

Abstract
Purpose.
Optic nerve (ON) ischemia associated with nonarteric anterior ischemic optic neuropathy (NAION) results in axon and myelin damage. Myelin damage activates the intraneural Ras homolog A (RhoA), contributing to axonal regeneration failure. We hypothesized that increasing extrinsic macrophage activity after ON infarct would scavenge degenerate myelin and improve postischemic ON recovery. We used the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) to upregulate ON macrophage activity, and evaluated GM-CSF's effects after ON ischemia in the NAION rodent model (rAION).
Methods.
Following rAION induction, GM-CSF was administered via intraventricular injection. Retinal ganglion cell (RGC) stereologic analysis was performed 1 month postinduction. The retinae and optic nerve laminae of vehicle- and GM-CSF-treated animals were examined immunohistochemically and ultrastructurally using transmission electron microscopy (TEM). RhoA activity was analyzed using a rhotekin affinity immunoanalysis and densitometry. Isolated ONs were analyzed functionally ex vivo by compound action potential (CAP) analysis.
Results.
Rodent NAION produces ON postinfarct demyelination and myelin damage, functionally demonstrable by CAP analysis and ultrastructurally by TEM. Granulocyte-macrophage colony-stimulating factor increased intraneural inflammation, activating and recruiting endogenous microglia, with only a moderate amount of exogenous macrophage recruitment. Treatment with GM-CSF reduced postinfarct intraneural RhoA activity, but did not neuroprotect RGCs after rAION.
Conclusions.
Sudden ON ischemia results in previously unrecognized axonal demyelination, which may have a clinically important role in NAION-related functional defects and recovery. Granulocyte-macrophage colony-stimulating factor is not neuroprotective when administered directly to the optic nerve following ON ischemia, and does not improve axonal regeneration. It dramatically increases ON-microglial activation and recruitment.
Keywords: optic nerve ischemia, GM-CSF, microglia, macrophages, immune regulation, naion, rodent models, postinfarct demyelination
Following optic nerve (ON) infarct, there is functional and histological evidence of ON demyelination and damage. Nonspecifically increasing macrophage activity is detrimental to ON recovery and blocks axonal regeneration. Increased nonspecific macrophage activity reduces RhoA activity, but does not improve retinal ganglion cell survival.
Introduction
Nonarteritic anterior ischemic optic neuropathy (NAION) is an optic nerve (ON) infarct, and the most common cause of sudden ON-related vision loss in the United States.1 No treatments to date have demonstrated unequivocally clinical effectiveness in reducing NAION damage. Following NAION onset, visual function declines further in most individuals, and then improves somewhat by 3 months postevent, although approximately 20% of individuals experience further loss of at least 3 lines of vision when measured at 3 months and then 2 years after the event.2 Nonarteritic anterior ischemic optic neuropathy–affected individuals also show a mild decline in mean visual acuity over years,2 suggesting that pathophysiological changes distant from the initial ischemic insult may have an important role in ON recovery.
