

Abstract
Over the past decade a lot of research has been performed towards the therapeutic use of mesenchymal stem cells (MSCs) in neurodegenerative and neuroinflammatory diseases. MSCs have shown to be beneficial in different preclinical studies of central nervous system (CNS) disorders due to their immunomodulatory properties and their capacity to secrete various growth factors. Nevertheless, most of the transplanted cells die within the first hours after transplantation and induce a neuroinflammatory response. In order to increase the efficacy of MSC transplantation, it is thus imperative to completely characterise the mechanisms mediating neuroinflammation and cell death following MSC transplantation into the CNS. Consequently, different components of these cell death- and neuroinflammation-inducing pathways can be targeted in an attempt to improve the therapeutic potential of MSCs for CNS disorders.
Introduction
Stem cell transplantation is hoped to become a promising therapy for regeneration of injured central nervous system (CNS) tissue as numerous preclinical studies have already demonstrated improved functional outcome following stem cell transplantation [1-4]. Several potential working mechanisms have been proposed to explain their clinical benefit [5]; these are based on (i) immunomodulation, (ii) stimulation of endogenous neural stem cells and/or endogenous regeneration-inducing mechanisms by (genetically modified) cellular grafts, or (iii) direct cell replacement. More recently, insights into neuroinflammatory processes induced by stem cell transplantation might further explain possible contributions of stem cell transplantation neuroprotection and/or neurorestoration.
Despite the observed beneficial effects of stem cell grafting into the CNS, which might be attributed to one or more of the above described mechanisms, little is known about the actual mechanism responsible for the beneficial effects observed in different CNS diseases (stroke, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, spinal cord and traumatic brain injuries, and multiple sclerosis). Functional outcome following cell grafting demonstrates very diverse functional and pathological results, which might be due to differences in disease model, cell source and dose, application route and time window [6-11]. Whereas in the past researchers looked mainly at the functional benefits following stem cell transplantation, attention is now being paid to the fate (based on cell labelling with particles and/or reporter genes) and physiology (based on differentiation capacity and secretion potential) of the transplanted cells in order to reach a better understanding of the underlying mechanism. Looking into the cell fate, the survival of transplanted cells was poorly investigated and found to be very low [12-16]. While intravenous injection is the most feasible administration route, stem cell survival is very poor following intravenous injection as the cells become entrapped in filter organs such as liver, spleen and lung [17], where they die via apoptosis (within hours to a few days) [18]. Highest cell survival has been observed following cell transplantation into the CNS [19,20], despite the latter being shown to induce neuroinflammation at the site of injection. The latter has mainly been characterised by the recruitment of microglia and astrocytes in both healthy [21] and diseased CNS [9,22]. Alternatively, other research groups reported a decreased activation of microglia and astrocytes at lesion sites [6,12], as well as the production of anti-inflammatory cytokines leading to disease improvement [23-25] following mesenchymal stem cell (MSC) transplantation into the CNS. Given the low cell survival after transplantation, it might be possible that the cells themselves are not the key players in regeneration, but rather cell death-induced responses and subsequent (immunological) responses following cell transplantation. Therefore, it is imperative to thoroughly characterise cell survival and neuroinflammation following MSC transplantation, in order to gain better insights into the physiological responses leading to disease improvement and to find specific targets for therapeutic intervention.
Besides their successful therapeutic application based on their intrinsic properties, MSCs also form an interesting cell source for the secretion of growth factors and cytokines, supporting CNS disease improvement [26]. Adopting this approach, the beneficial effect is induced by the secreted factors, which can support endogenous neurogenesis and/or neuroprotection, and its success is highly dependent on stem cell survival and their potential to secrete growth factors. Low cell survival, due to hypoxia and serum deprivation, has already been reported following stem cell transplantation in myocardial infarction [27], and these are most likely also the causal factors for the low cell survival observed after stem cell transplantation into the CNS. Therefore, the use of trophic factor-producing MSCs for CNS disease treatment might hold promise for developing strategies to improve stem cell survival after transplantation, in order to obtain highly viable, growth factor-producing stem cells at the site of injury. In addition to establishing better cell survival, reducing the neuroinflammation is also of interest, as MSCs become surrounded by an astrocytic scar [20], probably induced by the microglial neuroinflammatory response. Such glial scarring may prevent the secreted growth factors from reaching their target, thus possibly reducing the therapeutic benefits.
Improved functional outcome after MSC transplantation for CNS disorders is attributed to neuroprotection, immunomodulation, or improved endogenous neurogenesis induced by the immunomodulatory signalling cascade, or by growth factors secreted by the transplanted stem cells. However, the therapeutic application of MSCs in CNS disorders is challenged by low cell survival following transplantation, as well as by the presence of neuroinflammatory responses. Therefore, a comprehensive characterisation of both neuroinflammation and the mechanisms underlying low cell survival following MSC transplantation is absolutely crucial for identifying possible targets that can be modulated to improve the therapeutic potential of MSC application.
Neuroinflammation and neuroprotection
Neuroinflammation is a common characteristic of neurodegenerative and neuroinflammatory diseases, of which the presence of activated microglia is an important hallmark. It is imperative to recognise that microglia can be activated in different ways, resulting in distinct microglial functions during CNS disease. Below, we describe the general mechanisms behind the different types of microglia activation (classical versus alternative activation, and acquired deactivation), and discuss the neuroinflammation (as a type of microglia activation) observed after autologous transplantation of MSCs into the mouse brain.
Classical activation of microglia
In most CNS disorders, microglia become activated in a similar fashion to when they respond to the presence of pathogens (classical activation), where certain ligands characteristic for CNS disease (for example, amyloid beta plaques, α-synuclein) act as pathogen-associated molecular patterns and are recognised via pathogen recognition receptors (PRRs) [28]. Toll-like receptors (TLRs) 1 to 9 are well-known PRRs expressed on microglial membranes [29], and different compounds associated with cell death or CNS diseases are known to activate microglia via TLR signalling [30]. Activation of TLRs can have two downstream effects: it leads to either nuclear factor (NF)-κB signalling via myeloid differentiation primary response gene 88 (MyD88) recruitment, or interferon regulatory factor (IRF)3 triggering upon TRIF-related adapter molecule-dependent TIR-domain-containing adapter-inducing interferon-β (TRIF) signalling [31]. Activation of either NF-κB or IRF3 leads to the nuclear translocation of both transcription factors, an event that induces the transcription of inflammatory cytokines (IL6, IL12, IL18, TNFα, IL1β) [29] and type I interferon [32], elicits the production of reactive oxygen and nitrogen species [33], and stimulates phagocytosis [34]. The production of reactive oxygen and nitrogen species leads to neuronal death, thereby further augmenting the ongoing neuroinflammation [35]. In addition, ATP is released into the extracellular environment by dying neurons, activating microglia and astrocytes. ATP triggers the purinergic receptor P2X7, present on microglial membranes [36], resulting in the production of neurotoxic cytokines (IL1β and TNFα) upon NF-κB signalling, consequently sustaining neuronal death [37]. Classic activation of microglia has been widely studied in CNS disorders, and can easily be stimulated in vitro via activation of TLR4 upon lipopolysaccharide treatment [38] or via activation of the interferon γ receptor with interferon γ. In contrast to lipopolysaccharide, which activates NF-κB through TLR4, interferon γ triggers Janus kinase signal transducer and activator of transcription (Jak/STAT) signalling to induce the production of pro-inflammatory cytokines [39]. In general, this type of classical microglia activation is considered detrimental for the disease, although a few cases have been reported in which classically activated microglia can be supportive for remyelination [40] and microglial phagocytosis has a beneficial effect on Alzheimer’s disease [41].
Alternative activation of microglia
In addition to classical activation of microglia, which generally results in neuroinflammation, microglia can acquire a more neuroprotective, alternatively activated phenotype. Alternative activation can be obtained via cytokine (IL4 and IL13) signalling through their common IL4Rα receptor followed by Jak/STAT signalling and activation of the transcription factor STAT6 [42]. Subsequent nuclear translocation of STAT6 leads to the transcription of anti-inflammatory genes and the transcription factor peroxisome proliferator-activated receptor (PPAR)γ, which has an inhibitory effect on the expression of pro-inflammatory genes upon classical microglia activation [43]. Furthermore, the production of reactive nitrogen and oxygen species is reduced, whereas the release of anti-inflammatory cytokines (IL10, transforming growth factor (TGF)β) is promoted, resulting in neuroprotection. Although the spontaneous alternative activation (M2) of microglia has not been well-described in vivo for CNS disorders and is largely overruled by a neurotoxic M1 response [44], it is believed that the presence of M2 microglia is supportive in CNS disease and is able to suppress the neurotoxic effects of classically activated microglia.
