

The remarkable regenerative capacity of the fresh water planarian Schmidtea mediterranea is useful for studying stem cell identity and function. Several novel miRNAs have recently been identified in this organism, but their roles in neoblasts and during regeneration are unknown. Here, the authors use next-generation sequencing to identify miRNAs that are enriched in neoblast subpopulations and in regenerating tissue at different time points during regeneration. The findings provide new insight into the identities of the miRNAs that support planarian regeneration and neoblast function.
Keywords: microRNA, neoblast, planaria, regeneration
Abstract
In recent years, the planarian Schmidtea mediterranea has emerged as a tractable model system to study stem cell biology and regeneration. MicroRNAs are small RNA species that control gene expression by modulating translational repression and mRNA stability and have been implicated in the regulation of various cellular processes. Though recent studies have identified several miRNAs in S. mediterranea, their expression in neoblast subpopulations and during regeneration has not been examined. Here, we identify several miRNAs whose expression is enriched in different neoblast subpopulations and in regenerating tissue at different time points in S. mediterranea. Some of these miRNAs were enriched within 3 h post-amputation and may, therefore, play a role in wound healing and/or neoblast migration. Our results also revealed miRNAs, such as sme-miR-2d-3p and the sme-miR-124 family, whose expression is enriched in the cephalic ganglia, are also expressed in the brain primordium during CNS regeneration. These results provide new insight into the potential biological functions of miRNAs in neoblasts and regeneration in planarians.
INTRODUCTION
Planarians are fresh water invertebrates belonging to the phylum Platyhelminthes. They share common traits such as bilateral symmetry, triploblasticity, and encephalization with metazoans. Additionally, planarians possess well-defined anterior-posterior and dorsoventral axes. Planarians are well known for their enormous regenerative ability—a tiny body fragment can fully regenerate within a week. The regenerative prowess of planarians has been attributed to specialized pluripotent cells called neoblasts. Advances in cellular and molecular techniques have helped reveal the mechanisms that govern neoblast function and regeneration. Following amputation or injuries, planarians respond by wound healing, which involves wound closure by migration of adjacent epithelial cells, followed by the migration and rapid proliferation of neoblasts to form an undifferentiated tissue called blastema. The initial steps of regeneration also involve selective loss of old cells near the wound region via apoptosis, followed by remodeling of new and old tissues. Wound healing and blastema formation take place within 2–3 d after amputation. The later stages of regeneration involve repatterning of old and new tissues within 2 wk of post-amputation, resulting in complete restoration of normal morphology of the animal (Lobo et al. 2012). This complex phenomenon is controlled by a variety of genes that code for proteins involved in chromatin modifications, various signaling pathways, and post-transcriptional regulatory processes (Aboobaker et al. 2011; Reddien et al. 2011; King and Newmark 2012). Recently Wenemoser et al. (2012) have identified waves of gene expression profiles that peak within 30 min following amputation and gradually taper down by 24 h, indicating tight regulation of gene expression during regeneration.
MicroRNAs are short noncoding RNAs (18–24 nt) that play a major role in post-transcriptional gene regulation in metazoans. They usually bind to sequences in the 3′ UTRs of mRNAs and silence gene expression by either mRNA degradation or translational repression. miRNAs are involved in controlling diverse biological functions such as embryogenesis, development, and regeneration by fine tuning protein expression levels (Bartel et al. 2004). In a recent study, knockdown of Smed-Ago, a key factor involved in miRNA/siRNA biogenesis in planarians, led to a drastic reduction of the neoblast population and defects in regeneration, suggesting a role of miRNAs/siRNAs in neoblast function and regeneration (Rouhana et al. 2010; Li et al. 2011). Previous studies have identified several miRNAs that are expressed in neoblasts (Palakodeti et al. 2006; Friedländer et al. 2009; Lu et al. 2009), but little is known about the differential expression of miRNAs in proliferating neoblasts and neoblast progeny. Recently, Tian et al. (2012) and Qin et al. (2011) identified several miRNAs that are enriched in 3-d regenerating tissues. However, a systematic analysis of miRNA expression in anterior and posterior regenerating tissues at various time points has not been conducted.
Here, we studied miRNA expression in several purified subpopulations of neoblasts and during regeneration in Schmidtea mediterranea. Small RNA sequencing from various FACS-sorted neoblast populations identified miRNAs whose expression is enriched in proliferating neoblasts and neoblast progeny. Sequencing of small RNAs throughout regeneration revealed several miRNAs that are expressed within 3–24 h post-amputation and miRNAs that are asymmetrically expressed during regeneration. For example, members of the mir-124 family are up-regulated in anterior regenerating tissue but not in posterior regenerating tissue. We also found that the mature miRNAs from the sme-miR71b/miR2d/miR752/miR13 miRNA cluster are differentially enriched in proliferating neoblasts and neoblast progeny. These studies suggest that the abundance of mature miRNAs from the sme-miR71b/miR2d/miR752/miR13 miRNA cluster is post-transcriptionally regulated. Together, these results highlight miRNA-mediated post-transcriptional regulation as a key regulator of gene expression involved in stem cell function and regeneration in planarians.
