

Abstract
Several potential pathophysiologic phenomena, including “cerebral shutdown,” are postulated to be responsible for SUDEP. Since the evidence for a seizure-related mechanism is strong, a poor understanding of the physiology of human seizure termination is a major handicap. However, rather than a failure of a single homeostatic mechanism, such as postictal arousal, it may be a “perfect storm” created by the lining up of a several factors that lead to death.
There is strong evidence to suggest that sudden unexpected death in epilepsy (SUDEP) is a seizure-related phenomenon (1–5). The agonal pathophysiological processes in SUDEP are likely to begin during, or in the immediate aftermath of, a seizure. However, potential mechanisms are speculative. Epidemiological studies have consistently pointed to the generalized tonic–clonic seizure as the seizure type most commonly associated with SUDEP (6, 7), and almost all SUDEP reports of deaths during monitoring have occurred after generalized tonic–clonic seizures, (8–11) and often during sleep (3, 12). This seizure type is associated with more severe obtundation of sensorium in the postictal period and can be associated with cardiorespiratory dysfunction during this time. Several questions arise: How do seizures terminate, and how do termination mechanisms relate to postictal arousal? Is the extent of obtundation and subsequent postictal arousal different in the frequent generalized tonic–clonic seizures of patients with chronic intractable epilepsy at high risk of SUDEP? Does postictal arousal differ according to the sleep–wake state at the time of seizure onset? Is postictal failure of arousal the same as the “cerebral shutdown” phenomenon seen on EEG? These questions remain unanswered.
Seizure Termination and the Postictal State
The generalized tonic–clonic seizure is usually characterized by generalized tonic posturing, a phase of tremulousness or “vibration” followed by a generalized clonic phase and, usually, seizure termination within the first 2 minutes of the clinical onset of generalization (13). In some genetic generalized epilepsies (e.g., juvenile myoclonic epilepsy), there may be generalized myoclonic jerking before the tonic phase; in secondarily generalized seizures, there may be a pretonic phase where version or “figure 4” posturing occurs. However, given this relative stereotypy, there is considerable clinical as well as electrographic heterogeneity in the postictal period (14, 15). Some patients recover quickly, while others have varying periods of postictal stupor. Some have brief periods of focal or generalized EEG slowing, while others have prolonged postictal generalized EEG suppression (PGES) and recovery to baseline can sometimes take many hours. Whether this postictal electroclinical heterogeneity aids SUDEP risk-stratification is debated (9, 16). Undoubtedly, there are a number of potential influences, including those of medication effects, but none have been systematically studied.
Postictal Generalized EEG Suppression and “Cerebral Shutdown”
PGES occurs in between 8% of pediatric seizure patients (17) to 65% or more of adult patients with generalized motor seizures (9) and has been reported in several monitored SUDEP/near SUDEP cases (8–11, 18, 19) where some authors have used the term “cerebral shutdown” (19). Prolonged PGES may be an independent risk factor for SUDEP, but whether it is a surrogate marker or a direct index of cerebral and brainstem dysregulation is unclear. It has been shown that >50 seconds of PGES significantly increases the adjusted odds rations for SUDEP (5.22; 95% CI, 1.26–21.58; p < 0.05) and for each 1-second increase in the duration of PGES, the odds of SUDEP increases by a factor of 1.7% (95% CI, 1.005–1.027; p < 0.005) (9). A recent study examining sympathetic and parasympathetic changes in seizure patients measured by electrodermal activity and heart rate variability (20). An increase in electrodermal activity response amplitude and a decrease in parasympathetic-modulated high-frequency power of heart rate variability were directly correlated to prolonged PGES. Therefore, PGES may serve as a marker of postictal autonomic dysregulation, although how these parameters are pathophysiologically associated is unclear. In another study of 48 patients with generalized tonic–clonic seizures, 13 patients with PGES were compared to 12 random controls and one seizure analyzed per individual (21). Patients with PGES were significantly more likely to be motionless in the postictal period and to have simple resuscitative interventions performed (suction, oxygen administration, placed in recovery position, vital signs checked). PGES in such individuals may indicate deeper postictal coma, more delayed arousal and, at least hypothetically, a predisposition to SUDEP. One study compared secondarily generalized convulsive seizures with and without PGES, and found that oxygen desaturation duration and extent as well as peak end-tidal CO2 elevation was more marked in patients with PGES (22).
