like conformations. So we do not expect the shift in equilibrium between these conformations to have an effect on translation. This agrees with the results of a previous study, where the shift between the LDI and the BMH conformation did not show any effect on translation in vivo (Abbink et al. 2005). However, while Berkhout’s group reported an interaction of the DIS stem–loop with the poly(A) stem–loop (Huthoff and Berkhout 2001), our SHAPE data indicate a site of interaction of this DIS region within the U5 region, which while similar to that determined by Summers’ group (Lu et al. 2011), involves a different sequence within the U5 region. We have confirmed the interaction between DIS and U5 sequences through oligomer annealing specifically to the U5 sequence involved in this interaction and that resulted in an increase of dimerization, as expected (Fig. 5B). These variations in reported 5′-UTR conformations could be due to differences in experimental conditions associated with the different techniques used to determine the structure. For example, Berkhout’s group used chemical and enzymatic probes (Huthoff and Berkhout 2001) and later confirmed their structure using microarray scanning (Ooms et al. 2004a), while Summers’ group used NMR analysis (Lu et al. 2011).
In addition to the PBS sequence, tRNALys3 also binds to sites in gRNA upstream of the PBS sequence (Beerens et al. 2001; Wilkinson et al. 2008). Although our data indicate that annealing to the PBS sequence by cPBS is sufficient to induce dimerization in vitro, these other interactions might also promote dimerization in vivo. For example, annealing of DNA oligomers to parts of the PBS stem which include sites thought to interact with tRNALys3 were shown to affect the dimerization of the 5′ UTR (Reyes-Darias et al. 2012).
While annealed synthetic tRNALys3 promotes 5′-UTR dimerization, it does not do so as efficiently as the DNA oligomers (Fig. 5D), which do not require either heat or viral proteins to promote their annealing (Xing et al. 2011). The fact is that the conditions for the annealing of tRNALys3 to viral RNA in vivo are certain to be quite different from the in vitro annealing conditions used here, and previous evidence suggests that a number of different proteins such as Gag, GagPol, lysyl-tRNA synthetase, and RNA helicase A exist within a cytoplasmic protein complex with composition and architecture promoting this annealing (Saadatmand and Kleiman 2012). tRNALys3 annealing to HIV-1 RNA is multi-staged, being facilitated first by Gag in the cytoplasm, and, after Gag processing, by mature nucleocapsid (NCp7). Compared with NCp7-annealed tRNALys3, Gag-annealed tRNALys3 is less efficient at initiating reverse transcription (Cen et al. 2000) and binds more weakly to the viral RNA (Guo et al. 2009). We would predict that following the translation of Gag, Gag promotes the initial annealing of tRNALys3 to gRNA in the cytoplasm, and that both tRNALys3 annealing and Gag promote gRNA dimerization and its incorporation into the virus. Other factors, such as RNA helicases, may also contribute to this process (Xing et al. 2011). The 60% reduction in ΔPBS gRNA incorporation into HIV-1 shown in Figure 6 is consistent with tRNALys3 being one of the factors that promote dimerization and viral packaging of the gRNA.
MATERIALS AND METHODS
For the complete list of the oligomers used in this study please refer to the Supplemental Material (Supplemental Table S1).
RNA preparation
The DNA templates used for transcription of HIV-1 5′-UTR wild type, ΔPBS, and ΔDIS (Xing et al. 2011) were amplified by PCR from vectors coding for HIV-1 BH10 sequence with the corresponding mutations. 5′ and 3′ primers T7HIV-1 and HIV382–406, respectively, were used to produce the DNA templates for the 406-nt 5′ UTRs. The templates were sequenced to confirm the sequence of the constructs. The RNAs were transcribed in vitro using T7 Megascript kit from Ambion (Cat. #AM1334) and treated with Turbo DNase supplied with the kit according to the manufacturer’s recommendations. The newly synthesized RNAs were purified using RNA Clean & Concentrator-5 from Zymoresearch (Cat. #R1015).
Dimerization assays
One picomole of purified 5′-UTR RNA, with or without 4 pmol of DNA oligomer to be annealed, was added to a total volume of 10 μL containing 10 mM HEPES pH 8.0, 1 mM MgCl2, 10 mM NaCl, 150 mM KCl. The sample was heated to 68°C for 5 min, and then incubated at 37°C for 30 min. Following incubation, the products were loaded on 1.7% agarose gel containing ethidium bromide, and run in 1× TAE buffer pH 8.0, at 4°C for 60 min. The products were quantitated using ImageJ software (Schneider et al. 2012).