RESULTS
Deep sequencing of small RNAs from regenerating tissue and neoblast populations
Previous studies of planarian miRNAs have revealed several strain- and neoblast-specific miRNAs. Most of these studies involved comparing small RNA profiles between irradiated and nonirradiated animals or isolating a mixture of proliferating neoblasts and neoblast progeny (Friedländer et al. 2009; Lu et al. 2009). Thus, these studies lack information about miRNA expression in different neoblast subpopulations, which is essential to understand the function of miRNAs during proliferation and differentiation. To compare miRNA profiles in neoblast subpopulations, we FACS-separated proliferating neoblasts (X1), neoblast progeny (X2), and differentiated cells (Xins) using the methodology described previously (Supplemental Fig. 1A; Resch et al. 2012). Total RNA was isolated from each of the cell populations, and small RNA libraries were prepared. Systematic profiling of miRNAs was also performed at 3 h, 6 h, 12 h, 24 h, 3 d, 5 d, and 7 d after amputation from heads that were regenerating tails (posterior regenerating tissue) and separately from tails that were regenerating heads (anterior regenerating tissue) (Supplemental Fig. 1B). These time points were selected so that various regenerative processes, such as wound healing, neoblast proliferation, differentiation, and patterning were represented. Small RNA libraries were also prepared from unamputated animals, which served as a baseline control for miRNA expression levels. Deep sequencing of the small RNA libraries was performed on an Illumina HiSeq 1000/Illumina GAIIx. Consistent with previous studies (Palakodeti et al. 2008; Friedländer et al. 2009), we observed two distinct small RNA populations, one of 18–24 nt representing miRNAs and siRNAs, and a second of 31–32 nt representing piRNAs. Since our focus here is on miRNA expression, all subsequent analysis was restricted to the 18- to 24-nt population. We obtained 50–55 million 18- to 24-nt reads from the regenerating time point libraries and 2–3 million 18- to 24-nt reads from the FACS-purified cell population libraries (Supplemental Fig. 1C). The raw reads were aligned to the S. mediterranea draft genome using Bowtie (Langmead et al. 2009) without any mismatches. Approximately 80%–90% of the total raw reads aligned to the S. mediterranea genome (Supplemental Fig. 1C). The reads were also mapped to a database of known S. mediterranea miRNAs (miRbase). Interestingly, only 36%–40% of the reads obtained from the neoblast populations aligned to known miRNAs, whereas 45%–55% of the reads obtained from the regenerating tissue aligned to known miRNAs (Supplemental Fig. 1C). The unaligned reads could be novel miRNAs, siRNAs, or degradation fragments of larger RNA species. We next used miRDeep2 (Friedländer et al. 2012) to identify novel miRNAs. After filtering the miRDeep2 predicted list using a miRDeep2 score cutoff of +10 and a Randfold P-value cutoff of <0.05, we identified 15 potential new miRNA loci. Of these, two are new members of the sme-miR-754 and sme-miR-2182 miRNA families, while the 13 others appear to be novel planarian-specific miRNAs (Supplemental Table1; Supplemental Fig. 4B). Only 0.01% of the aligned small RNA reads map to the 15 new miRNA loci. The majority of the reads that failed to map to the miRNAs aligned to regions of the genome to which piRNAs align, suggesting that these reads could either be the degradation products of piRNAs or processed products of piRNA precursors (data not shown).
miRNAs enriched in the X1, X2, and Xins populations of S. mediterranea
Recent studies have identified miRNAs that are expressed in neoblasts. These studies either compared miRNA expression between irradiated and nonirradiated animals (Lu et al. 2009) or examined miRNA expression in total neoblasts, which included both the X1 and X2 cell subpopulations (Friedländer et al. 2009). However, neither study provided information regarding miRNAs that are specifically enriched in the X1, X2, and Xins subpopulations. To address this issue, we compared the relative abundance of miRNAs within the X1, X2, and Xins subpopulations. Though most miRNAs were expressed equally in all three cell populations, 61 miRNAs were enriched in one of the three populations. Twenty-six miRNAs were expressed at least two- to threefold higher in the X1 and/or X2 populations, and 35 miRNAs were specifically enriched in the Xins cells (Fig. 1A; Supplemental Table 2). Among the 26 X1- or X2-enriched miRNAs, six were expressed two- to threefold higher in X1 cells, 10 were enriched in X2 cells, and the remaining 10 were enriched in both X1 and X2 cell populations (Fig. 1A; Supplemental Table 2). Although biological replicates were not generated for these data, significant differences in relative miRNA abundance were detected between the X1, X2, and Xins populations (P < 0.0001, ANOVA; P < 0.01, Tukey's HSD test: X1 vs. Xins and X2 vs. Xins). Our data also confirmed that nine of the 10 previously reported neoblast-specific miRNAs (Friedländer et al. 2009) were expressed in the X1 and/or X2 populations. The remaining miRNA, sme-miR-752, was detected in our data but was excluded from subsequent analysis due to the low number of read counts in each population (X1:20, X2:32, and Xins:10).
FIGURE 1.
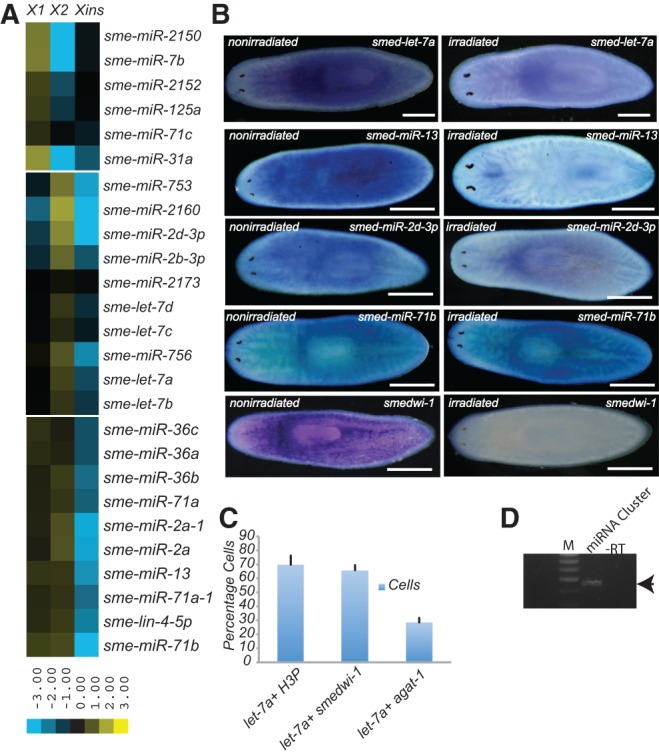
Neoblast enriched miRNAs. (A) miRNAs that are expressed twofold higher in neoblasts (X1) and neoblast progeny (X2) compared to differentiated cells (Xins) were identified, and the expression ratios were log2 transformed and depicted as a heat map. (B) In situ hybridization showing expression of miRNAs in nonirradiated and irradiated animals. The miRNAs sme-miR-7a (8/8), sme-miR-13 (8/8), sme-miR-2d-3p (8/8), sme-miR-71b (8/8), and smedwi-1 (8/8) displayed mesenchymal staining. The expression of all the above-mentioned miRNAs except sme-miR-71b was substantially reduced 3 d post-irradiation. Smedwi-1 is a neoblast-specific gene used as a positive control for the experiment. The scale bars indicate 500 µ. Numbers (x/x) indicate the number of animals that showed similar staining patterns (numerator) versus the total number of animals used for in situs (denominator). (C) Histogram depicting the percentage of sme-let-7a positive cells that colocalize with H3PS10, smedwi-1, and agat-1 cells. H3PS10 and smedwi-1 stain neoblast cells, and agat-1 is a category 3 progenitor cell marker. Error bars are calculated from the biological duplicates. (D) RT-PCR shows expression of the miRNA cluster sme-miR71b/miR2d/miR752/miR13. Minus RT is used as a negative control for RT-PCR. Arrow mark shows the band on the agarose gel.