At least one study has questioned the association between PGES and SUDEP, although it has important methodological differences (16). This study, which examined the EEG records of 17 SUDEP cases and matched controls, found no significant differences in either presence or duration of PGES between the two groups. However, it was carried out mainly in temporal lobe epilepsy patients undergoing presurgical evaluations for temporal lobectomy. SUDEP cases, therefore, are more likely to have had intractable epilepsy compared to surgically seizure-free controls whose risk of SUDEP was artificially removed but who were potentially biologically indistinct from cases. To negate these biases, a much larger number of controls with actively intractable epilepsy and a broader representation of different epilepsy syndromes are necessary.
Seizure Termination
Does postictal electroclinical heterogeneity reflect differences in seizure termination mechanisms in etiologically and syndromically diverse epilepsies? What bearing do these have on SUDEP? Clinical and EEG patterns of seizure termination suggest that different mechanisms operate in different situations, although none of these have been convincingly explained in humans. The 3–4 Hz generalized spike–wave discharges of the typical absence seizure terminate abruptly with an almost immediate clinical and electrographic return to the pre-seizure baseline. It has been hypothesized that the seizure termination mechanism here involves a switch-like desynchronization rather than exhaustion of neurotransmitter release, the latter potentially inducing longer lasting neuronal dysfunction than observed in absence seizures (23). Such neuronal dysfunction is usually evident in the postictal state of generalized convulsions in the form of PGES or high amplitude regional/generalized slowing of the EEG and the attendant clinical features of confusion, cognitive slowing, and even postictal coma. The focal status epilepticus of epilepsia partialis continua is an interesting phenomenon where there is failure of seizure termination but a concurrent containment of the ictal discharge that prevents secondary generalization. It is likely that different mechanisms account for each process (23).
In the generalized tonic–clonic seizure, the clonic phase EEG record shows generalized EEG suppression in between the epileptiform bursts that drive clonus. Seizure end then comprises an abrupt cessation of these bursts, followed by a more continuous PGES. Does this intermittent suppression toward the end of the clonic phase (Figure 1) indicate an actively accruing seizure termination process, and can the characterization of this throw light on subsequent PGES, particularly the prolongation associated with SUDEP? Asymmetric seizure endings (Figure 2) in some patients with generalized tonic clonic seizure patients suggest that there is no central switch or generalized desynchronization mechanism in at least the generalized tonic clonic seizures of focal epilepsy. At present, whether neurotransmitter-mediated seizure termination mechanisms comprise active inhibition or passive depletion is unknown; depolarization block, electrogenic pumps, ionic fluxes, and pH changes have all been implicated. GABA, adenosine, neuropeptide Y, endocannabinoids, and endogenous opioids have also been identified as candidate neurotransmitter systems in animal studies (24). In particular, adenosine, GABA, and opioids merit close study. Ultimately, are over-efficient inhibitory processes that drive seizure termination responsible for SUDEP? At the moment, this remains simplistic speculation.
FIGURE 1.
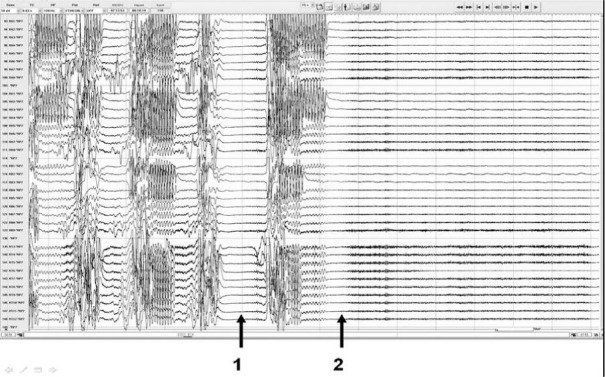
Intracranial EEG of the generalized tonic–clonic seizure of a patient with MRI negative left temporal lobe epilepsy showing 1. onset of profound EEG suppression even before the end of the generalized clonic phase and 2. onset of postictal generalized EEG suppression.
FIGURE 2.