Acquired deactivation
Following classical and alternative activation of microglia, the quiescent phenotype of microglia can be restored through a process of microglial deactivation [45,46]. This acquired microglia deactivation has been studied thoroughly in tumour models in which the tumours are a source of anti-inflammatory cytokines (IL4, IL6, IL10 and TGFβ) [47] that induce microglia deactivation through signalling via their receptors (IL4R, IL6R, IL10R and TGFβR), leading to the suppression of immune responses against the tumour [48]. IL4R ligand binding triggers the Jak/STAT signalling cascade and subsequent transcription of PPARγ, as discussed previously (section on alternative microglia activation). In addition, IL10 and TGFβ, cytokines produced upon alternative activation of microglia, give rise to a quiescent microglia phenotype through signalling via their respective receptors, IL10R and TGFβR [49]. IL10, TGFβ and corticosteroids are also upregulated upon phagocytosis of apoptotic cells [50]. Therefore, both classical and alternative activation of microglia, each characterised by distinct cell functions and cytokine production, might induce microglia deactivation [49] and subsequently a reduction of neuroinflammation.
Neuroinflammation following mesenchymal stem cell transplantation
Although MSC transplantation into the CNS has been ascribed beneficial effects in numerous preclinical studies of neurodegenerative and neuroinflammatory disorders [1], a minority of studies report stem cell survival and its influence on neighbouring tissue upon transplantation into the brain. However, when a follow-up of stem cell behaviour after transplantation was performed, the survival of MSCs was found to be very low (between 0 and 30%) and was highly dependent on the injection route/site (intravenous, intracerebral or intrathecal) and cell source (auto-, allo- or xenogeneic) [2-6]. Our research group previously demonstrated the ability of autologous MSCs to survive after transplantation into the CNS [20,51], whereas no cell survival was observed after transplantation into the muscle [51] or following intravenous administration [18]. Nevertheless, cell survival was only observed for autologous, but not for allogeneic or xenogeneic stem cell grafts, which are rejected via a T cell-independent mechanism [33]. By accurately quantifying the survival of autologous CNS-implanted MSCs, we demonstrated that only a fraction of the initially implanted cells are able to survive in both healthy [21] and diseased [52] CNS. In addition to the low cell survival after transplantation, an increased neuroinflammatory response, characterised by microgliosis and astrogliosis, was observed after MSC transplantation in healthy CNS [21,53] (Figure 1i). We noted that microglia have the capacity to recognise the presence of autologous MSCs and become activated in a classical way, demonstrated by the expression of inducible nitrogen oxide synthase (Figure 1ii). Although MSCs have been shown to have neuroprotective features when administered in models of CNS disease [6,12,54-57], they are able to induce pro-inflammatory responses at the site of injection in healthy [21] or diseased animals [52]. Consequently, this inflammatory cascade induced by stem cell application needs to be considered and possibly even modified in the event of stem cell transplantation, in order to prevent possible neurotoxic effects and optimise the therapeutic potential of stem cells.
Figure 1.
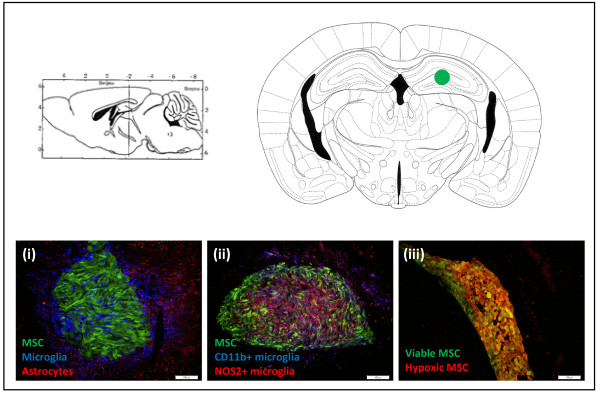
Characterisation of responses following mesenchymal stem cell transplantation. The upper panel of Figure 1 demonstrates a transveral and coronal brain slice at the site where mesenchymal stem cells (MSCs) were transplanted (green dot). The bottom row of Figure 1 demonstrates immunofluorescent pictures of cryosections from FVB mice transplanted with 2 × 105 enhanced green fluorescent protein (eGFP)- and luciferase-expressing MSCs into the striatum. (i) Transplanted MSCs are recognised by the brain’s immune system, demonstrating Iba1+ microglia (blue) invading and activated GFAP + astrocytes (red) surrounding the eGFP-expressing MSC implant (green) at week 1 post-implantation. (ii) A proportion of the Iba1+ microglia are classically activated and demonstrate CD11b (blue) and NOS2 (red) expression, corresponding to a neuroinflammatory microglial phenotype. (iii) At 6 h post-transplantation a lot of hypoxic (red) cells are found within the eGFP-positive (green) MSC transplant, leading to death of about half of the transplanted cells. The representative pictures were taken at a magnification of 20 × .
Below, we describe different strategies to modify neuroinflammation, which may be adopted to eliminate undesirable immune responses induced by stem cell applications.
Strategies for the modulation of neuroinflammation
In general, brain injury results in microglia activation (either classical or alternative), which can be beneficial or detrimental for different types of CNS diseases [58]. Directed modification of the microglial activation status could positively influence the disease course, thereby offering great potential as a therapeutic strategy. Moreover, this approach also holds promise for altering the immunological responses observed after stem cell transplantations, with the aim of decreasing neurodestructive immune responses at the site of injection. In order to use the adequate approach (stimulation or inhibition of neuroinflammation and/or neuroprotection), it is imperative to start by defining the activation status of microglia and their function in CNS disease or following stem cell application. Exact specification of the type of microglia activation may prove challenging, as it is highly dependent on the type of stimulated PRR, the location of microglia in the CNS and the time point during the disease course. Applying this concept to an example such as stroke, cytotoxic microglia are observed in the striatum, whereas subventricular zone microglia promote neurogenesis [59]. Furthermore, mouse models for amyotrophic lateral sclerosis exhibit neuroprotective microglia at disease onset, which are gradually transformed into cells with a more neurotoxic phenotype at the end of the disease [60]. Therefore, therapeutic intervention at the right place and time is vital to achieve success. Strategies can either focus on the inhibition of microglia activation by maintaining tissue homeostasis, or the modification of the activation status (classical or alternative activation or acquired deactivation) of microglia in order to attain neuroprotection. Based on the complex microglial activation signalling, modulation of neuroinflammation can be exerted at different levels of the signalling pathway: PPARs, TLRs, NF-κB signalling, Jak/STAT signalling, ATP signalling via purinoreceptors. Within the scope of this review we will, however, only focus on neuro-immunomodulating targets that hold great potential to be influenced via the application of (un)modified MSCs.
Mesenchymal stem cells for the inhibition of microglia activation
MSCs are generally believed to induce immunosuppression via the production of different immunomodulatory factors (for example, nitric oxide, indoleamine 2,3-dioxygenase, TGFβ) that can inhibit T-cell proliferation, induce tolerogenic dendritic cells or prevent/polarise T-cell activation [61]. Although the immunomodulatory potential of MSCs and its relation to cells from the peripheral immune system has well been described, so far little is known about the role of MSCs in neuroinflammation. Recently, it became clear that MSCs also have the potential to influence neuroinflammation, both on the level of inhibiting microglia activation as well as modulation of microglia activation. Within the scope of this review we will only describe the effect of cytokines produced by MSCs that have an effect on neuroinflammation mediated by microglia.
Under steady state conditions, microglia display a ramified morphology and exert surveillance of the brain in order to accurately respond to danger signals [62]. Different mechanisms involving microglia-neuron signalling are able to maintain these steady state conditions, while the disruption of this cross-talk results in microglia activation [49].
CX3CR1-CX3CL1
Fractalkine (CX3CL1) is known to be an important compound in microglia-neuron interaction. CX3CL1 exists both as a membrane-bound and as a secreted protein [63]. The secreted form functions as a chemoattractant for inflammatory cells while the membrane-bound form induces the adhesion of cells expressing the fractalkine receptor (CX3CR1) [64]. The binding of CX3CL1, expressed on neurons, to its receptor suppresses microglia activation [49]. Following CNS injury or disease, CX3CR1-CX3CL1 homeostasis is frequently disturbed, leading to either increased neuroprotection or disease worsening.
While neurons secrete higher levels of CX3CL1 upon CNS injury [65], leading to neuroprotection via a downregulation of pro-inflammatory cytokine production (nitric oxide, IL6, TNFα) by activated microglia [66], decreased neuroinflammation has been described for Alzheimer’s disease [67,68] and stroke [69] in CX3CR1−/− mice. An explanation for the latter can be found in the chemotactic properties of fractalkine resulting in the absence of migration of activated microglia towards CNS lesions and subsequent neuroprotection upon CX3CR1 silencing. Alternatively, deletion of CX3CR1 leads to enhanced phagocytosis of amyloid beta plaques by microglia in Alzheimer’s disease, requiring the microglia to be present at the lesion sites [70].