For dimerization assays using radiolabeled products, tRNALys3 was transcribed in vitro and 3′-end-labeled with 32pCp using RNA ligase as previously described (Xing et al. 2011). Oligomers cGag(369–386) and cU5(115–124) were labeled with 32P, using T4-Polynucleotide Kinase from Thermo Scientific (Cat. #EK0031) according to the manufacturer’s recommendations. Annealing was performed as described above, using 0.01 pmol of radiolabeled tRNALys3 or oligomers. The reactions were electrophoretically resolved using 5% non-denaturing acrylamide gels run in Tris-Borate buffer, pH 8.0, at 4°C for 60 min. Radiolabeled RNAs were visualized and quantitated using a PhosphorImager and autoradiography.
SHAPE analysis
Six tubes contained 1 pmol of 5′-UTR RNA in 100 µL of 10 mM HEPES pH 8.0, 1 mM MgCl2, 10 mM NaCl, 150 mM KCl. Eight picomoles of cPBS (182–199) oligomer was added to each of two of these tubes. All reactions were then heated to 68°C for 5 min, followed by incubation at 37°C for 30 min. Acylation of RNA was then performed as previously described (Mortimer and Weeks 2009). To two of the tubes, one with oligomer and one without, 2 µL of 1 M HEPES pH 8.0 buffer was added, followed by 4 µL of 130 mM NMIA (N-methylisatoic anhydride). The solution of NMIA was prepared and stored as described (Wilkinson et al. 2006). Two other tubes, one with added oligomer and one without, served as negative controls, i.e., only 2 µL of 1 M HEPES pH 8.0 buffer and 4 µL of DMSO were added. Two other tubes (without oligomer) served as sequencing ladders and were treated the same way as the negative controls. All tubes were incubated for 60 min at 37°C. The RNA from all tubes was treated with phenol and purified using RNA Clean & Concentrator-5 from Zymoresearch (Cat. #R1015).
The RNA was reverse-transcribed using Superscript III Reverse Transcriptase kit from Invitrogen according to the recommendations of the manufacturer (Cat. #18080–044), using the primer HIV382–406 labeled with Vic-fluorescent dye from Applied Biosystems. For the tubes that served as sequence ladders, ddTTP or ddGTP were added to the transcription reactions. The products were purified on Sephadex G-50 columns and precipitated as described (Pang et al. 2011).
The products were resolved by capillary electrophoresis on a DNA analyser ABI 3730 at the IRIC (Institut de Recherche en Immunologie et Cancérologie) at University of Montreal. The data were analyzed as described (Pang et al. 2011). The signal was first analyzed using peakScanner (freely available from ABI) to perform base line adjustment and peak area integration. The load error, signal decay, and reactivites were calculated using FAST software freely provided by the Jeffrey Glenn laboratory. The alignment of the fragments and the ladders was inspected and corrected manually when necessary. The reactivities and the sequence of the RNA were input into RNAstructure software, freely provided by the Dr. David Mathews laboratory. Because the reverse-transcribed reactions were purified on sephadex G50 columns, we eliminated 50 nt from the 3′ end. Reactivites of the first 4 nt from the 5′ end (position 1–4) and the last 5 nt from the 3′ end (position 351–355) were not included because the alignment was ambiguous at the beginning and at the end of the analyzed sequence. The reactivities were calculated from three independent experiments for the unannealed conformation and four independent experiments for the annealed conformation. The averages and standard deviations reactivities for both conformations are supplied in the Supplemental Material.
Purification of virus and viral RNA, and quantitation of gRNA, tRNALys3, and 7SL RNA
HEK293-T cells were grown in complete Dulbecco’s modified Eagle’s medium (DMEM) and transfected with BH10 wild-type and ΔPBS mutant plasmids using Lipofectamine 2000 from Invitrogen. Supernatant was collected 48 h post-transfection and filtered through 0.2-µm filters. The viruses were pelleted from the culture medium by centrifugation at 35,000 rpm for 1 h in a Beckman Ti-45 rotor. The RNA was then purified using Trizol reagent from Life technologies. Viral gnomic RNA (gRNA), tRNALys3, and 7SL RNA were quantitated by dot blot hybridization as previously described (Cen et al. 2000), using probes 5′-end-labeled with 32pCp. The probe sequences are listed in Supplemental Table S1.