Recently, early inflammation components were identified in clinical NAION and its models, including blood–brain barrier (BBB) breakdown and extrinsic macrophage invasion.3–5 Similar to other central nervous system (CNS) infarct and spinal cord injury models,6,7 NAION and sudden ON ischemia result in early cytokine mediated changes,8 followed by sequential inflammatory cellular activation and infiltration.5 In the rodent NAION (rAION) model, extrinsic macrophage invasion typically begins within 3 days postinduction,3 and postinfarct demyelination and oligodendrocyte death follow days after ON ischemia.9,10 While axonal regeneration has been demonstrated in a number of ON trauma models,11,12 demyelination generates release of soluble factors that inhibit axonal regeneration. These factors include NOGO66 and myelin-associated glycoprotein (MAG), which activate the axonal membrane protein complex leucine rich repeat and Ig domain containing 1 (LINGO-1).13 LINGO-1 activates the axonal kinase RAS homolog A (RhoA) by GTP addition,14 which directly inhibits actin cytoskeleton polymerization, resulting in axonal growth cone collapse.13
Macrophage activity can be either neurodegenerative and/or neuroprotective.15 While macrophage activity can block axonal regeneration,16 extrinsic macrophage activation can enhance remyelination,12,17 eliminate degenerate myelin,18 and improve axonal regeneration and neuronal survival.19 Unfortunately, inflammation also can generate myelin “pores,” which can functionally disturb neural impulse propagation as well as result in demyelination.20 Nevertheless, we hypothesized that recruitment of extrinsic macrophages following ischemic ON injury could improve regeneration and postinsult function, by eliminating degenerate myelin and reducing active RhoA levels.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine that induces phagocytic differentiation of hematopoietic bone marrow precursors and recruits extrinsic macrophages to tissues.21 Studies have suggested intraperitoneal or direct local application of GM-CSF administration can be neuroprotective following CNS trauma and ischemia.22,23 Intravenous GM-CSF can reduce infarct damage, and increase vascular collateralization and revascularization following carotid occlusion and CNS infarct,24,25 and is neuroprotective for neurons and oligodendrocytes following spinal cord injury.26,27 Granulocyte-macrophage colony-stimulating factor is directly neuroprotective in neural cell culture.28 These previous reports suggested that direct GM-CSF administration could improve long-term outcomes following sudden ON ischemia. We wanted to determine whether local GM-CSF administration following ON infarct would be neuroprotective for retinal ganglion cells and their axons.
To measure ON function, visual evoked potentials (VEPs) are used most commonly. However, VEP measurements are based on cortical potentials, and are an end-measure of the entire visual system, rather than a direct analysis of ON changes. We decided to compare ON function directly, using compound action potentials (CAPs) in isolated ON tissue.29,30 We evaluated ON function, and confirmed with morphologic and ultrastructural analysis. To support our hypothesis that extrinsic macrophage recruitment could reduce degenerate myelin levels and minimize RhoA activity, we assayed active RhoA levels with rhotekin.31,32 Rhotekin protein binds only to active RhoA, and, thus, can be used to measure directly relative RhoA activity following different conditions.31
Methods
Animals
All animal protocols were approved by the institutional animal care and use committee (IACUC), and all animals were handled in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Male Sprague-Dawley rats (120–150 g) were obtained from Harlan Laboratories (Indianapolis, IN). Rose bengal (90% purity) was purchased from Sigma Chemicals (St. Louis, MO). Anesthesia was induced with a mixture of ketamine (100 mg/kg)/xylazine (4 mg/kg) administered intraperitoneally. Eyes were dilated with 1% cyclopentolate and 2.5% phenylephrine. Anesthetized animals were placed on heating pads until recovery from anesthesia and then returned to their cages. After intracranial surgery, animals were dosed with buprenorphine (0.03 mg/kg) every 12 hours for 3 days. We used a total of 46 animals (23 animals per treatment group).
rAION Induction
An ON infarct (rAION) was induced in the right eye of each test animal. Eyes of anesthetized animals were dilated with tropicamide 1%, and a 7-mm custom fundus contact lens with a flat front surface was used to visualize the retina and ON. Sterile rose bengal (2.5 mM/ml, 1 mL/kg) was injected intravenously via tail vein. At 30 seconds postinjection, the capillaries of the optic nerve were illuminated using 532 nm laser light/500 um spot size/50 mW power for 12 1-second pulses, using a frequency doubled neodymium aluminum garnet (nd-YAG) laser (Iridex, Mountain View, CA). This procedure generates ON head ischemia without direct thermal damage, with a resulting loss of 40% to 55% of retinal ganglion cells and their ON axons by 30 days postinduction.33
GM-CSF Injection
Initial studies revealed that GM-CSF is not transported to the ON when injected into the superior colliculus, unlike FluoroGold (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or horseradish peroxidase (data not shown). Intraventricular injection results in general GM-CSF distribution throughout the cerebrospinal fluid compartment, which bathes the optic nerves. We therefore evaluated the effect of intraventricularly administered GM-CSF following rodent NAION (rAION) induction. Animals in two treatment groups were rAION-induced (n = 9/group).