On the other hand, the complete depletion of CX3CR1 has been demonstrated to worsen Parkinson’s disease, amyotrophic lateral sclerosis [71], experimental autoimmune encephalomyelitis (EAE) [72] and Alzheimer’s disease [73]. In addition, CX3CR1 deficiency leads to severe cognitive deficits in healthy animals [74]. As demonstrated above, the chemotactic properties of fractalkine might play an important role in microglia recruitment towards CNS injured sites. Complete elimination of activated microglia may prove to be detrimental as for some CNS diseases the presence of (neuroprotective) microglia is absolutely necessary in order to obtain a clinical benefit [58].
Since the above-mentioned studies show contradictory results about CX3CR1 signalling in Alzheimer’s disease, and describe a dual role for CX3CR1-CX3CL1 signalling in neuroinflammation, attention needs to be paid while modifying CX3CR1 or CX3CL1 expression in order to improve disease outcome. Whereas increased recruitment of activated microglia towards a site of CX3CL1 expression can be desirable in some situations (by the intracerebral injection of fractalkine in rodent brain [75]), a complete lack of CX3CL1 signalling may be preferred in other instances (using a CX3CR1 antagonist, which inhibits the CX3CL1-induced chemotaxis [76]).
For future applications, MSCs hold great potential as a strategy to modulate neuroinflammation via CX3CR1-CX3CL1 signalling, as in vitro experiments demonstrated that they are able to produce CX3CL1, thereby directing microglia towards a more neuroprotective phenotype [56,77]. Neuroprotection induced by CX3CL1 production by MSCs was mediated by the downregulation of neuroinflammatory cytokine production (IL1β and TNFα) and upregulation of TREM2, CX3CR1 and CD200R expression on microglia. In addition, in vivo MSC transplantation has been shown to reduce stroke-related neuroinflammation via the downregulation of the expression of the pro-inflammatory inducible nitrogen oxide synthase gene and decreased accumulation of microglia and astrocytes at the site of ischaemia, possibly induced via the production of fractalkine [56].
In conclusion, for CX3CR1 to be used as a target to influence neuroinflammation, the approach will need to be adapted to each type of CNS disease. In this regard, it is very important to determine if the presence of deactivated/alternatively activated microglia or complete absence of microglia will be supportive for obtaining neuroprotection. Therefore, treatment strategies need to focus on microglia deactivation, in which case the re-establishment of CX3CR1-CX3CL1 homeostasis is pursued, or on microglia elimination, by counteracting the chemotactic properties of CX3CL1 [76].
CD200R-CD200
Microglia activation can also be suppressed upon interaction of various other microglia receptors (CD172, CD200 receptor (CD200R) and CD45) with markers expressed on neurons (CD47, CD200 and CD22) [78]. So far, a substantial amount of research has been performed towards elucidating the role of CD200-CD200R signalling in neuroinflammation, which generally results in increased microglia activation and neuroinflammation upon decreased CD200-CD200R interaction.
Increased numbers of hyperactive microglia were observed in CD200−/− mice [79] and in aged animals exhibiting a lower CD200 expression [80]. Moreover, CD200 deficiency leads to an earlier EAE onset [79] but can nevertheless be helpful in the clearance of certain parasites via increased inflammation [81]. In contrast, upregulation of CD200 expression on neurons in wld mutant mice results in a decreased susceptibility to EAE [82], and CD200 upregulation upon kainic acid-induced neurodegeneration might play a role in alternative microglia activation [83].
In general, physiological CD200-CD200R signalling should be either maintained or stimulated, via the increased expression of CD200 and CD200R, to promote neuroprotection. For example, IL4 treatment promotes CD200 expression on microglia, suppressing their activation [80,84,85], thereby creating novel possibilities for neuroprotection in Alzheimer’s disease [86]. Stimulation of CD200-CD200R signalling via an agonistic anti-CD200R antibody can be beneficial in different autoimmune diseases, particularly for the resolution of experimental autoimmune uveoretinitis [87]. Additionally, microglia on their own are able to express CD200 upon excitotoxic lesioning, which was not observed in healthy conditions. Therefore, microglia themselves are able to contribute to neuroprotection and tissue homeostasis through the expression of both CD200 and CD200R [85]. Alternatively, increased numbers of activated microglia as well as an upregulation of the production of pro-inflammatory cytokines was observed upon blocking of CD200-CD200R signalling with a CD200R blocking antibody [88].
In vitro studies have demonstrated that MSCs are able to express CD200, and consequently can induce a CD200 expression level-dependent reduction in TNFα production by macrophages, resulting in increased neuroprotection [89]. As TNFα is a well-known cytokine produced by classically activated macrophages and microglia that contributes to neuroinflammation, downregulation of its production is a highly recommended strategy to modulate neuroinflammation. Despite the evidence of CD200 as a promising target molecule in immune-modulating strategies, to date there are no reports on the modulation of CD200-CD200R signalling upon MSC transplantation into the CNS.
Mesenchymal stem cells for the modification of microglia activation
While the above-described approaches aim at preventing microglia activation, neuroprotection can also be stimulated via the reorientation of the microglia neurotoxic activation state (classical activation) towards a more neuroprotective activation state (alternative activation). Despite the fact that neuroinflammation occurs following MSC transplantation in healthy [21] and diseased [52] brain, many researchers ascribe neuroprotective effects to MSCs [90]. In vitro, MSCs have been shown to reduce the TNFα and nitric oxide production of lipopolysaccharide-stimulated microglia [91,92] and to suppress the production of pro-inflammatory cytokines in co-culture with astrocytes [93]. Therefore, the application of MSCs on their own might hold great potential as a strategy to modify neuroinflammation in vivo. Following this approach, neuroprotection can be induced via alternative activation of microglia following MSC transplantation in spinal cord injury [25], multiple sclerosis and Alzheimer’s disease [23,24,94]. In addition, MSCs can be used as carriers to secrete IL4, changing the microglial activation state towards a more neuroprotective phenotype in EAE [95]. The latter approach might also be an ideal immunomodulating strategy to treat other CNS disorders.
Mesenchymal stem cell applications for trophic factor delivery
Previously, the CNS was believed to be immune privileged due to the presence of the blood brain barrier and the absence of a lymphatic system [96,97], suggesting the possible long-term survival of CNS-transplanted stem cells. However, it recently became widely accepted that specific immune responses, orchestrated by microglia and astrocytes, are able to fight foreign tissue (such as stem cells) in the brain [98,99]. Consequently, research reoriented towards the follow-up of stem cell survival and fate after transplantation into the CNS, including stem cell labelling with particles or dyes and/or reporter genes. Despite the fact that reporter gene labelling enables us to follow up the cell fate very accurately, only a minor number of studies included the determination of cell survival in a quantitative manner [12-16], demonstrating low cell survival (0 to 30%) upon transplantation. Although the invasion of the brain’s innate immune cells, starting on day 3 following autologous transplantation of MSCs into the CNS, represents a possible trigger for cell death [21], most of the transplanted cells die during the first few hours after transplantation, at a time when immune cells are not yet involved. As the transplanted MSCs become hypoxic within the first 24 hours following transplantation (Figure 1iii), it is most likely that hypoxia and serum deprivation are the major contributors to early cell death following transplantation. Although low cell survival due to hypoxia and serum deprivation has already been widely investigated for myocardial infarction [100], this observed low cell survival following autologous MSC transplantation into the CNS has not yet been thoroughly explored. In culture conditions, cells are provided with oxygen and nutrients in order to allow them to grow and proliferate. However, when cultured stem cells are used for transplantation experiments, they are removed from their oxygen- and nutrient-rich environment and placed in an injured (hypoxic) tissue environment without adequate oxygen and nutrient supply, leading to cell death via apoptosis. The success of stem cells as carriers for growth factors in therapeutic approaches for CNS diseases relies on their ability to survive and engraft in the surrounding tissue, in order to sustain growth factor production. The production of brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), vascular endothelial growth factor (VEGF) and/or neurotrophin 3 (NT3) can be very supportive in stroke [101-103], Huntington’s disease [104], Parkinson’s disease [26,105,106], spinal cord injury [107,108], and traumatic brain injury [109,110]. Therefore, it is important to obtain cells with high survivability following transplantation in order to obtain high expression levels of the supportive growth factors in vivo. Further research on the roles of hypoxia and serum deprivation in cell death after transplantation into the CNS will be necessary in order to develop adequate strategies to circumvent the problem of low cell survival. Below, we give an overview of strategies that can be adopted to face these problems of hypoxia and serum deprivation, thereby increasing the therapeutic potential of stem cell applications.
Strategies for the improvement of cell survival
How to deal with hypoxia?