Reverse transcription using c(115–124) as primer
The oligomer c(115–124) was 5′-labeled with γ–32P-ATP, and annealed to the 5′ UTR, as described above. The products were ethanol-precipitated and dissolved in reverse transcription buffer (Bio Basic). The annealed RNA was reverse-transcribed using M-MLV reverse transcriptase (Bio Basic) according to the manufacturer’s recommendations. One fourth of the radiolabeled product was loaded next to a radiolabeled 100-bp DNA size marker (Invitrogen).
SUPPLEMENTAL MATERIAL
Supplemental Figure S1 is provided, which shows nucleotide reactivities in regions of the 5′ UTR where the changes in reactivities are highest. Also included is a complete list of sequences of the DNA oligomers used in this study (Supplemental Table S1), and a file containing the averages and standard deviations of 5′-UTR SHAPE reactivities for both unannealed and annealed conformations as described in the Materials and Methods section (Supplemental Table S2).
ACKNOWLEDGMENTS
We thank Dr. Jeffrey Glenn’s laboratory for kindly providing FAST software, and Dr. David Mathews’s laboratory for generously making available RNAstructure software. We would like also to highlight the excellent technical assistance provided by the IRIC (Institut de Recherche en Immunologie et Cancérologie) technical team and Dr. Franz Lang’s laboratory at University of Montreal. This work was funded by a grant from the Canadian Institutes of Health Research.
REFERENCES
- Abbink TE, Berkhout B 2003. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J Biol Chem 278: 11601–11611 [DOI] [PubMed] [Google Scholar]
- Abbink TE, Berkhout B 2008. RNA structure modulates splicing efficiency at the human immunodeficiency virus type 1 major splice donor. J Virol 82: 3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbink TE, Ooms M, Haasnoot PC, Berkhout B 2005. The HIV-1 leader RNA conformational switch regulates RNA dimerization but does not regulate mRNA translation. Biochemistry 44: 9058–9066 [DOI] [PubMed] [Google Scholar]
- Aldovini A, Young RA 1990. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J Virol 64: 1920–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts EJ, Ghosh M, Jacques PS, Ehresmann B, LeGrice SFJ 1996. Restoration of tRNA3Lys-primed (−)-strand DNA synthesis to an HIV-1 reverse transcriptase mutant with extended tRNAs. J Biol Chem 271: 9054–9061 [DOI] [PubMed] [Google Scholar]
- Beerens N, Groot F, Berkhout B 2001. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J Biol Chem 276: 31247–31256 [DOI] [PubMed] [Google Scholar]
- Berkhout B, Ooms M, Beerens N, Huthoff H, Southern E, Verhoef K 2002. In vitro evidence that the untranslated leader of the HIV-1 genome is an RNA checkpoint that regulates multiple functions through conformational changes. J Biol Chem 277: 19967–19975 [DOI] [PubMed] [Google Scholar]
- Cen S, Khorchid A, Gabor J, Rong L, Wainberg MA, Kleiman L 2000. Roles of Pr55gag and NCp7 in tRNA3Lys genomic placement and the initiation step of reverse transcription in human immunodeficiency virus type 1. J Virol 74: 10796–10800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F, Orenstein JM 1990. A mutant of human immunodeficiency virus with reduced RNA packaging and abnormal particle morphology. J Virol 64: 5230–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman N, Lever A 2000. Comparison of viral genomic RNA sorting mechanisms in human immunodeficiency virus type 1 (HIV-1), HIV-2, and Moloney murine leukemia virus. J Virol 74: 11413–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Saadatmand J, Niu M, Kleiman L 2009. Roles of Gag and NCp7 in facilitating tRNA3Lys annealing to viral RNA in human immunodeficiency virus type 1. J Virol 83: 8099–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng X, Kharytonchyk S, Garcia EL, Lu K, Divakaruni SS, LaCotti C, Edme K, Telesnitsky A, Summers MF 2012. Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J Mol Biol 417: 224–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H, Berkhout B 2001. Two alternating structures of the HIV-1 leader RNA. RNA 7: 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isel C, Marquet R, Keith G, Ehresmann C, Ehresmann B 1993. Modified nucleotides of tRNA3Lys modulate primer/template loop-loop interaction in the initiation complex of HIV-1 reverse transcription. J Biol Chem 268: 25269–25272 [PubMed] [Google Scholar]
- Klasens BI, Thiesen M, Virtanen A, Berkhout B 1999. The ability of the HIV-1 AAUAAA signal to bind polyadenylation factors is controlled by local RNA structure. Nucleic Acids Res 27: 446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman L, Halwani R, Javanbakht H 2004. The selective packaging and annealing of primer tRNALys3 in HIV-1. Curr HIV Res 2: 163–175 [DOI] [PubMed] [Google Scholar]