Previously, it was reported that let-7a and let-7b were enriched in neoblasts (Friedländer et al. 2009; Lu et al. 2009). Interestingly, our study revealed that let-7a and let-7b show fold-change enrichment in the X2 population (X1: 1.004, X2: 1.429, Xins: 0.56), which predominantly contains neoblast progeny rather than proliferating neoblasts (X1) and differentiated cells (Xins). Neoblast progeny cells are G1-cycling cells that could potentially differentiate into the progenitors. To test whether sme-let-7a expression is enriched in the G1-cycling cells or the progenitors, we performed colocalization studies on cell macerates using in situ probes for smedwi-1 and agat-1, which are the markers for the neoblast population and category 3 progenitors (Eisenhoffer et al. 2008), respectively. Colocalization studies were also performed using antibodies against H3PS10, which stains neoblasts in the G2/M stage of the cell cycle. Our results clearly showed that let-7a strongly colocalizes with H3P (69.32%) and smedwi-1 (65.35%) compared to agat-1 (27.7%), confirming that let-7a expression is more prevalent in neoblasts than in category 3 progenitors (Fig. 1C; Supplemental Fig. 5). We observed a slight discrepancy between the miRNA-seq and whole animal macerate results for sme-let-7a expression in the X1 and X2 populations. Analysis of miRNA-seq data revealed a 1.4-fold enrichment of sme-let-7a in the X2 population compared to X1, while the colocalization studies using whole animal macerates indicated similar levels of sme-let-7a expression in the X1 and X2 populations. Although we cannot entirely explain the slight difference in these results, we believe the incongruity may be attributed to subtle differences in the nature and sensitivity of the experiments. Nonetheless, the results clearly show that sme-let-7a expression is enriched in the neoblast population as opposed to progenitor cells. Thus, the abundance of let-7a in the neoblast population suggests a potential role for these miRNAs in neoblast function.
It was previously reported that mature miRNAs from the sme-miR-71b/2d/752/13 miRNA cluster were expressed in neoblast populations (Friedländer et al. 2009; Lu et al. 2009). Interestingly, we found that miR-71b and miR-13 were enriched in both the X1 and X2 cell populations, while sme-miR-2d-3p was enriched in the X2 population (Fig. 1A). It is thought that miRNA clusters are expressed as a single pri-miRNA transcript containing all of the miRNAs in the cluster that are then processed by Drosha to generate multiple pre-miRNAs (Baskerville and Bartel 2005). To demonstrate the polycistronic nature of the sme-miR71b/miR2d/miR752/miR13 cluster, we performed RT-PCR using primers that amplify the miRNA cluster. This experiment generated an ∼500-bp fragment indicating that the sme-miR71b/miR2d/miR752/miR13 cluster is indeed expressed as a polycistronic precursor (Fig. 1D). Thus, the differential enrichment of the individual miRNAs encoded in the sme-miR-71b/2d/752/13 cluster in different cell populations suggests that expression levels of the individual miRNAs may be regulated by differential processing by Drosha or Dicer. We performed whole mount in situ hybridization using LNA oligonucleotides to study the expression of miRNAs sme-miR-13, miR-71b, and miR-2d-3p from the sme-miR-71b/2d/752/13 cluster. In addition to the mesenchymal expression pattern, we also observed expression of miR-13 and miR-2d-3p in germline and CNS tissues, respectively (Supplemental Fig. 2A), which confirmed differential processing of the sme-miR-71b/2d/752/13 cluster. We observed a twofold enrichment of sme-miR-124b in Xins cells indicating expression of this miRNA in differentiated tissue (Fig. 2A). Sme-miR-124b was previously shown to be expressed in the cephalic ganglion (González-Estévez et al. 2009; Pearson et al. 2009), and our whole mount LNA based in situ hybridization experiment confirmed this expression pattern (Supplemental Fig. 2C).
FIGURE 2.

miRNAs enriched in differentiated cells. (A) miRNAs that are expressed twofold higher in differentiated cells (Xins) compared to neoblasts (X1) and neoblast progeny (X2) were identified, and the expression ratios were log2 transformed and depicted as a heat map. (B) In situ hybridization showing the expression of miRNAs in irradiated and nonirradiated animals. The miRNA sme-miR-124c (8/8) is expressed in the cephalic ganglion (CG) and ventral nerve cord (VNC). sme-miR-lin4-3p (4/4) is expressed in the intestine (I) and pharynx (P) whereas sme-miR-1c (8/8) expression is restricted to the pharynx (P). Numbers (x/x) indicate the number of animals that showed similar staining patterns (numerator) versus the total number of animals used for in situs (denominator).
We performed in situ hybridization to study the expression of sme-miR-1c and sme-lin-4-3p, which we found to be enriched in Xins cells from our sequencing data. Both sme-mir-1c and sme-lin-4-3p are localized to the pharyngeal region, though sme-lin4-3p is also expressed in intestinal tissue (Fig. 2B). Since X1 and X2 cells are highly sensitive to irradiation, miRNAs expressed in those cells should be significantly reduced upon irradiation, while miRNAs enriched in Xins cells remain unchanged. Whole mount in situ hybridization revealed that sme-let-7a, sme-miR-13, and sme-miR-2d-3p were significantly reduced after irradiation confirming their expression in neoblasts (Fig. 1B). Interestingly, although sme-mir-71b is expressed in the neoblast population, its staining intensity does not decrease upon irradiation (Fig. 1B). This is in agreement with published data showing unperturbed expression of sme-mir-71b upon irradiation (Lu et al. 2009). Sme-miR-124c, which is expressed in the CNS, and Smedwi-1, which is used as a neoblast marker, were included as negative and positive controls, respectively, for these irradiation studies.
miRNAs enriched in regenerating tissue
To identify miRNAs enriched in regenerating tissue, we examined the small RNA sequence data obtained from the regeneration time course described above. The fold-change in the expression of each miRNA at each time point of regeneration was calculated with respect to the miRNA levels in intact (nonregenerating) animals. For this analysis, only miRNAs with >50 reads in one or more of the regeneration time points were considered (Supplemental Table 3). For each time point, we constructed a vector of the above-described gene expression fold-changes across 191 miRNAs. These vectors were used to assemble a matrix of correlation coefficients between time points. This matrix was subjected to hierarchical clustering along both axes. We observed four distinct clusters. The first cluster consists of regeneration time points from 3 h to 24 h for both anterior and posterior regenerating tissues, with a particularly strong correlation between 3-h and 6-h anterior and posterior regenerating tissue. This suggests that most of the miRNAs within these regeneration time points share a similar expression pattern, and some might be essential during the initial stages of regeneration such as wound healing and neoblast migration. A second cluster includes the 5-d and 7-d posterior regenerating tissue, and a third cluster comprises the 5-d and 7-d anterior regenerating tissues. miRNAs within these clusters might be essential for later stages of regeneration such as lineage specification and tissue formation (Supplemental Fig. 4A). These also support the assertion that distinction between anterior and posterior regeneration establishes itself during the later stages of regeneration and that this can be observed from the expression profiles of miRNAs.