Three days postinduction, each treatment group received either an intraventricular injection of GM-CSF or vehicle (0.1% rat serum albumen in normal saline). Animals were anesthetized with ketamine/xylazine, the subcutaneous space over the skull infiltrated with 1% lidocaine, and the animal immobilized in a Stoelting stereotactic frame with digital readout. Lateral ventricle access was obtained by unilateral craniotomy at the appropriate coordinates.34 Coordinates were 1.30 mm rostral to bregma and 1.8 mm temporal to the midline, with a depth of 2.6 mm. Treatment group animals were injected in the lateral ventricle with 2 μL of 50 ng/μL GM-CSF (R&D Systems, Minneapolis, MN). Postinjection, craniotomies were closed using stainless steel wound clips.
Rhotekin Affinity Analysis
Rhotekin is a bacterial protein that selectively binds only to the active (GTP bound) form of RhoA and, thus, can be used to evaluate levels of RhoA activation.32,35 A construct containing the active portion of the rhotekin peptide linked to glutathione-S-transferase (Rhotekin-GST) was purchased from Cytoskeleton (Denver, CO). Active RhoA immunohistochemical analysis was performed on ONs 7 days postinduction from perfusion-fixed (PF) animals that received intraventricular treatment 3 days after induction with either GM-CSF or vehicle. Following tissue isolation, nerves additionally were postfixed in 2% PF-PBS sectioned at 10 μm thickness. Sections then were reacted with the soluble rhotekin construct and cross-linked using 0.5% glutaraldehyde. Rhotekin localization was evaluated by confocal microscopy, using primary mouse anti-GST antibody and labeled secondary donkey anti-mouse antibody.
Tissue for Stereology and Immunohistochemistry
Nine animals were used in each treatment group for long-term retinal ganglion cell stereology and late ON morphology. At 35 days after induction, these animals were anesthetized with ketamine/xylazine to deep surgical plane, and then perfused transcardially with 4% paraformaldehyde (PFA) in 0.05 M phosphate buffered saline (PF-PBS). Eyes were removed and postfixed in 4% PF-PFA overnight, and transferred to fresh PBS before retinal isolation. The ONs were isolated and the distal segment postfixed in glutaraldehyde-paraformaldehyde buffer (4FIG) for transmission electron microscopy (TEM) ultrastructural analysis. The rest of the ON was postfixed overnight in PF-PBS, cryoprotected in 30% sucrose in 0.05 M PBS and embedded in OCT. The ONs were frozen sectioned at 10 μm thickness.
An additional group of animals (n = 3/group) were analyzed for early ON immune cell infiltration. Animals in the early groups were perfused 7 days postinduction, and ON and retinae isolated and fixed in PF-PBS. The intrascleral ON region was dissected along with the adjacent sclera, as well as the adjoining anterior 2 mm of the ON. This tissue was then cryoprotected and embedded in OCT, and sectioned at 10 μm. The perfused brains were isolated, and the region surrounding the lateral ventricles cryoprotected and embedded in OCT, and sectioned at 30 μm. Following incubation with the appropriate primary antibody, tissues were reacted with fluorescently labeled donkey secondary antibodies (Jackson Immunoresearch, West Point, PA), specific for the primary antibody species. Tissues were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA). The ON and brain immune cell quantification was performed using three adjacent sections for each condition.
RGC Stereology
Quantitative RGC stereology was performed on flat-mounted whole retinae from animals 35 days after rAION induction, during which time >90% of RGC loss takes place.4,36 Eye cups were equilibrated in 0.5% Triton X-100 in PBS (PBST), and then incubated for 4 hours in hyaluronidase (1:500, Sigma H3506; Sigma Chemicals) in PBST. Postincubation, retinae then were incubated overnight at 4°C in primary antibody consisting of goat polyclonal Brn-3a (SC-31984; Santa Cruz Biotechnology, Inc.) 1:500 dilution. Retinae then were isolated from the eye cups and washed extensively with PBS, then incubated with Donkey anti-goat Cy3–labeled secondary antibody. Tissue was washed in PBS, incised to form a maltese cross pattern, and flat mounted.