Hypoxic preconditioning can be applied to MSCs in culture prior to transplantation, in order to adapt the cells to the environmental conditions they will need to handle after transplantation. Cell culture in hypoxic conditions (1 to 3% oxygen) will increase their expression of pro-survival and pro-angiogenic genes [111], which can be beneficial for the survival of the cells as they are better adapted to reside in a more hypoxic environment, for example, brain tissue. The upregulation of hypoxia-inducible factor (HIF)-1α is a general consequence of hypoxic culture conditions and leads to enhanced expression of stromal-derived factor (SDF)-1α [112] and CXCR4 [113]. This attributes a special role to the SDF-1α/CXCR4 axis in cell survival [114,115] via the inhibition of caspase 3 activity through increased Akt phosphorylation (Table 1). Therefore, stimulation of the SDF-1α/CXCR4 signalling axis via hypoxic preconditioning or genetic modification of MSCs in order to induce secretion of SDF-1α/CXCR4 has been shown to enhance MSC survival [116,117]. So far, it has already been described that transplanted MSCs are able to survive better upon hypoxic preconditioning in mouse models of myocardial infarction [118,119], stroke [120,121] or renal ischaemia [116]. In addition to HIF-1α, other pro-angiogenic factors, such as erythropoietin and VEGF, also become upregulated upon hypoxic cell preconditioning. Consequently, additional administration of one of these factors can be applied as an alternative strategy to expensive hypoxic preconditioning. While erythropoietin already demonstrated a beneficial effect on the survival of different kinds of progenitor cells after transplantation [122-124], HIF-1α-transduced MSCs significantly improved myocardial infarction [125] and VEGF-overexpressing neural stem cells demonstrated higher cell survival in an injured spinal cord [126]. Attention needs to be paid to additional genetic or phenotypic alterations that might be introduced accidentally upon modification of MSCs in order to produce hypoxia-resistant cells. Therefore, research efforts should be focused on the development of oxygen-generating biomaterials [127] or oxygen-permeable chips [128] that enable an increased oxygen supply to the graft without further adaptations to the graft.
Table 1.
Overview of different approaches for mesenchymal stem cell modulation in order to increase cell survival
| General effect | Strategy | Reference |
|---|---|---|
| Upregulation of different pro-survival and pro-angiogenic genes (HIF1α, SDF1α/CXCR4, EPO, VEGF, BDNF, GDNF) |
Hypoxic preconditioning |
[112,113] |
| Addition of factors to the culture medium |
[122,124,133] |
|
| Genetic modification to induce gene overexpression |
[125,126,129-132] |
|
| Physical protection of MSCs against hypoxia |
Oxygen supply via oxygen generating scaffolds and biomaterials |
[127,128] |
| Stimulation of PI3K/Akt pathway to prevent apoptosis |
Treatment with chemokines (for example, SDF1α) |
[138] |
| Knockout of TLR4 |
[137] |
|
| Overexpression of genes involved in apoptosis |
[139] |
|
| Downregulation of caspase 3 activity to prevent apoptosis |
Treatment with compounds (carvedilol, salvianolic acid) that block the activity |
[140,141] |
| Decreased apoptosis | Down- or upregulation of microRNA | [142] |
BDNF, brain-derived neurotrophic factor; EPO, erythropoietin; GDNF, glial cell line-derived neurotrophic factor; HIF, hypoxia-inducible factor; MSC, mesenchymal stem cell; PI3K, phosphoinositide 3-kinase; SDF, stromal-derived factor; TLR, Toll-like receptor; VEGF, vascular endothelial growth factor.
How to deal with serum deprivation?
In vitro, the survival of certain cell types (for example, neural stem cells) is highly dependent on the availability of growth factors in the culture medium. Upon transplantation, however, these cells are deprived of these compounds necessary for survival and undergo cell death. Genetic modification of MSCs [129-132] or cell treatment with minocycline [133] in order to overexpress some of these essential growth factors (GDNF, BDNF, VEGF, survivin; Table 1) have already been applied in cell transplantation experiments for CNS disorders and has been shown to induce functional improvement of the disease. However, only few studies [134,135] reported increased survival of the transplanted cells upon growth factor treatment. Therefore, further research is imperative in order to understand how serum deprivation can be tackled with the aim of favouring cell survival.
The general consequence of hypoxia and serum deprivation is cell death mediated via apoptosis. Therefore, aside from manipulating the oxygen and serum supplies themselves, it is also possible to interfere with apoptosis via the stimulation of the phosphoinositide 3-kinase (PI3K)/Akt pathway. In this context, it is known that TLR4 contributes to hypoxia-mediated apoptosis [136], and that TLR4 knockout in MSCs leads to improved survival after hypoxic treatment [137]. Additionally, inhibition of hypoxia- and serum deprivation-induced apoptosis via activation of PI3K/Akt can also be obtained via cell treatment with chemokines [138] or by means of overexpressing (or downregulating) genes regulating the apoptotic process [139] (Table 1). Inhibition of caspase 3 activity via cell treatment with carvedilol [140] or salvianolic acid [141] leads to decreased apoptosis and subsequent improved cell survival (Table 1). Although the underlying mechanism is still unknown, an important role has been ascribed to the up- or downregulation of microRNAs in preventing apoptosis [142] (Table 1).
To summarise, many targets can be used in order to make MSCs more tolerant to hypoxic conditions, including those that (i) have a direct effect on the PI3K/Akt signalling pathway, (ii) modulate caspase 3 activity, (iii) enhance cell survival via different growth promoting factors (indirect and direct cell treatment or genetic modification) or (iv) act via the modification of microRNA expression.
Although some of the above-described strategies have the potential to ameliorate cell survival upon transplantation, research on cell survival after transplantation into the CNS and subsequent modification strategies is still in its infancy. A more profound characterisation of the cell death-inducing mechanisms will help us to understand how to appropriately prevent cell death following transplantation and obtain efficient carriers to deliver supportive growth factors.
Drawbacks of mesenchymal stem cell modification to improve their potential to treat central nervous system disorders
While many of the above described strategies hold promise to ameliorate the capacities of MSCs to prevent neuroinflammation or hypoxia-mediated cell death upon grafting in the CNS, we need to take into account that several of these approaches require genetic modification of MSCs. One of the major concerns is that these genetic alterations might alter general properties of the MSCs, possibly leading to tumour formation following transplantation [143]. On the other hand, we also should assess the impact of different in vitro manipulations of stem cells, in order to increase cytokine and growth factor (BDNF, VEGF) secretion, on the direct surrounding of the transplanted stem cells in vivo. The secreted growth factors might on their own lead to aberrant tissue formation, possibly leading to tumour formation arising from endogenous cells. Profound long-term preclinical evaluation after transplantation of modified MSCs is therefore absolutely necessary in order to claim a clinical cell product is safe.
Conclusion
Over the past decade there has been a growing interest in the use of MSCs to treat different neurodegenerative and neuroinflammatory diseases. Although various research groups ascribe neuroprotective and immunomodulatory properties to MSCs, their survival upon transplantation remains very poor. Moreover, conflicting data regarding their neuroprotective effect in CNS disorders have emerged in which MSCs have been described to induce a pro-inflammatory immune response, characterised by the presence of activated microglia and astrocytes, at implantation sites.
In order to optimise the therapeutic potential of MSC applications for neurodegenerative and inflammatory diseases, it thus becomes very important to completely characterise (and possibly modulate) neuroinflammation in CNS diseases as well as following MSC transplantation, and improve cell survival upon transplantation. As it is known that microglia are the major key players in neuroinflammation in many CNS disorders as well as following MSC transplantation, even without the contribution of T cells and other peripheral immune cells, new insights into microglia activation and the underlying mechanisms of neuroinflammation will be helpful in understanding the role of the brain’s innate immune system in CNS disease. Consequently, modulation of neuroinflammation can be employed, whether or not through the use of MSC transplantation, in order to ameliorate different neuroinflammatory diseases or to reduce the neurotoxic effect induced upon MSC transplantation itself. Finally, research on hypoxia and serum deprivation following stem cell transplantation into the CNS will broaden our knowledge on efficient approaches for increasing cell survival and optimising therapeutic stem cell-based implementations.
While applying different approaches in order to ameliorate stem cell-based beneficial effects on CNS disorders, we need to take into account the differences between animal models and human diseases, as well as potential drawbacks of genetic modification of transplanted stem cells before application in clinical trials.
Abbreviations
BDNF: Brain-derived neurotrophic factor; CD-R: Cluster of differentiation receptor; CNS: Central nervous system; CX3CL1: CX3C chemokine ligand 1 (fractalkine); CX3CR1: CX3C chemokine receptor 1 (fractalkine receptor); CXCR4: C-X-C chemokine receptor type 4; EAE: Experimental autoimmune encephalomyelitis; GDNF: Glial cell line-derived neurotrophic factor; HIF: Hypoxia-inducible factor; IL: Interleukin; IL-R: Interleukin receptor; IRF: Interferon regulatory factor; Jak/STAT: Janus kinase signal transducer and activator of transcription; MSC: Mesenchymal stem cell; NF: Nuclear factor; PI3K: Phosphoinositide 3-kinase; PPAR: Peroxisome proliferator-activated receptor; PRR: Pathogen recognition receptor; SDF: Stromal-derived factor; TGF: Transforming growth factor; TGFβR: Transforming growth factor β receptor; TLR: Toll-like receptor; TNF: Tumour necrosis factor; VEGF: Vascular endothelial growth factor.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Nathalie De Vocht, Email: nathalie.devocht@ua.ac.be.