Next, we classified miRNA expression according to patterns of enrichment or depletion across the regeneration time points outlined above. Relative miRNA abundance was used to calculate fold-change expression of miRNA across all time points from regenerating tissue compared to intact (nonregenerating) tissue. Fold-change enrichment for the miRNAs in regenerating tissue was calculated as the ratio between the normalized reads for each miRNA from the regenerating tissue at a specific time point and the normalized reads for that miRNA in intact animals. Fold-change values were used to classify miRNAs into the following three categories using hierarchical clustering: (1) miRNAs that were enriched or depleted in the regenerating tissue at all time points during regeneration; (2) miRNAs that were enriched during the initial time points (3–24 h) of regeneration; and (3) miRNAs enriched during later time points in regeneration (3–7 d). We identified 53 miRNAs that belong to category 1, 30 of which showed increased expression and 23 that showed decreased expression in both the anterior and posterior regenerating samples at all time points compared to intact animals (Supplemental Fig. 2A). The miRNAs that were up-regulated at all time points may have roles in wound healing, neoblast proliferation, differentiation, and repatterning of the blastema. In contrast, miRNAs that were down-regulated at all time points may play a role in homeostasis rather than regeneration. In category 2, we identified 20 miRNAs that were enriched within 3–24-h time points following amputation and showed similar patterns of expression for both anterior and posterior regenerating samples (Fig. 3A). These miRNAs may play a role in wound healing, the initial phase of neoblast proliferation, or blastema formation. Among the 20 miRNAs in this category, seven were up-regulated approximately greater than twofold at 3 h and 6 h following amputation (Fig. 3A). Recent studies have shown that wound healing is initiated within 30 min of injury or amputation, which is followed by the migration of neoblasts to the site of injury (Wenemoser and Reddien 2010; Wenemoser et al. 2012). Thus, the miRNAs that are up-regulated at 3 h and 6 h following amputation are the best candidates for investigating the role of miRNAs in wound healing and neoblast migration. Among miRNAs classified as category 3, we identified 22 candidates where miRNAs are expressed in the blastema at later stages of regeneration, 3–7 d post-amputation (Fig. 4A). Interestingly, category 3 miRNAs were predominantly expressed in anterior regenerating tissue, with the exception of sme-miR-96a, sme-miR-96b, and sme-miR-2d-3p that were enriched by 1.7-fold in the blastema of posterior regenerating tissue (Fig. 4A). Consistent with previous studies showing that sme-miR-124c and sme-miR-124b are expressed in the CNS, we found that expression of the miR-124 family (miR-124a, miR-124b, miR-124c, and miR-124e) was enriched in the anterior regenerating tissue 3–7 d following amputation (Fig. 4A), suggesting a possible role in CNS development. Statistical significance of the expression of category 2 and 3 miRNAs was evaluated using DESeq (Anders and Huber 2010) and EgdeR (Robinson et al. 2010). Due to the absence of biological replicates, we considered regeneration time points from category 2 and 3 miRNAs that showed strong correlation (3 h, 6 h [r= 0.89; Pearson's correlation coefficient], and 5 d, 7 d [r =0.91; Pearson's correlation coefficient] from anterior regenerating tissue) as proxies for biological replicates to calculate significance. The majority of miRNAs classified as category 2 (miRNAs enriched during the initial stages of regeneration; 17/20) and category 3 (miRNAs enriched during the later stages of regeneration; 22/22) based on our fold-change analysis also appear to be significantly differentially expressed (P < 0.05; corrected for multiple hypothesis testing) according to our DESeq and EdgeR analysis (Supplemental Table 4).
FIGURE 3.

miRNAs enriched in the regenerating tissue 3–24 h post-amputation. (A) Heat map showing miRNAs enriched greater than twofold during the initial phase of regeneration (3–24 h) in both anterior and posterior regenerating blastema. The heat map indicates log2 fold-change in expression of miRNAs in the regenerative blastema compared to intact animals. (B) Histogram depicting the fold-change in expression of sme-miR-2169-5p 3h and 3d post-amputation in both posterior (top) and anterior (bottom) regenerating blastema as monitored by qRT-PCR. Error bars are calculated from the biological duplicates. (C) In situ hybridization analysis of the expression of sme-mir-2169-5p in regenerating tissue. Sme-miR-2169-5p (6/8) is most abundantly expressed in both the anterior and posterior regenerating blastema 3 h and 6 h post-amputation. Arrows indicate the regenerating blastema. Numbers (x/x) indicate the number of animals that showed similar staining patterns at each time point (numerator) versus the number of animals used for in situs at each time point (denominator).
FIGURE 4.

miRNAs enriched at late stages of regeneration. (A) Heat map showing miRNAs that were enriched greater than twofold 3–7 d post-amputation in both anterior and posterior regenerating blastema. The heat map displays the log2 fold-change of miRNA expression in the regenerative blastema compared to intact animals. (B) Histogram shows the fold-change of sme-miR-124c expression 3 h, 3 d, and 7 d post-amputation in anterior (top) and posterior (bottom) regenerating blastema based on the qRT-PCR results. Error bars are calculated from the biological duplicates. (C) In situ hybridization showing expression of sme-miR-124c (5/5) in regenerating animals. Sme-miR-124c is expressed in the regenerating cephalic ganglion 3 d post-amputation in anterior regenerating tissue. Arrows represent the regenerating cephalic ganglion. Numbers (x/x) indicate the number of animals that showed similar staining patterns at each time point (numerator) versus the number of animals used for in situs at each time point (denominator).
We also analyzed the expression of mature miRNAs derived from the sme-miR-71b/2d/752/13 cluster at various regeneration time points. Interestingly, we found that sme-miR-13, sme-miR-2d-5p, and sme-miR-752-5p expression is up-regulated in the blastema during all time points of regeneration (category 1) in both anterior and posterior regenerating tissue (Supplemental Fig. 3A), while sme-miR-2d-3p is expressed only in the blastema of anterior regenerating tissue 3–7 d post-amputation (Fig. 4A). It is interesting that both sme-miR-2d-3p and sme-miR-2d-5p, which are derived from the same precursor, are enriched in regenerating tissue at different time points during regeneration.