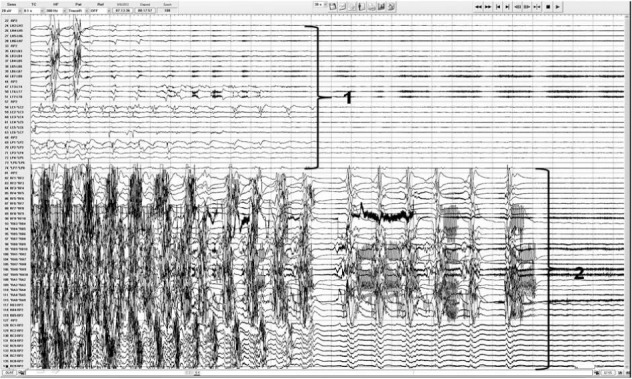
Intracranial EEG recordings showing asymmetric generalized tonic–clonic seizure termination beginning in the left hemisphere (1) followed 11 seconds later by seizure end in the right hemisphere.
Effect of Medication on Postictal Recovery
Postictal EEG changes have been reported to last a mean of 275 seconds in a cohort of patients with focal epilepsy (25). Only two patients in this cohort had generalized tonic–clonic seizures, each lasting 1990 and 2450 seconds, respectively, an observation of a phenomenon true to more recent studies (9, 16, 21). The only study that has looked at postictal EEG in relation to medication is a 2006 levetiracetam (11 patients) versus placebo (12 patients) study (26). The mean time for recovery of the postictal record to baseline was 90 and 130 seconds, respectively, in the two patient groups before randomization to either placebo or levetiracetam. Whereas the placebo group remained at 90 seconds, the recovery time decreased in the treatment group from 130 to 4 seconds—an apparently dramatic effect. In a small sample with subsequently unstudied and therefore unreplicated results, it is perhaps a stretch to extrapolate this to a protective effect against SUDEP, but there is a suggestion here that the postictal EEG deserves scrutiny.
Sleep, Arousal, and Serotonin
Is SUDEP, in some or all cases, a critical failure of arousal mechanisms, and do seizures in the sleep state particularly compromise arousal? Most SUDEP deaths appear to have occurred during sleep (3, 12), and the majority of eyewitness accounts and monitored SUDEP case reports support this observation (3, 7, 8, 10, 12, 27, 28). Conversely, nocturnal supervision has been reported to decrease SUDEP risk (7, 28, 29), although one recent report suggests that supervision and even trained intervention immediately after a seizure may not make a difference in some individuals (29). A systematic analysis of a SUDEP cohort of 154 patients found that those with sleep-related SUDEP were more likely to have a history of nocturnal seizures than were controls, and the presence of nocturnal seizures appeared to be a significant risk factor (OR 2.6, 95% CI 1.3–5.0) (27). However, SUDEP also occurs in patients awake at the outset; whether these patients are biologically distinct is unknown.
Serotonin predominantly plays a part in wakefulness and in the inhibition of REM sleep but may have other complex roles in normal sleep physiology. Serotonin is also thought to enhance respiration in response to hypercarbia, which can occur during seizures in humans. Abnormalities in the medullary serotonergic system have been well described in the sudden infant death syndrome (SIDS), a phenomenon with which it is tempting to draw parallels to SUDEP (30). Increased polymorphism frequencies have been identified in 25 genes in SIDS infants compared to controls, 3 of which are serotonin genes (31). Tissue receptor autoradiographic studies of brainstem tissue in SIDS patients have shown arcuate nucleus (30, 32) and more widespread brainstem reduction in 5HT1A binding, increased 5HT cells and reduction in 5HT transporter binding (32, 33), suggesting abnormalities in a system prominently involved in cardiorespiratory homeostasis. Such studies have not been carried out in SUDEP brains.
Additional information on the serotonin theme comes from animal studies: mice with an audiogenic seizure disorder due to a genetic deletion of the 5HT2c receptor die in the postictal period if unresuscitated (34). DBA/2 strain mice, which suffer audiogenic seizures, have marked susceptibility to postictal respiratory arrest, which can be reduced when pretreated with selective serotonin reuptake inhibitors (SSRIs) (35). Prevention of seizure-induced sudden death in mice by semichronic administration of a selective serotonin reuptake inhibitor has been reported by the same group (36). When fluoxetine was administered, it significantly reduced incidences of respiratory arrest, appearing to support a subsequent report on SSRIs on respiration in patients with epilepsy (37).
Conclusion
In the end, as eloquently described in the triple-risk model of SIDS (30), it may be the sum of a number of unfortunate concurrences that comprise the perfect storm that ends in SUDEP. For example, a generalized tonic–clonic seizure in a mature brain, genetically or otherwise predisposed to neurotransmitter-driven dysregulation of postictal arousal mechanisms, with cardiorespiratory homeostasis compromised by the prone position, and a subsequent agonal cycle of cerebral and cardiorespiratory dysfunction that ends in death. Without the depth and sophistication of current SIDS research, all this remains speculative.