- Laughrea M, Jetté L 1994. A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry 33: 13464–13474 [DOI] [PubMed] [Google Scholar]
- Lever A, Göttlinger H, Haseltine W, Sodroski J 1989. Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J Virol 63: 4085–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Rong L, Morin N, Cherry E, Huang Y, Kleiman L, Wainberg MA 1997. The roles of human immunodeficiency virus type 1 Pol protein and the primer binding site in the placement of tRNA3Lys onto viral genomic RNA. J Virol 71: 9075–9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, Dorjsuren B, Kulandaivel G, Jones S, Hiremath A, et al. 2011. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334: 242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak J, Jiang M, Wainberg MA, Hammarskjold M-L, Rekosh D, Kleiman L 1994. Role of Pr160gag-pol in mediating the selective incorporation of tRNALys into human immunodeficiency virus type 1 particles. J Virol 68: 2065–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M 2005. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res 33: W577–W581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquet R, Baudin F, Gabus C, Darlix J-L, Mougel M, Ehresmann C, Ehresmann B 1991. Dimerization of human immunodeficiency virus (type 1) RNA: Stimulation by cations and possible mechanism. Nucleic Acids Res 19: 2349–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Fu W, Nikolaitchik O, Chen J, Ptak RG, Hu WS 2007. Dimer initiation signal of human immunodeficiency virus type 1: Its role in partner selection during RNA copackaging and its effects on recombination. J Virol 81: 4002–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Nikolaitchik OA, Chen J, Hammarskjold ML, Rekosh D, Hu WS 2009. Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS Pathog 5: e1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM 2009. Time-resolved RNA SHAPE chemistry: Quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat Protoc 4: 1413–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muesing MA, Smith DH, Capon DJ 1987. Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell 48: 691–701 [DOI] [PubMed] [Google Scholar]
- Onafuwa-Nuga AA, Telesnitsky A, King SR 2006. 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12: 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Huthoff H, Russell R, Liang C, Berkhout B 2004a. A riboswitch regulates RNA dimerization and packaging in human immunodeficiency virus type 1 virions. J Virol 78: 10814–10819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Verhoef K, Southern E, Huthoff H, Berkhout B 2004b. Probing alternative foldings of the HIV-1 leader RNA by antisense oligonucleotide scanning arrays. Nucleic Acids Res 32: 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PS, Elazar M, Pham EA, Glenn JS 2011. Simplified RNA secondary structure mapping by automation of SHAPE data analysis. Nucleic Acids Res 39: e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Darias JA, Sanchez-Luque FJ, Berzal-Herranz A 2012. HIV RNA dimerisation interference by antisense oligonucleotides targeted to the 5′ UTR structural elements. Virus Res 169: 63–71 [DOI] [PubMed] [Google Scholar]
- Rosen CA, Sodroski JG, Haseltine WA 1985. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 41: 813–823 [DOI] [PubMed] [Google Scholar]
- Saadatmand J, Kleiman L 2012. Aspects of HIV-1 assembly that promote primer tRNALys3 annealing to viral RNA. Virus Res 169: 340–348 [DOI] [PubMed] [Google Scholar]
- Sakuragi J, Ueda S, Iwamoto A, Shioda T 2003. Possible role of dimerization in human immunodeficiency virus type 1 genome RNA packaging. J Virol 77: 4060–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuragi J, Sakuragi S, Shioda T 2007. Minimal region sufficient for genome dimerization in the human immunodeficiency virus type 1 virion and its potential roles in the early stages of viral replication. J Virol 81: 7985–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuragi J, Ode H, Sakuragi S, Shioda T, Sato H 2012. A proposal for a new HIV-1 DLS structural model. Nucleic Acids Res 40: 5012–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripkin E, Paillart J-C, Marquet R, Ehresmann B, Ehresmann C 1994. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci 91: 4945–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrolijk MM, Ooms M, Harwig A, Das AT, Berkhout B 2008. Destabilization of the TAR hairpin affects the structure and function of the HIV-1 leader RNA. Nucleic Acids Res 36: 4352–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1: 1610–1616 [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Liang C, Kleiman L 2011. Coordinate roles of Gag and RNA helicase A in promoting the annealing of tRNA3Lys to HIV-1 RNA. J Virol 85: 1847–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]