Recently, Qin et al. (2011) identified eight miRNAs that were enriched ≥1.3-fold and five miRNAs that were depleted ≥1.3-fold in regenerating tissue 3 d post-amputation. Four of the miRNAs identified by Qin et al. (sme-miR-13, sme-miR-2d-3p, sme-miR-745, and sme-miR-748) were also observed to be differentially expressed in our study. The remaining four miRNAs were not detected by our analysis, and this difference may be attributed to the fact that Qin et al. (2011) profiled miRNAs from a mixture of both anterior and posterior regenerating tissue or that different thresholds were used to classify miRNAs as differentially expressed.
We validated our sequencing data using real time qRT-PCR and LNA-based in situ hybridizations to detect mature miRNAs. We used qRT-PCR to validate expression of sme-miR-2169-5p, which was 2.5-fold up-regulated at 3 h post-amputation and 2.5-fold down-regulated at 3 d post-amputation in posterior regenerating tissue. In anterior regenerating tissue, sme-miR-2169-5p was 1.6-fold up-regulated and fivefold down-regulated 3 h and 3 d post-amputation, respectively (Fig. 3B). We observed a strong correlation between the sequencing and qRT-PCR results for sme-miR-2152, which was up-regulated at least twofold at all time points post-amputation in both anterior and posterior regenerating tissues (Supplemental Fig. 3B). We validated the expression of sme-miR-124c, which was up-regulated at least 3.5-fold 3 d and 7 d post-amputation in anterior regenerating tissue. In posterior regenerating tissue, sme-miR-124c was down-regulated at all time points (3 h, 6 h, 3 d, and 7 d) as in the sequencing data (Fig. 4B). We also verified expression of sme-miR-2152, sme-miR-2169-5p, sme-miR124c, and sme-miR-2d-3p by LNA-based in situ hybridization. Sme-miR-2152 was highly and ubiquitously expressed at all time points in regenerating animals and showed weak expression in intact animals (Supplemental Fig. 3C). Sme-miR-2169-5p expression was up-regulated at 3 h and 6 h post-amputation in both anterior and posterior regenerating tissues. Most of the sme-miR-2169-5p expression was restricted to the blastema and gradually decreased in expression from 3 h to 7 d post-amputation (Fig. 3C). Sme-miR-124c is known to be expressed in the CNS, and we observed expression in the blastema of anterior regenerating tissue beginning 3 d post-amputation in the primordial brain, and by day 7 expression was distributed throughout the cephalic ganglion and ventral nerve cord (Fig. 4C). Sme-miR-2d-3p, which was previously shown to be expressed in neoblast populations, was also expressed in anterior regenerating tissue in the cephalic ganglion 3 d post-amputation (Supplemental Fig. 2B). These data suggest a possible role of the miR-124 family and sme-miR-2d-3p in planarian CNS regeneration.
DISCUSSION
Several miRNAs are differentially enriched in neoblast subpopulations
Neoblasts are pluripotent cells that constitute 30% of the planarian cell population. They are extremely sensitive to irradiation and are characterized by dense nuclei and little cytoplasm. Recent transplantation studies revealed that a single clonogenic neoblast is sufficient to rescue irradiated animals demonstrating neoblast pluripotency (Wagner et al. 2011). Several groups have developed FACS-based methodologies to isolate different cell populations into the following three categories based on nuclear density and sensitivity to irradiation: X1 cells that primarily consist of proliferating neoblasts (∼90%); X2 cells comprised of a heterogeneous mixture of neoblast progeny (∼70%) and early and late progenitors (∼30%); and Xins cells that mostly consist of terminally differentiated cells (Hayashi et al. 2006; Eisenhoffer et al. 2008). Comparative transcriptome analyses have identified several genes that are commonly expressed in planarian neoblasts and embryonic stem cells from higher metazoans, suggesting that the pathways that regulate or confer pluripotency may be highly conserved (Labbé et al. 2012; Onal et al. 2012; Resch et al. 2012; Solana et al. 2012). Recent studies based on small RNA sequencing from irradiated animals and from neoblast populations identified miRNAs that are specifically enriched in neoblasts (Friedländer et al. 2009; Lu et al. 2009). A limitation of these studies is that they did not identify miRNAs that are specifically enriched in the X1, X2, and Xins cell populations. Here, we sequenced small RNA libraries prepared from purified X1, X2, and Xins cell populations to identify a set of miRNAs that are differentially expressed across these cell populations. Previous studies identified 10 miRNAs (sme-let7a, sme-let7b, sme-miR-36b, sme-miR-2a, sme-miR-2d-3p, sme-miR-13, sme-miR-71b, sme-miR-752, sme-miR-756, and sme-miR-2160) whose expression was enriched in neoblasts (Friedländer et al. 2009; Lu et al. 2009). The results reported here reconfirmed the enrichment of these 10 miRNAs in neoblasts and identified 17 additional neoblast-enriched miRNAs. Furthermore, our data revealed that these 27 miRNAs are specifically enriched in the X1 and/or X2 populations. For instance, sme-let7a, sme-let7b, sme-miR-2d-3p, sme-miR-756, and sme-miR-2160 are specifically enriched in the X2 population (Fig. 1A). The X2 population predominantly consists of neoblast progeny, which are the G1 cycling neoblast cells that could potentially differentiate into progenitors. Since the X2 population is a heterogeneous population consisting of neoblasts and progenitors, there is a possibility that sme-let-7a might either be expressed in neoblasts or progenitor cells. To resolve the issue of let-7a expression, we performed colocalization studies for let-7a on the cell macerates expressing smedwi-1, which is expressed in neoblasts, agat-1, which is a category 3 progenitor marker, and H3PS10, which stains G2/M cycling neoblasts. Our results clearly demonstrated that, unlike in mammals, sme-let-7a is enriched in neoblasts rather than progenitor cells (Fig. 1C). Nonetheless, it is important to note that agat-1- expressing progenitors constitute only a few among several characterized neoblast progenitors. Currently, we cannot rule out the possibility that sme-let-7a is expressed in other progenitor cell types that were not investigated in this study. We also observed enrichment of sme-miR-71b, sme-miR-36b, sme-miR-2a, and sme-miR-13 in both the X1 and X2 cell populations (Fig. 1A), suggesting a possible role in neoblast maintenance and transformation of proliferating neoblasts to neoblast progeny during differentiation.