Jelle Praet, Email: jelle.praet@ua.ac.be.
Kristien Reekmans, Email: kristien.reekmans@ua.ac.be.
Debbie Le Blon, Email: debbie.leblon@ua.ac.be.
Chloé Hoornaert, Email: chloe.hoornaert@ua.ac.be.
Jasmijn Daans, Email: jasmijn.daans@ua.ac.be.
Zwi Berneman, Email: zwi.berneman@ua.ac.be.
Annemie Van der Linden, Email: annemie.vanderlinden@ua.ac.be.
Peter Ponsaerts, Email: Peter.Ponsaerts@ua.ac.be.
Acknowledgements
This work was supported by research grants G.0136.11 and G.0130.11 (granted to ZB, AvDL and PP) of the Fund for Scientific Research-Flanders (FWO-Vlaanderen, Belgium), in part by a Methusalem research grant from the Flemish government (granted to ZB) and in part by funding received from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement number 278850 (INMiND) (granted to AvDL). Nathalie De Vocht holds a PhD-fellowship from the FWO-Vlaanderen. Debbie Le Blon holds a PhD-fellowship from the Flemish Institute for Science and Technology (IWT-Vlaanderen). All authors approved the final version of the manuscript.
References
- Huang B, Tabata Y, Gao JQ. Mesenchymal stem cells as therapeutic agents and potential targeted gene delivery vehicle for brain diseases. J Control Release. 2012;162:464–473. doi: 10.1016/j.jconrel.2012.07.034. [DOI] [PubMed] [Google Scholar]
- Joyce N, Annett G, Wirthlin L, Olson S, Bauer G, Nolta JA. Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen Med. 2010;5:933–946. doi: 10.2217/rme.10.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlacchio A, Bernardi G, Martino S. Stem cells: an overview of the current status of therapies for central and peripheral nervous system diseases. Curr Med Chem. 2010;17:595–608. doi: 10.2174/092986710790416272. [DOI] [PubMed] [Google Scholar]
- Uccelli A, Benvenuto F, Laroni A, Giunti D. Neuroprotective features of mesenchymal stem cells. Best Pract Res Clin Haematol. 2011;24:59–64. doi: 10.1016/j.beha.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Kan I, Melamed E, Offen D. Autotransplantation of bone marrow-derived stem cells as a therapy for neurodegenerative diseases. Handb Exp Pharmacol. 2007;180:219–242. doi: 10.1007/978-3-540-68976-8_10. [DOI] [PubMed] [Google Scholar]
- Cristofanilli M, Harris VK, Zigelbaum A, Goossens AM, Lu A, Rosenthal H, Sadiq SA. Mesenchymal stem cells enhance the engraftment and myelinating ability of allogeneic oligodendrocyte progenitors in dysmyelinated mice. Stem Cells Dev. 2011;20:2065–2076. doi: 10.1089/scd.2010.0547. [DOI] [PubMed] [Google Scholar]
- Fisher-Shoval Y, Barhum Y, Sadan O, Yust-Katz S, Ben-Zur T, Lev N, Benkler C, Hod M, Melamed E, Offen D. Transplantation of placenta-derived mesenchymal stem cells in the EAE mouse model of MS. J Mol Neurosci. 2012;48:176–184. doi: 10.1007/s12031-012-9805-6. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lv H, Huang S, Tan H, Zhang Y, Li H. Bone marrow mesenchymal stem cells can improve the motor function of a Huntington’s disease rat model. Neurol Res. 2011;33:331–337. doi: 10.1179/016164110X12816242542571. [DOI] [PubMed] [Google Scholar]
- Rossignol J, Boyer C, Leveque X, Fink KD, Thinard R, Blanchard F, Dunbar GL, Lescaudron L. Mesenchymal stem cell transplantation and DMEM administration in a 3NP rat model of Huntington’s disease: morphological and behavioral outcomes. Behav Brain Res. 2011;217:369–378. doi: 10.1016/j.bbr.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Steiner B, Roch M, Holtkamp N, Kurtz A. Systemically administered human bone marrow-derived mesenchymal stem home into peripheral organs but do not induce neuroprotective effects in the MCAo-mouse model for cerebral ischemia. Neurosci Lett. 2012;513:25–30. doi: 10.1016/j.neulet.2012.01.078. [DOI] [PubMed] [Google Scholar]
- Xia G, Hong X, Chen X, Lan F, Zhang G, Liao L. Intracerebral transplantation of mesenchymal stem cells derived from human umbilical cord blood alleviates hypoxic ischemic brain injury in rat neonates. J Perinat Med. 2010;38:215–221. doi: 10.1515/jpm.2010.021. [DOI] [PubMed] [Google Scholar]
- Park HJ, Bang G, Lee BR, Kim HO, Lee PH. Neuroprotective effect of human mesenchymal stem cells in an animal model of double toxin-induced multiple system atrophy parkinsonism. Cell Transplant. 2011;20:827–835. doi: 10.3727/096368910X540630. [DOI] [PubMed] [Google Scholar]
- Sadan O, Shemesh N, Barzilay R, Dadon-Nahum M, Blumenfeld-Katzir T, Assaf Y, Yeshurun M, Djaldetti R, Cohen Y, Melamed E, Offen D. Mesenchymal stem cells induced to secrete neurotrophic factors attenuate quinolinic acid toxicity: a potential therapy for Huntington’s disease. Exp Neurol. 2012;234:417–427. doi: 10.1016/j.expneurol.2011.12.045. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Honmou O, Radtke C, Kocsis JD. Development of a middle cerebral artery occlusion model in the nonhuman primate and a safety study of i.v. infusion of human mesenchymal stem cells. PLoS One. 2011;6:e26577. doi: 10.1371/journal.pone.0026577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang AJ, Hong SQ, Xu Q, Wang HY, Yang Y, Wang ZF, Xu BN, Jiang XD, Xu RX. NT-3-secreting human umbilical cord mesenchymal stromal cell transplantation for the treatment of acute spinal cord injury in rats. Brain Res. 2011;1391:102–113. doi: 10.1016/j.brainres.2011.03.019. [DOI] [PubMed] [Google Scholar]
- Walker PA, Harting MT, Jimenez F, Shah SK, Pati S, Dash PK, Cox CS Jr. Direct intrathecal implantation of mesenchymal stromal cells leads to enhanced neuroprotection via an NFkappaB-mediated increase in interleukin-6 production. Stem Cells Dev. 2010;19:867–876. doi: 10.1089/scd.2009.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detante O, Moisan A, Dimastromatteo J, Richard MJ, Riou L, Grillon E, Barbier E, Desruet MD, De Fraipont F, Segebarth C, Jaillard A, Hommel M, Ghezzi C, Remy C. Intravenous administration of 99mTc-HMPAO-labeled human mesenchymal stem cells after stroke: in vivo imaging and biodistribution. Cell Transplant. 2009;18:1369–1379. doi: 10.3727/096368909X474230. [DOI] [PubMed] [Google Scholar]
- Reekmans KP, Praet J, De Vocht N, Tambuyzer BR, Bergwerf I, Daans J, Baekelandt V, Vanhoutte G, Goossens H, Jorens PG, Ysebaert DK, Chatterjee S, Pauwels P, Van Marck E, Berneman ZN, Van der Linden A, Ponsaerts P. Clinical potential of intravenous neural stem cell delivery for treatment of neuroinflammatory disease in mice? Cell Transplant. 2011;20:851–869. doi: 10.3727/096368910X543411. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Evans A, Yu G, Hess DC. Limitations of intravenous human bone marrow CD133+ cell grafts in stroke rats. Brain Res. 2005;1048:116–122. doi: 10.1016/j.brainres.2005.04.087. [DOI] [PubMed] [Google Scholar]
- De Vocht N, Bergwerf I, Vanhoutte G, Daans J, De Visscher G, Chatterjee S, Pauwels P, Berneman Z, Ponsaerts P, Van der Linden A. Labeling of Luciferase/eGFP-expressing bone marrow-derived stromal cells with fluorescent micron-sized iron oxide particles improves quantitative and qualitative multimodal imaging of cellular grafts in vivo. Mol Imaging Biol. 2011;13:1133–1145. doi: 10.1007/s11307-011-0469-3. [DOI] [PubMed] [Google Scholar]
- De Vocht N, Lin D, Praet J, Hoornaert C, Reekmans K, Le Blon D, Daans J, Pauwels P, Goossens H, Hens N, Berneman Z, Van der Linden A, Ponsaerts P. Quantitative and phenotypic analysis of mesenchymal stromal cell graft survival and recognition by microglia and astrocytes in mouse brain. Immunobiology. 2012;218:696–705. doi: 10.1016/j.imbio.2012.08.266. [DOI] [PubMed] [Google Scholar]