Footnotes
Editor's Note: Authors have a Conflict of Interest disclosure which is posted under the Supplemental Materials (208.1KB, docx) link.
References
- 1.Surges R, Sander JW. Sudden unexpected death in epilepsy: Mechanisms, prevalence, and prevention. Curr Opin Neurol. 2012;25:201–207. doi: 10.1097/WCO.0b013e3283506714. [DOI] [PubMed] [Google Scholar]
- 2.Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: Risk factors and potential pathomechanisms. Nat Rev Neurol. 2009;5:492–504. doi: 10.1038/nrneurol.2009.118. [DOI] [PubMed] [Google Scholar]
- 3.Nashef L, Garner S, Sander JW, Fish DR, Shorvon SD. Circumstances of death in sudden death in epilepsy: Interviews of bereaved relatives. J Neurol Neurosurg Psychiatry. 1998;64:349–352. doi: 10.1136/jnnp.64.3.349. PMCID: 2170013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nashef L, Ryvlin P. Sudden unexpected death in epilepsy (SUDEP): Update and reflections. Neurol Clin. 2009;27:1063–1074. doi: 10.1016/j.ncl.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: Current knowledge and future directions. Lancet Neurol. 2008;7:1021–1031. doi: 10.1016/S1474-4422(08)70202-3. [DOI] [PubMed] [Google Scholar]
- 6.Nilsson L, Farahmand BY, Persson PG, Thiblin I, Tomson T. Risk factors for sudden unexpected death in epilepsy: A case-control study. Lancet. 1999;353:888–893. doi: 10.1016/s0140-6736(98)05114-9. [DOI] [PubMed] [Google Scholar]
- 7.Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology. 2005;64:1131–1133. doi: 10.1212/01.WNL.0000156352.61328.CB. [DOI] [PubMed] [Google Scholar]
- 8.Bird JMD, Dembny KAT, Sandeman D, Butler S. Sudden unexplained death in epilepsy. Epilepsia. 1997;38(suppl):S52–S6. [Google Scholar]
- 9.Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68:787–796. doi: 10.1002/ana.22101. [DOI] [PubMed] [Google Scholar]
- 10.Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: Report on two deaths in video-EEG-monitored patients. Epilepsia. 2010;51:916–920. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- 11.Tao JX, Qian S, Baldwin M, Chen XJ, Rose S, Ebersole SH, Ebersole JS. SUDEP, suspected positional airway obstruction, and hypoventilation in postictal coma. Epilepsia. 2010;51:2344–2347. doi: 10.1111/j.1528-1167.2010.02719.x. [DOI] [PubMed] [Google Scholar]
- 12.Nobili L, Proserpio P, Rubboli G, Montano N, Didato G, Tassinari CA. Sudden unexpected death in epilepsy (SUDEP) and sleep. Sleep Med Rev. 2011;15:237–246. doi: 10.1016/j.smrv.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Gastaut HB. Epileptic Seizures: Clinical and Electrographic Features, Diagnosis and Treatment. Springfield, IL: Thomas; 1972. [Google Scholar]
- 14.So NK, Blume WT. The postictal EEG. Epilepsy Behav. 2010;19:121–126. doi: 10.1016/j.yebeh.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 15.Theodore WH. The postictal state: Effects of age and underlying brain dysfunction. Epilepsy Behav. 2010;19:118–120. doi: 10.1016/j.yebeh.2010.06.031. PMCID: 2952737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surges R, Strzelczyk A, Scott CA, Walker MC, Sander JW. Postictal generalized electroencephalographic suppression is associated with generalized seizures. Epilepsy Behav. 2011;21:271–274. doi: 10.1016/j.yebeh.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Kim AJ, Kuroda MM, Nordli DR., Jr. Abruptly attenuated terminal ictal pattern in pediatrics. J Clin Neurophysiol. 2006;23:532–550. doi: 10.1097/01.wnp.0000229045.28725.5c. [DOI] [PubMed] [Google Scholar]