It has been reported that miRNAs from the same cluster are coexpressed and coordinately regulate cellular pathways (Xu and Wong 2008; Kim et al. 2009). However, our results showed that while the sme-miR-13/71b/2d/752 cluster is expressed as a single polycistronic transcript, the mature miRNAs are differentially enriched in either the X1 or X2 populations (Fig. 1A). Interestingly, in situ hybridization revealed that the mature miRNAs from the sme-miR-13/71b/2d/752 cluster are expressed in different tissue types in addition to their expression in mesenchymal tissue (Supplemental Fig. 2A). Since the X1 and X2 populations are extremely sensitive to irradiation, miRNAs expressed in these populations should show coordinately decreased expression upon irradiation. Although the levels of most miRNAs expressed in the X1 and X2 cells decreased after irradiation, the levels of sme-miR-71b surprisingly showed very little change (Fig. 1A,B). Together, these results suggest that the abundance of the mature miRNAs from the sme-miR-13/71b/2d/752 cluster is regulated post-transcriptionally. A similar observation was recently reported in Drosophila by Ryazansky et al. (2011), where the individual mature miRNAs from a single cluster showed different expression profiles from one another.
miRNAs show distinct expression patterns during regeneration
Regeneration in planarians is characterized by cellular events such as wound healing, neoblast proliferation, differentiation, and repatterning to regenerate complete animals or lost tissues. Recently, Wenemoser et al. (2012) identified four categories of wound-induced gene expression patterns, which differ in their timing and location of expression. They also identified waves of gene expression, where the first wave (W1) of expression is initiated within 30 min of injury and tapers down within 24 h post-injury. Most of the W1 genes show homology with immediate early response genes in mammals. The second and third waves of gene expression peak 6–12 h post-injury and are essential for neoblast proliferation and regeneration. Wenemoser and Reddien (2010) also reported that neoblast proliferation during regeneration happens in two phases: phase one is initiated immediately after wound healing and decreases 24 h post-amputation, and phase two is characterized by neoblast proliferation (essential for regeneration and growth of the blastema) and occurs 3 d post-amputation. These studies suggest that transcriptional and post-transcriptional regulation of gene expression is essential for regeneration and neoblast function. Though several genes and pathways that regulate neoblast proliferation and differentiation have been identified, the key signals that drive neoblasts to differentiate into specific tissues and organs are not known. Recent studies have revealed expression of miRNAs in regenerating animals, but a comprehensive analysis of miRNA expression in regenerating tissue at different time points has not been conducted (Qin et al. 2011; Tian et al. 2012; Xu et al. 2013). In the current study, we have performed deep sequencing of small RNA libraries prepared from regenerating tissue at various regeneration time points and detected several miRNAs that showed temporal expression in anterior and posterior regenerating tissue. Computational analysis identified miRNAs enriched between 3 and 24 h post-amputation (category 2 miRNAs) in both anterior and posterior regenerating tissue. These miRNAs resemble the W1 category of genes in their expression pattern and might be involved in wound healing, neoblast migration, and the initial phase of neoblast proliferation. In contrast, category 3 miRNA expression is observed 3 d post-amputation and is sustained throughout regeneration. Interestingly, this expression pattern is only observed in anterior regenerating tissue. Several of the miRNAs that display this expression pattern are members of the miR-124 family which is known to be expressed in the CNS and may, therefore, play a role in CNS regeneration. In contrast to what was observed in vertebrates, recent studies in Drosophila revealed a role for miR-124 in the proliferation of neuroblasts rather than differentiation (Weng and Cohen 2012). The expression of miR-124 3 d post-amputation specifically in the brain primordium suggests a role for mir-124 in differentiation and patterning rather than proliferation, though the latter cannot be ruled out. We also observed differential expression of sme-miR-124a, b, and c, which were similarly enriched in regenerating tissue. For instance, sme-miR-124a and b were predominantly expressed in the cephalic ganglion, whereas sme-miR-124c was primarily expressed in the cephalic ganglion and ventral nerve cord (Fig. 2B; Supplemental Fig. 2C). This difference in the expression pattern suggests potential functional differences within the miR-124 family of miRNAs. Further, sme-miR-2d-3p, which is expressed in the neoblast progeny and cephalic ganglion (Fig. 4A; Supplemental Fig. 2A) is also enriched in regenerating tissue in a manner similar to the miR-124 family. In situ hybridization revealed that sme-miR-2d-3p, like miR-124, is expressed in the brain primordium 3 d post-amputation throughout the remainder of the differentiation process of the blastema into the completely regenerated cephalic ganglion (Supplemental Fig. 2B). Interestingly, both miR-2d-3p and miR-124 miRNAs are not expressed in the posterior regenerating blastema, indicating that these miRNAs may not be essential for ventral nerve cord regeneration. The fact that miR-2d-3p is enriched in the neoblast progeny and the regenerating brain indicates that it may drive the differentiation of neoblast progeny to the brain primordium. Confirmation of this would require studies in which sme-miR-2d-3p is inactivated, something not currently possible in planarians. Supporting our observation that sme-miR-2d-3p may be involved in brain regeneration, recent studies in Drosophila have shown that the mRNA targets regulated by the miR-2 family are enriched for genes essential for neural development (Marco et al. 2012). We also observed the enrichment of sme-miR-96a and b 3 d post-amputation in anterior regenerating blastema. The mir-96 family of miRNAs are vertebrate-specific miRNAs, and recent studies have shown that mutations in the seed region of these miRNAs leads to defects in inner hair cell formation and deafness in mammals (Kuhn et al. 2011). Intriguingly, we did not identify any miRNAs that were enriched in posterior regenerating blastema 3 d post-amputation, suggesting that few if any miRNAs are specifically involved in the regeneration of posterior regions of the planarians.
In conclusion, our study has identified several miRNAs that are specifically expressed in different neoblast populations and in the blastema at different time points of regeneration, highlighting the importance of miRNAs in fine tuning gene expression during planarian regeneration. Future goals include identifying the mRNA targets that are directly regulated by these miRNAs, which will increase our understanding of the miRNA-mediated post-transcriptional regulatory networks essential for regeneration and stem cell function in planarians.
MATERIALS AND METHODS
Whole mount in situ hybridization
In situ hybridization was performed as described by Pearson et al. (2009) with slight modifications based on the size of the animals. For animals bigger than 4 mm, N-acetyl cysteine concentration was increased to 10%. Fixation and proteinase K treatment was increased by 10 min each. Hybridization was done at 52°C for >16 h. To reduce nonspecific staining, the antibody dilution was increased to 1:2500 (anti-Dig AP conjugated). Digoxigenin-labeled miRCURY LNA (locked nucleic acid) probes synthesized by Exiqon were used to carry out the miRNA specific in situs. Wi-1 probe was synthesized using the DIG RNA labeling kit (Roche). BM purple (Roche) was used as a substrate for alkaline phosphatase instead of NBT/BCIP.