- Khoo ML, Tao H, Meedeniya AC, Mackay-Sim A, Ma DD. Transplantation of neuronal-primed human bone marrow mesenchymal stem cells in hemiparkinsonian rodents. PLoS One. 2011;6:e19025. doi: 10.1371/journal.pone.0019025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Lee JK, Lee H, Carter JE, Chang JW, Oh W, Yang YS, Suh JG, Lee BH, Jin HK, Bae JS. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol Aging. 2012;33:588–602. doi: 10.1016/j.neurobiolaging.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Lee JK, Jin HK, Endo S, Schuchman EH, Carter JE, Bae JS. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells. 2010;28:329–343. doi: 10.1002/stem.277. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Uchida K, Guerrero AR, Watanabe S, Sugita D, Takeura N, Yoshida A, Long G, Wright KT, Johnson WE, Baba H. Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. J Neurotrauma. 2012;29:1614–1625. doi: 10.1089/neu.2011.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney TC, Rooney GE, Barry FP, Howard L, Dowd E. Potential of rat bone marrow-derived mesenchymal stem cells as vehicles for delivery of neurotrophins to the Parkinsonian rat brain. Brain Res. 2010;1359:33–43. doi: 10.1016/j.brainres.2010.08.040. [DOI] [PubMed] [Google Scholar]
- Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol. 2001;33:907–921. doi: 10.1006/jmcc.2001.1367. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends Neurosci. 2008;31:176–182. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl) 2006;84:712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambuyzer BR, Bergwerf I, De Vocht N, Reekmans K, Daans J, Jorens PG, Goossens H, Ysebaert DK, Chatterjee S, Van Marck E, Berneman ZN, Ponsaerts P. Allogeneic stromal cell implantation in brain tissue leads to robust microglial activation. Immunol Cell Biol. 2009;87:267–273. doi: 10.1038/icb.2009.12. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- Sim JA, Young MT, Sung HY, North RA, Surprenant A. Reanalysis of P2X7 receptor expression in rodent brain. J Neurosci. 2004;24:6307–6314. doi: 10.1523/JNEUROSCI.1469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Debetto P, Giusti P. The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J. 2010;24:337–345. doi: 10.1096/fj.09-138883. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, Hong JS, Block ML. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. 2007;82:237–243. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. 2005;18:166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Reichert F, Rotshenker S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol Dis. 2003;12:65–72. doi: 10.1016/s0969-9961(02)00008-6. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwerf I, De Vocht N, Tambuyzer B, Verschueren J, Reekmans K, Daans J, Ibrahimi A, Van Tendeloo V, Chatterjee S, Goossens H, Jorens PG, Baekelandt V, Ysebaert D, Van Marck E, Berneman ZN, Linden AV, Ponsaerts P. Reporter gene-expressing bone marrow-derived stromal cells are immune-tolerated following implantation in the central nervous system of syngeneic immunocompetent mice. BMC Biotechnol. 2009;9:1. doi: 10.1186/1472-6750-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praet J, Reekmans K, Lin D, De Vocht N, Bergwerf I, Tambuyzer B, Daans J, Hens N, Goossens H, Pauwels P, Berneman Z, Van der Linden A, Ponsaerts P. Cell type-associated differences in migration, survival and immunogenicity following grafting in CNS tissue. Cell Transplant. 2012;21:1867–1881. doi: 10.3727/096368912X636920. [DOI] [PubMed] [Google Scholar]
- Coyne TM, Marcus AJ, Reynolds K, Black IB, Woodbury D. Disparate host response and donor survival after the transplantation of mesenchymal or neuroectodermal cells to the intact rodent brain. Transplantation. 2007;84:1507–1516. doi: 10.1097/01.tp.0000288185.09601.4d. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Lee JK, Lee H, Shin JW, Carter JE, Sakamoto T, Jin HK, Bae JS. The therapeutic potential of human umbilical cord blood-derived mesenchymal stem cells in Alzheimer’s disease. Neurosci Lett. 2010;481:30–35. doi: 10.1016/j.neulet.2010.06.045. [DOI] [PubMed] [Google Scholar]
- Ryu HH, Kang BJ, Park SS, Kim Y, Sung GJ, Woo HM, Kim WH, Kweon OK. Comparison of mesenchymal stem cells derived from fat, bone marrow, Wharton’s jelly, and umbilical cord blood for treating spinal cord injuries in dogs. J Vet Med Sci. 2012;74:1617–1630. doi: 10.1292/jvms.12-0065. [DOI] [PubMed] [Google Scholar]
- Sheikh AM, Nagai A, Wakabayashi K, Narantuya D, Kobayashi S, Yamaguchi S, Kim SU. Mesenchymal stem cell transplantation modulates neuroinflammation in focal cerebral ischemia: contribution of fractalkine and IL-5. Neurobiol Dis. 2011;41:717–724. doi: 10.1016/j.nbd.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Zeng X, Zeng YS, Ma YH, Lu LY, Du BL, Zhang W, Li Y, Chan WY. Bone marrow mesenchymal stem cells in a three dimensional gelatin sponge scaffold attenuate inflammation, promote angiogenesis and reduce cavity formation in experimental spinal cord injury. Cell Transplant. 2011;20:1881–1899. doi: 10.3727/096368911X566181. [DOI] [PubMed] [Google Scholar]
- Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Dev Neurosci. 2011;33:199–209. doi: 10.1159/000328989. [DOI] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, Lindvall O. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57:835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Desforges NM, Hebron ML, Algarzae NK, Lonskaya I, Moussa CE. Fractalkine mediates communication between pathogenic proteins and microglia: implications of anti-inflammatory treatments in different stages of neurodegenerative diseases. Int J Alzheimers Dis. 2012;2012:345472. doi: 10.1155/2012/345472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong N, Perry SW, Zhang Q, James HJ, Guo H, Brooks A, Bal H, Kinnear SA, Fine S, Epstein LG, Dairaghi D, Schall TJ, Gendelman HE, Dewhurst S, Sharer LR, Gelbard HA. Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J Immunol. 2000;164:1333–1339. doi: 10.4049/jimmunol.164.3.1333. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011;286:32713–32722. doi: 10.1074/jbc.M111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC, Gemma C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31:16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia. 2002;37:314–327. [PubMed] [Google Scholar]
- Dorgham K, Ghadiri A, Hermand P, Rodero M, Poupel L, Iga M, Hartley O, Gorochov G, Combadiere C, Deterre P. An engineered CX3CR1 antagonist endowed with anti-inflammatory activity. J Leukoc Biol. 2009;86:903–911. doi: 10.1189/jlb.0308158. [DOI] [PubMed] [Google Scholar]
- Giunti D, Parodi B, Usai C, Vergani L, Casazza S, Bruzzone S, Mancardi G, Uccelli A. Mesenchymal stem cells shape microglia effector functions through the release of CX3CL1. Stem Cells. 2012;30:2044–2053. doi: 10.1002/stem.1174. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert M, Sedgwick JD, Fischer E, Schluter D. Regulation of microglial cell responses in murine Toxoplasma encephalitis by CD200/CD200 receptor interaction. Acta Neuropathol. 2006;111:548–558. doi: 10.1007/s00401-006-0062-z. [DOI] [PubMed] [Google Scholar]
- Chitnis T, Imitola J, Wang Y, Elyaman W, Chawla P, Sharuk M, Raddassi K, Bronson RT, Khoury SJ. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am J Pathol. 2007;170:1695–1712. doi: 10.2353/ajpath.2007.060677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol. 2009;85:352–370. doi: 10.1189/jlb.0608385. [DOI] [PubMed] [Google Scholar]
- Lyons A, McQuillan K, Deighan BF, O’Reilly JA, Downer EJ, Murphy AC, Watson M, Piazza A, O’Connell F, Griffin R, Mills KH, Lynch MA. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. Brain Behav Immun. 2009;23:1020–1027. doi: 10.1016/j.bbi.2009.05.060. [DOI] [PubMed] [Google Scholar]
- Yi MH, Zhang E, Kang JW, Shin YN, Byun JY, Oh SH, Seo JH, Lee YH, Kim DW. Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus. Brain Res. 2012;1481:90–96. doi: 10.1016/j.brainres.2012.08.053. [DOI] [PubMed] [Google Scholar]
- Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copland DA, Calder CJ, Raveney BJ, Nicholson LB, Phillips J, Cherwinski H, Jenmalm M, Sedgwick JD, Dick AD. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. 2007;171:580–588. doi: 10.2353/ajpath.2007.070272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, Zhang YJ, Ding JQ, Chen SD. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:154. doi: 10.1186/1742-2094-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietila M, Lehtonen S, Tuovinen E, Lahteenmaki K, Laitinen S, Leskela HV, Natynki A, Pesala J, Nordstrom K, Lehenkari P. CD200 positive human mesenchymal stem cells suppress TNF-alpha secretion from CD200 receptor positive macrophage-like cells. PLoS One. 2012;7:e31671. doi: 10.1371/journal.pone.0031671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- Ooi YY, Ramasamy R, Rahmat Z, Subramaiam H, Tan SW, Abdullah M, Israf DA, Vidyadaran S. Bone marrow-derived mesenchymal stem cells modulate BV2 microglia responses to lipopolysaccharide. Int Immunopharmacol. 2010;10:1532–1540. doi: 10.1016/j.intimp.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Zhou C, Zhang C, Chi S, Xu Y, Teng J, Wang H, Song Y, Zhao R. Effects of human marrow stromal cells on activation of microglial cells and production of inflammatory factors induced by lipopolysaccharide. Brain Res. 2009;1269:23–30. doi: 10.1016/j.brainres.2009.02.049. [DOI] [PubMed] [Google Scholar]
- Schafer S, Calas AG, Vergouts M, Hermans E. Immunomodulatory influence of bone marrow-derived mesenchymal stem cells on neuroinflammation in astrocyte cultures. J Neuroimmunol. 2012;249:40–48. doi: 10.1016/j.jneuroim.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Lee JK, Schuchman EH, Jin HK, Bae JS. Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid beta ameliorates Alzheimer’s disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells. 2012;30:1544–1555. doi: 10.1002/stem.1125. [DOI] [PubMed] [Google Scholar]
- Payne NL, Dantanarayana A, Sun G, Moussa L, Caine S, McDonald C, Herszfeld D, Bernard CC, Siatskas C. Early intervention with gene-modified mesenchymal stem cells overexpressing interleukin-4 enhances anti-inflammatory responses and functional recovery in experimental autoimmune demyelination. Cell Adh Migr. 2012;6:179–189. doi: 10.4161/cam.20341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker CF, Billingham RE. Immunologically privileged sites. Adv Immunol. 1977;25:1–54. [PubMed] [Google Scholar]
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwerf I, Tambuyzer B, De Vocht N, Reekmans K, Praet J, Daans J, Chatterjee S, Pauwels P, Van der Linden A, Berneman ZN, Ponsaerts P. Recognition of cellular implants by the brain’s innate immune system. Immunol Cell Biol. 2011;89:511–516. doi: 10.1038/icb.2010.141. [DOI] [PubMed] [Google Scholar]
- Reekmans K, Praet J, Daans J, Reumers V, Pauwels P, Van der Linden A, Berneman ZN, Ponsaerts P. Current challenges for the advancement of neural stem cell biology and transplantation research. Stem Cell Rev. 2012;8:262–278. doi: 10.1007/s12015-011-9266-2. [DOI] [PubMed] [Google Scholar]
- Zhu W, Chen J, Cong X, Hu S, Chen X. Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells. Stem Cells. 2006;24:416–425. doi: 10.1634/stemcells.2005-0121. [DOI] [PubMed] [Google Scholar]
- Bao X, Feng M, Wei J, Han Q, Zhao H, Li G, Zhu Z, Xing H, An Y, Qin C, Zhao RC, Wang R. Transplantation of Flk-1+ human bone marrow-derived mesenchymal stem cells promotes angiogenesis and neurogenesis after cerebral ischemia in rats. Eur J Neurosci. 2011;34:87–98. doi: 10.1111/j.1460-9568.2011.07733.x. [DOI] [PubMed] [Google Scholar]
- Bao X, Wei J, Feng M, Lu S, Li G, Dou W, Ma W, Ma S, An Y, Qin C, Zhao RC, Wang R. Transplantation of human bone marrow-derived mesenchymal stem cells promotes behavioral recovery and endogenous neurogenesis after cerebral ischemia in rats. Brain Res. 2011;1367:103–113. doi: 10.1016/j.brainres.2010.10.063. [DOI] [PubMed] [Google Scholar]
- Zheng W, Honmou O, Miyata K, Harada K, Suzuki J, Liu H, Houkin K, Hamada H, Kocsis JD. Therapeutic benefits of human mesenchymal stem cells derived from bone marrow after global cerebral ischemia. Brain Res. 2010;1310:8–16. doi: 10.1016/j.brainres.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Dey ND, Bombard MC, Roland BP, Davidson S, Lu M, Rossignol J, Sandstrom MI, Skeel RL, Lescaudron L, Dunbar GL. Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington’s disease. Behav Brain Res. 2010;214:193–200. doi: 10.1016/j.bbr.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Blandini F, Cova L, Armentero MT, Zennaro E, Levandis G, Bossolasco P, Calzarossa C, Mellone M, Giuseppe B, Deliliers GL, Polli E, Nappi G, Silani V. Transplantation of undifferentiated human mesenchymal stem cells protects against 6-hydroxydopamine neurotoxicity in the rat. Cell Transplant. 2010;19:203–217. doi: 10.3727/096368909X479839. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Schafer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, Buadze M, Lourhmati A, Klopfer T, Schaumann F, Schmid B, Koehle C, Proksch B, Weissert R, Reichardt HM, van den Brandt J, Buniatian GH, Schwab M, Gleiter CH, Frey WH 2nd. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011;14:3–16. doi: 10.1089/rej.2010.1130. [DOI] [PubMed] [Google Scholar]
- Hu SL, Luo HS, Li JT, Xia YZ, Li L, Zhang LJ, Meng H, Cui GY, Chen Z, Wu N, Lin JK, Zhu G, Feng H. Functional recovery in acute traumatic spinal cord injury after transplantation of human umbilical cord mesenchymal stem cells. Crit Care Med. 2010;38:2181–2189. doi: 10.1097/CCM.0b013e3181f17c0e. [DOI] [PubMed] [Google Scholar]
- Kang ES, Ha KY, Kim YH. Fate of transplanted bone marrow derived mesenchymal stem cells following spinal cord injury in rats by transplantation routes. J Korean Med Sci. 2012;27:586–593. doi: 10.3346/jkms.2012.27.6.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SQ, Zhang HT, You J, Zhang MY, Cai YQ, Jiang XD, Xu RX. Comparison of transdifferentiated and untransdifferentiated human umbilical mesenchymal stem cells in rats after traumatic brain injury. Neurochem Res. 2011;36:2391–2400. doi: 10.1007/s11064-011-0567-2. [DOI] [PubMed] [Google Scholar]
- Zanier ER, Montinaro M, Vigano M, Villa P, Fumagalli S, Pischiutta F, Longhi L, Leoni ML, Rebulla P, Stocchetti N, Lazzari L, De Simoni MG. Human umbilical cord blood mesenchymal stem cells protect mice brain after trauma. Crit Care Med. 2011;39:2501–2510. doi: 10.1097/CCM.0b013e31822629ba. [DOI] [PubMed] [Google Scholar]
- Chacko SM, Ahmed S, Selvendiran K, Kuppusamy ML, Khan M, Kuppusamy P. Hypoxic preconditioning induces the expression of prosurvival and proangiogenic markers in mesenchymal stem cells. Am J Physiol Cell Physiol. 2010;299:C1562–C1570. doi: 10.1152/ajpcell.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Surviving ischemia: adaptive responses mediated by hypoxia-inducible factor 1. J Clin Invest. 2000;106:809–812. doi: 10.1172/JCI11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung SC, Pochampally RR, Hsu SC, Sanchez C, Chen SC, Spees J, Prockop DJ. Short-term exposure of multipotent stromal cells to low oxygen increases their expression of CX3CR1 and CXCR4 and their engraftment in vivo. PLoS One. 2007;2:e416. doi: 10.1371/journal.pone.0000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Yao Y, Chen L, Li Y, Sheng Z, Ma G. Hypoxic preconditioning improves survival of cardiac progenitor cells: role of stromal cell derived factor-1alpha-CXCR4 axis. PLoS One. 2012;7:e37948. doi: 10.1371/journal.pone.0037948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu S, Li Y, Wang X, Xue W, Ge G, Luo X. The role of SDF-1-CXCR4/CXCR7 axis in the therapeutic effects of hypoxia-preconditioned mesenchymal stem cells for renal ischemia/reperfusion injury. PLoS One. 2012;7:e34608. doi: 10.1371/journal.pone.0034608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Xue W, Ge G, Luo X, Li Y, Xiang H, Ding X, Tian P, Tian X. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1alpha in MSCs. Biochem Biophys Res Commun. 2010;401:509–515. doi: 10.1016/j.bbrc.2010.09.076. [DOI] [PubMed] [Google Scholar]