- 18.So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: Evidence from near-SUDEP incident. Epilepsia. 2000;41:1494–1497. doi: 10.1111/j.1528-1157.2000.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 19.McLean BN, Wimalaratna S. Sudden death in epilepsy recorded in ambulatory EEG. J Neurol Neurosurg Psychiatry. 2007;78:1395–1397. doi: 10.1136/jnnp.2006.088492. PMCID: 2095624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poh MZ, Loddenkemper T, Reinsberger C, Swenson NC, Goyal S, Madsen JR, Picard RW. Autonomic changes with seizures correlate with postictal EEG suppression. Neurology. 2012;78:1868–1876. doi: 10.1212/WNL.0b013e318258f7f1. PMCID: 3369522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semmelroch M, Elwes RD, Lozsadi DA, Nashef L. Retrospective audit of postictal generalized EEG suppression in telemetry. Epilepsia. 2012;53:e21–24. doi: 10.1111/j.1528-1167.2011.03296.x. [DOI] [PubMed] [Google Scholar]
- 22.Seyal M, Hardin KA, Bateman LM. Postictal generalized EEG suppression is linked to seizure-associated respiratory dysfunction but not postictal apnea. Epilepsia. 2012;53:825–831. doi: 10.1111/j.1528-1167.2012.03443.x. [DOI] [PubMed] [Google Scholar]
- 23.Engel J. Basic mechanisms of human epilepsy. In: Engel J, editor. Epilepsy, a Comprehensive Textbook. Philadelphia: Lippincott, Williams and Wilkins; 2008. pp. 495–508. In. ed. [Google Scholar]
- 24.Lado FA, Moshe SL. How do seizures stop? Epilepsia. 2008;49:1651–1664. doi: 10.1111/j.1528-1167.2008.01669.x. PMCID: 2738747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaibara M, Blume WT. The postictal electroencephalogram. Electroencephalogr Clin Neurophysiol. 1988;70:99–104. doi: 10.1016/0013-4694(88)90109-5. [DOI] [PubMed] [Google Scholar]
- 26.Tilz C, Stefan H, Hopfengaertner R, Kerling F, Genow A, Wang-Tilz Y. Influence of levetiracetame on ictal and postictal EEG in patients with partial seizures. Eur J Neurol. 2006;13:1352–1358. doi: 10.1111/j.1468-1331.2006.01516.x. [DOI] [PubMed] [Google Scholar]
- 27.Lamberts RJ, Thijs RD, Laffan A, Langan Y, Sander JW. Sudden unexpected death in epilepsy: People with nocturnal seizures may be at highest risk. Epilepsia. 2012;53:253–257. doi: 10.1111/j.1528-1167.2011.03360.x. [DOI] [PubMed] [Google Scholar]
- 28.Nashef L, Fish DR, Garner S, Sander JW, Shorvon SD. Sudden death in epilepsy: A study of incidence in a young cohort with epilepsy and learning difficulty. Epilepsia. 1995;36:1187–1194. doi: 10.1111/j.1528-1157.1995.tb01061.x. [DOI] [PubMed] [Google Scholar]
- 29.Swinghamer J, Devinsky O, Friedman D. Can post-ictal intervention prevent sudden unexpected death in epilepsy? A report of two cases. Epilepsy Behav. 2012;24:377–379. doi: 10.1016/j.yebeh.2012.04.122. [DOI] [PubMed] [Google Scholar]
- 30.Kinney HC, Randall LL, Sleeper LA, Willinger M, Belliveau RA, Zec N, Rava LA, Dominici L, Iyasu S, Randall B, Habbe D, Wilson H, Mandell F, McClain M, Welty TK. Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol. 2003;62:1178–1191. doi: 10.1093/jnen/62.11.1178. [DOI] [PubMed] [Google Scholar]
- 31.Hunt CHF, Hauck FR. Gene and gene-environment risk factors in sudden unexpected death in infants. In: Mitchell EA, editor. Current Pediatric Reviews. Oak Park, IL: Bentham Science Publishers; 2010. pp. 56–62. In. ed. [Google Scholar]
- 32.Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, Foley E, White WF, Kinney HC. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol. 2000;59:377–384. doi: 10.1093/jnen/59.5.377. [DOI] [PubMed] [Google Scholar]
- 33.Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, Chadwick AE, Krous HF, Kinney HC. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124–2132. doi: 10.1001/jama.296.17.2124. [DOI] [PubMed] [Google Scholar]
- 34.Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- 35.Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–26. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 36.Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 37.Bateman LM, Li CS, Lin TC, Seyal M. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]