In situ hybridization on macerates
For in situs on dissociated cells, cell maceration was performed as previously described (Hayashi et al. 2006; Resch et al. 2012). Approximately 200,000 cells were dried overnight at room temperature on 0.1% gelatin-chromalum-coated slides, followed by a PBS wash and fixation for 25 min in 4% formaldehyde. Cells were then rinsed with 100% methanol for 5 min, followed by washes with a methanol dilution series (75%, 50%, 25%) in PBT(1× PBS+0.1% Tween-20) and finally two PBT washes for 10 min each. Cells were permeabilized with proteinase K solution for 10 min at room temperature, then refixed and washed twice with PBT. Prehybridization and hybridization were done as mentioned earlier but at 45°C. Post-hybridization washes were done with 1:1 hyb solution: 2× SSC for 5 min, 2× SSC for 5 min, and 2× SSCT (2× SSC +0.1% Tween-20) wash for 15 min at hyb temperature. Two stringent washes were given with 0.2× SSCT for 15 min each at 65°C, followed by PBT washes at room temperature. Antibody incubation and tyramide amplification were performed as described in King and Newmark (2013). The antibodies used were anti-dig POD (1:2000) and streptavidin-HRP (1:1000). FITC and Cy3 tyramide were used at a dilution of 1:2000. Images were taken using a Zeiss LSM 700 confocal microscope. Images for quantitation were taken using a 40× oil objective, and the representative images were taken at 63× oil immersion. Quantitation was done manually by counting the number of cells that were positive for DAPI, DAPI+Let7a, DAPI+H3PS10, and DAPI+Smedwi1. The no-probe control and no-primary-antibody control were used as negative controls. Correction for the nonspecific background fluorescence was performed using negative controls.
Cell sorting
Fluorescent activated cell sorting (FACS) was performed as described (Hayashi et al. 2006; Resch et al. 2012) in a Becton and Dickenson FACS ARIA cell sorter.
Exposure to γ irradiation
Animals were given a lethal dose of 9000 Rads (blood irradiator 2000, BRIT) and cultured at 20°C for 3 d before fixing for the experiments.
Total RNA isolation
RNA was isolated using Trizol according to the manufacturer's instructions (Invitrogen). Animals were cut anterior of the pharynx, such that all the posterior regenerating pieces were devoid of pharynx. The blastema was systematically collected at different time points (3 h, 6 h, 12 h, 24 h, 3 d, 5 d, 7 d) using microdissection under an Olympus SZ61 stereo microscope and stored in Trizol. Total RNA was isolated using Trizol, after collecting the blastema from all time points.
Small RNA library preparation
Small RNA libraries were prepared using the Illumina TruSeq Small RNA kit as described by the manufacturer (Illumina). Two hundred fifty nanograms of total RNA were used from each sample for the library preparation. 5′ and 3′ Small RNA adaptors were ligated to the RNA, and the ligated products were reverse-transcribed using SuperScript II reverse transcriptase (Invitrogen). The RT products were then amplified by polymerase chain reaction and resolved on an 8% polyacryamide gel. Bands corresponding to 140–160 nt were gel-eluted. The size and integrity of each library was verified using the Bioanalyzer. The libraries were sequenced on an Illumina HiSeq 1000.
Computational analysis
Reads were aligned to both the draft assembly of the S. mediterranea genome and the known S. mediterranea miRNAs from miRBase using Bowtie-0.12.1 (Langmead et al. 2009). Reads that mapped to miRNAs were normalized to the total number of mapped reads. Fold-change enrichment of miRNAs in the X1, X2, and Xins cell populations was calculated as the ratio between the normalized reads for each miRNA and the average normalized reads for that miRNA across all the three samples. In contrast, fold-change enrichment for the miRNAs in regenerating tissue was calculated as the ratio between the normalized reads for each miRNA from the regenerating tissue at a specific time point and the normalized reads for that miRNA in intact animals. Hierarchical clustering was used to cluster fold-change values using cluster3.0 (de Hoon et al. 2004). Heat maps were constructed using Java TreeView (Saldanha 2004).
Real-time PCR
Real-time PCR was performed following the protocol described previously (Chen et al. 2005). One hundred fifty nanograms of total RNA were used for each reaction. The primers for reverse transcription were designed to form a double-stranded stem–loop structure with the 5′ end being complementary with a universal reverse primer and last 8 nt at the 3′ end being complementary to the 3′ end of the miRNA. PCR was carried out using the universal primer and an miRNA specific primer. To normalize for variations that may occur during reverse transcription and PCR, we used a synthetic RNA sequence that lacks complementarity with the S. mediterranea genome. All RNA samples were spiked with 0.5 pg of the synthetic RNA control prior to reverse transcription. cDNA was prepared using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR was carried out using SYBR green (Applied Biosystems) as the fluorescent detector on an Applied Biosystems 7900HT machine.
miRNA-specific primers
sme-miR-2169-5p
RT primer: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATCAGATC
FR primer: ACACTCCAGCTGGGAACTTTGAAATTC
sme-miR-2152
RT primer: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAAAGAGAG
FR primer: ACACTCCAGCTGGGGGGTTCGAAGTATG
sme-miR-124c
RT primer: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGCATTC
FR primer: ACACTCCAGCTGGGTAAGGCACGCGGT
Universal Reverse primer: TGGTGTTCGTGGAGTCGGCAATTCAGTTG
Solexa RT primer: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGATCGTCG
Solexa FR primer: ACACTCCAGCTGGGGTTCAGAGTTCTACAG
Solexa RNA adaptor sequence: GUUCAGAGUUCUACAGCGACGAUC
DATA DEPOSITION
The small RNA sequencing data have been deposited at the NCBI Sequence Read Archive (SRA) at http://www.ncbi.nlm.nih.gov/sra (accession number SRA065477).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article, and codes of LS-RSMR are available at http://sse.tongji.edu.cn/yingshen/RSMR/LS_RSMR.html.