- Hu X, Yu SP, Fraser JL, Lu Z, Ogle ME, Wang JA, Wei L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg. 2008;135:799–808. doi: 10.1016/j.jtcvs.2007.07.071. [DOI] [PubMed] [Google Scholar]
- Rosova I, Dao M, Capoccia B, Link D, Nolta JA. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells. 2008;26:2173–2182. doi: 10.1634/stemcells.2007-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theus MH, Wei L, Cui L, Francis K, Hu X, Keogh C, Yu SP. In vitro hypoxic preconditioning of embryonic stem cells as a strategy of promoting cell survival and functional benefits after transplantation into the ischemic rat brain. Exp Neurol. 2008;210:656–670. doi: 10.1016/j.expneurol.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Wei L, Fraser JL, Lu ZY, Hu X, Yu SP. Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol Dis. 2012;46:635–645. doi: 10.1016/j.nbd.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Hu R, Lv L, Ling L, Jiang S. Erythropoietin improves the efficiency of endothelial progenitor cell therapy after myocardial infarction in mice: effects on transplanted cell survival and autologous endothelial progenitor cell mobilization. J Surg Res. 2012;176:e47–e55. doi: 10.1016/j.jss.2012.04.047. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Oh JS, An SS, Pennant WA, Gwak SJ, Kim AN, Han PK, Yoon DH, Kim KN, Ha Y. Hypoxia-specific GM-CSF-overexpressing neural stem cells improve graft survival and functional recovery in spinal cord injury. Gene Ther. 2012;19:513–521. doi: 10.1038/gt.2011.137. [DOI] [PubMed] [Google Scholar]
- Pavlica S, Milosevic J, Keller M, Schulze M, Peinemann F, Piscioneri A, De Bartolo L, Darsow K, Bartel S, Lange HA, Bader A. Erythropoietin enhances cell proliferation and survival of human fetal neuronal progenitors in normoxia. Brain Res. 2012;1452:18–28. doi: 10.1016/j.brainres.2012.02.043. [DOI] [PubMed] [Google Scholar]
- Cerrada I, Ruiz-Sauri A, Carrero R, Trigueros C, Dorronsoro A, Sanchez-Puelles JM, Diez-Juan A, Montero JA, Sepulveda P. Hypoxia-inducible factor 1 alpha contributes to cardiac healing in mesenchymal stem cells-mediated cardiac repair. Stem Cells Dev. 2012;22:501–511. doi: 10.1089/scd.2012.0340. [DOI] [PubMed] [Google Scholar]
- Lian Jin H, Pennant WA, Hyung Lee M, Su S, Ah Kim H, Lu Liu M, Soo Oh J, Cho J, Nyun Kim K, Heum Yoon D, Ha Y. Neural stem cells modified by a hypoxia-inducible VEGF gene expression system improve cell viability under hypoxic conditions and spinal cord injury. Spine (Phila Pa 1976) 2011;36:857–864. doi: 10.1097/BRS.0b013e3181e7f34b. [DOI] [PubMed] [Google Scholar]
- Pedraza E, Coronel MM, Fraker CA, Ricordi C, Stabler CL. Preventing hypoxia-induced cell death in beta cells and islets via hydrolytically activated, oxygen-generating biomaterials. Proc Natl Acad Sci USA. 2012;109:4245–4250. doi: 10.1073/pnas.1113560109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anada T, Fukuda J, Sai Y, Suzuki O. An oxygen-permeable spheroid culture system for the prevention of central hypoxia and necrosis of spheroids. Biomaterials. 2012;33:8430–8441. doi: 10.1016/j.biomaterials.2012.08.040. [DOI] [PubMed] [Google Scholar]
- Liu N, Zhang Y, Fan L, Yuan M, Du H, Cheng R, Liu D, Lin F. Effects of transplantation with bone marrow-derived mesenchymal stem cells modified by Survivin on experimental stroke in rats. J Transl Med. 2011;9:105. doi: 10.1186/1479-5876-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney TC, Dockery P, Windebank AJ, Barry FP, Howard L, Dowd E. Survival and immunogenicity of mesenchymal stem cells from the green fluorescent protein transgenic rat in the adult rat brain. Neurorehabil Neural Repair. 2010;24:645–656. doi: 10.1177/1545968309357745. [DOI] [PubMed] [Google Scholar]
- Tao J, Ji F, Liu B, Wang F, Dong F, Zhu Y. Improvement of deficits by transplantation of lentiviral vector-modified human amniotic mesenchymal cells after cerebral ischemia in rats. Brain Res. 2012;1448:1–10. doi: 10.1016/j.brainres.2012.01.069. [DOI] [PubMed] [Google Scholar]
- Xiong N, Zhang Z, Huang J, Chen C, Jia M, Xiong J, Liu X, Wang F, Cao X, Liang Z, Sun S, Lin Z, Wang T. VEGF-expressing human umbilical cord mesenchymal stem cells, an improved therapy strategy for Parkinson’s disease. Gene Ther. 2011;18:394–402. doi: 10.1038/gt.2010.152. [DOI] [PubMed] [Google Scholar]
- Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, Narasimhan P, Maier CM, Nishiyama Y, Chan PH. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci. 2012;32:3462–3473. doi: 10.1523/JNEUROSCI.5686-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix GJ, Garbayo E, Sindji L, Thomas O, Vanpouille-Box C, Schiller PC, Montero-Menei CN. The therapeutic potential of human multipotent mesenchymal stromal cells combined with pharmacologically active microcarriers transplanted in hemi-parkinsonian rats. Biomaterials. 2011;32:1560–1573. doi: 10.1016/j.biomaterials.2010.10.041. [DOI] [PubMed] [Google Scholar]
- Yan-Wu G, Yi-Quan K, Ming L, Ying-Qian C, Xiao-Dan J, Shi-Zhong Z, Wang-Ming Z, Chuan-Zhi D. Human umbilical cord-derived Schwann-like cell transplantation combined with neurotrophin-3 administration in dyskinesia of rats with spinal cord injury. Neurochem Res. 2011;36:783–792. doi: 10.1007/s11064-011-0402-9. [DOI] [PubMed] [Google Scholar]
- Mkaddem SB, Bens M, Vandewalle A. Differential activation of Toll-like receptor-mediated apoptosis induced by hypoxia. Oncotarget. 2010;1:741–750. doi: 10.18632/oncotarget.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster BD, Rouch JD, Wang M, Meldrum DR. Toll-like receptor 4 ablation improves stem cell survival after hypoxic injury. J Surg Res. 2012;177:330–333. doi: 10.1016/j.jss.2012.04.042. [DOI] [PubMed] [Google Scholar]
- Yin Q, Jin P, Liu X, Wei H, Lin X, Chi C, Liu Y, Sun C, Wei Y. SDF-1alpha inhibits hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells through PI3K/Akt and ERK1/2 signaling pathways. Mol Biol Rep. 2011;38:9–16. doi: 10.1007/s11033-010-0071-9. [DOI] [PubMed] [Google Scholar]
- Deng J, Han Y, Yan C, Tian X, Tao J, Kang J, Li S. Overexpressing cellular repressor of E1A-stimulated genes protects mesenchymal stem cells against hypoxia- and serum deprivation-induced apoptosis by activation of PI3K/Akt. Apoptosis. 2010;15:463–473. doi: 10.1007/s10495-009-0434-7. [DOI] [PubMed] [Google Scholar]
- Hassan F, Meduru S, Taguchi K, Kuppusamy ML, Mostafa M, Kuppusamy P, Khan M. Carvedilol enhances mesenchymal stem cell therapy for myocardial infarction via inhibition of caspase-3 expression. J Pharmacol Exp Ther. 2012;343:62–71. doi: 10.1124/jpet.112.196915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Guo XW, Chen K, Xu RX, Zheng HZ, Wang J. Inhibition of hypoxia and serum deprivation-induced apoptosis by salvianolic acid in rat mesenchymal stem cells. J Tradit Chin Med. 2012;32:222–228. doi: 10.1016/s0254-6272(13)60015-1. [DOI] [PubMed] [Google Scholar]
- Nie Y, Han BM, Liu XB, Yang JJ, Wang F, Cong XF, Chen X. Identification of microRNAs involved in hypoxia- and serum deprivation-induced apoptosis in mesenchymal stem cells. Int J Biol Sci. 2011;7:762–768. doi: 10.7150/ijbs.7.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronsyn MW, Daans J, Spaepen G, Chatterjee S, Vermeulen K, D’Haese P, Van Tendeloo VF, Van Marck E, Ysebaert D, Berneman ZN, Jorens PG, Ponsaerts P. Plasmid-based genetic modification of human bone marrow-derived stromal cells: analysis of cell survival and transgene expression after transplantation in rat spinal cord. BMC Biotechnol. 2007;7:90. doi: 10.1186/1472-6750-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]