ACKNOWLEDGMENTS
We thank Dr. Yashoda Ghanekar and Palakodeti lab members for proofreading and helpful comments. We also thank Prof. Phil Newmark for supplying animals (Schmidtea mediterranea). High-throughput sequencing was performed at the Next-Generation Sequencing facility at the Centre for Cellular and Molecular Platform (C-CAMP), Bangalore. FACS analysis was done at the CIFF facility, NCBS, Bangalore. A.S. was supported by Ramanujam Fellowship from DST. This work was generously supported by the DBT-Wellcome Trust India Alliance. D. Palakodeti is supported by funding from a DBT-Wellcome Trust Intermediate Fellowship awarded by DBT-Wellcome Trust India Alliance.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Aboobaker AA 2011. Planarian stem cells: A simple paradigm for regeneration. Trends Cell Biol 21: 304–311 [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP 2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. 2005. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res 33: e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S 2004. Open source clustering software. Bioinformatics 20: 1453–1454 [DOI] [PubMed] [Google Scholar]
- Eisenhoffer GT, Kang H, Sánchez Alvarado A 2008. Molecular analysis of stem cells and their descendants during cell turnover and regeneration in the planarian Schmidtea mediterranea. Cell Stem Cell 3: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Adamidi C, Han T, Lebedeva S, Isenbarger TA, Hirst M, Marra M, Nusbaum C, Lee WL, Jenkin JC, et al. 2009. High-resolution profiling and discovery of planarian small RNAs. Proc Natl Acad Sci 106: 11546–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Estévez C, Arseni V, Thambyrajah RS, Felix DA, Aboobaker AA 2009. Diverse miRNA spatial expression patterns suggest important roles in homeostasis and regeneration in planarians. Int J Dev Biol 53: 493–505 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Asami M, Higuchi S, Shibata N, Agata K 2006. Isolation of planarian X-ray-sensitive stem cells by fluorescence-activated cell sorting. Dev Growth Differ 48: 371–380 [DOI] [PubMed] [Google Scholar]
- Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, Hur K, Yoo MW, Lee HJ, Yang HK, et al. 2009. Functional links between clustered microRNAs: Suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res 37: 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RS, Newmark PA 2012. The cell biology of regeneration. J Cell Biol 196: 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RS, Newmark PA 2013. In situ hybridization protocol for enhanced detection of gene expression in the planarian Schmidtea mediterranea. BMC Dev Biol 12: 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn S, Johnson SL, Furness DN, Chen J, Ingham N, Hilton JM, Steffes G, Lewis MA, Zampini V, Hackney CM, et al. 2011. miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc Natl Acad Sci 108: 2355–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé RM, Irimia M, Currie KW, Lin A, Zhu SJ, Brown DD, Ross EJ, Voisin V, Bader GD, Blencowe BJ, et al. 2012. A comparative transcriptomic analysis reveals conserved features of stem cell pluripotency in planarians and mammals. Stem Cells 30: 1734–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YQ, Zeng A, Han XS, Wang C, Li G, Zhang ZC, Wang JY, Qin YW, Jing Q 2011. Argonaute-2 regulates the proliferation of adult stem cells in planarian. Cell Res 21: 1750–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo D, Beane WS, Levin M 2012. Modeling planarian regeneration: A primer for reverse-engineering the worm. PLoS Comput Biol 8: e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Smielewska M, Palakodeti D, Lovci MT, Aigner S, Yeo GW, Graveley BR 2009. Deep sequencing identifies new and regulated microRNAs in Schmidtea mediterranea. RNA 15: 1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Hooks KB, Griffiths-Jones S 2012. Evolution and function of the extended miR-2 microRNA family. RNA Biology 9: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onal P, Grün D, Adamidi C, Rybak A, Solana J, Mastrobuoni G, Wang Y, Rahn HP, Chen W, Kempa S, et al. 2012. Gene expression of pluripotency determinants is conserved between mammalian and planarian stem cells. EMBO J 31: 2755–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palakodeti D, Smielewska M, Graveley BR 2006. MicroRNAs from the planarian Schmidtea mediterranea: A model system for stem cell biology. RNA 12: 1640–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palakodeti D, Smielewska M, Lu YC, Yeo GW, Graveley BR 2008. The PIWI proteins SMEDWI-2 and SMEDWI-3 are required for stem cell function and piRNA expression in planarians. RNA 14: 1174–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, Eisenhoffer GT, Gurley KA, Rink JC, Miller DE, Sánchez Alvarado A 2009. Formaldehyde-based whole-mount in situ hybridization method for planarians. Dev Dyn 238: 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin YF, Fang HM, Tian QN, Bao ZX, Lu P, Zhao JM, Mai J, Zhu ZY, Shu LL, Zhao L, et al. 2011. Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica. Genomics 97: 364–371 [DOI] [PubMed] [Google Scholar]
- Reddien PW 2011. Constitutive gene expression and the specification of tissue identity in adult planarian biology. Trends Genet 27: 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch AM, Palakodeti D, Lu YC, Horowitz M, Graveley BR 2012. Transcriptome analysis reveals strain-specific and conserved stemness genes in Schmidtea mediterranea. PLoS One 7: e34447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK 2010. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhana L, Shibata N, Nishimura O, Agata K 2010. Different requirements for conserved post-transcriptional regulators in planarian regeneration and stem cell maintenance. Dev Biol 341: 429–443 [DOI] [PubMed] [Google Scholar]
- Ryazansky SS, Gvozdev VA, Berezikov E 2011. Evidence for post-transcriptional regulation of clustered microRNAs in Drosophila. BMC Genomics 12: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Solana J, Kao D, Mihaylova Y, Jaber-Hijazi F, Malla S, Wilson R, Aboobaker A 2012. Defining the molecular profile of planarian pluripotent stem cells using a combinatorial RNAseq, RNA interference and irradiation approach. Genome Biol 13: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian QN, Bao ZX, Lu P, Qin YF, Chen SJ, Liang F, Mai J, Zhao JM, Zhu ZY, Zhang YZ, et al. 2012. Differential expression of microRNA patterns in planarian normal and regenerative tissues. Mol Biol Rep 39: 2653–2658 [DOI] [PubMed] [Google Scholar]
- Wagner DE, Wang IE, Reddien PW 2011. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 13: 811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenemoser D, Reddien PW 2010. Planarian regeneration involves distinct stem cell responses to wounds and tissue absence. Dev Biol 344: 979–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenemoser D, Lapan SW, Wilkinson AW, Bell GW, Reddien PW 2012. A molecular wound response program associated with regeneration initiation in planarians. Genes Dev 26: 988–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng R, Cohen SM 2012. Drosophila miR-124 regulates neuroblast proliferation through its target anachronism. Development 139: 1427–1434 [DOI] [PubMed] [Google Scholar]
- Xu J, Wong C 2008. A computational screen for mouse signaling pathways targeted by microRNA clusters. RNA 14: 1276–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Chen M, Ren Z, Zhang N, Xu H, Liu X, Tian G, Song L, Yang H 2013. Deep sequencing identifies regulated small RNAs in Dugesia japonica. Mol Biol Rep 40: 4075–4081 [DOI] [PubMed] [Google Scholar]