The RGC quantification was performed using an ×60 air objective on an Olympus E900 microscope (Olympus Corporation, Tokyo, Japan), coupled to a motor-driven stage driven by a stereological software package; Stereoinvestigator (MBF Bioscience, Burlington, VT). This method generates random fields (frames) of up to 30 cell nuclei for counting, although the number typically was far fewer per frame. The retinal counting area was outlined, with a 29 to 47 mm2 retinal area used for each eye (mean area = 39 mm2), and the RGC layer defined as Brn3a(+) cells within 30 μm depth. More than 1000 cells were counted per retina control retinae typically yielded between 1500 and 1700 marker cells in at least 300 counting frames, which is greater than the number (600) required by the Schmitz-Hof equation37 for statistical validity.
ON Immunohistochemistry
The ONs were evaluated using confocal microscopy via an Olympus E300 4-channel laser microscope (Olympus Corporation). Tissue was evaluated for inflammation and damage distribution pattern using antibodies specific for axonal neurofilaments (SMI 312), general inflammation (IBA1, 1:1000; Dako, Carpinteria, CA), and extrinsic macrophages (CD68/clone ED1, 1:1000; Abd-Serotec, Raleigh, NC; Jackson Immunoresearch) generated to the appropriate primary species. All samples were counterstained with DAPI for nuclear identification.
ON Ultrastructural Analysis
The PF-PBS perfusion fixed tissues were postfixed in buffer containing glutaraldehyde and paraformaldehyde (4FIG), and postfixed with osmium tetroxide, followed by 1% uranyl acetate, and infiltrated with Durcupan resin and polymerized at 60°C. Specimens were sectioned at 70 nm and examined using a Tecnai transmission EM at ×1650, ×4400, and ×6500 magnification. Myelin damage was analyzed as a fraction of the axonal circumference, for the three axon sizes: small (<3000 nm circumference), medium (<5000 nm circumference), and large (>5000 nm circumference) fibers.38
Generation of Compound Action Potentials (CAP)
The ONs for CAP measurements were obtained from 4 animals in each treatment group euthanized at 35 days after rAION induction. The ONs were dissected and immediately submerged in ice-cold (4°C) Locke solution of the following composition (in mM): 136 NaCl, 5.6 KCl, 14.3 NaHCO3, 1.2 NaH2PO4, 2.2 CaCl2, 1.2 MgCl2, 11 dextrose, equilibrated continuously with 95% O2, 5% CO2, pH 7.2 to 7.4. Nerves were pinned to the Sylgard (Dow Corning, Midland, MI)–coated floor of a recording chamber (∼0.25 mL volume) and superfused (3–5 mL/min) with oxygenated Locke solution at 35°C to 37°C. The CAPs were recorded with a glass suction electrode connected to the input stage of an AC-coupled differential preamplifier (0.1–1 kHz; model DAM-5A; WPI, Sarasota, FL). Data were filtered at 2 kHz and sampled at 10 kHz. The CAPs were evoked with electrical pulses (0.1–0.5 msec in duration) elicited at 0.2 Hz using a second glass suction electrode. Stimulus strength was two to three times that necessary to evoke a maximum CAP response. The CAPs were digitized via a Digidata 1200 A/D converter (Axon Instruments, Sunnyvale, CA) and stored on a PC. Ten CAPs were averaged for analysis. Data acquisition and storage were controlled via pClamp 9.1 (Axon Instruments), and analyzed with Clampfit 9.2 software (Axon Instruments). Following CAP analysis, ONs were postfixed in a mixture of glutaraldehyde-formaldehyde buffer, and analyzed for ultrastructure, using TEM.
Results
GM-CSF Effects on Intracerebral Microglial Activity in Uninduced Nonischemic Tissue
We evaluated GM-CSF's potential for generalized (CNS) inflammatory upregulation at 4 days after injection. Frozen sections of periventricular brain regions (4 regions/animal) from vehicle- and GM-CSF-treated animals were analyzed by confocal immunohistochemistry (Fig. 1) for immune cells; IBA1(+) cells were quantified (Fig. 1C). There was a slight trend towards increased microglial numbers in GM-CSF-treated animals (6.56 ± 1.6 cells/field in vehicle-treated versus 7.1 ± 1.9 cells/field in GM-CSF-treated tissue). This trend was statistically nonsignficant (2-tailed t-test: P = 0.33).
Figure 1.
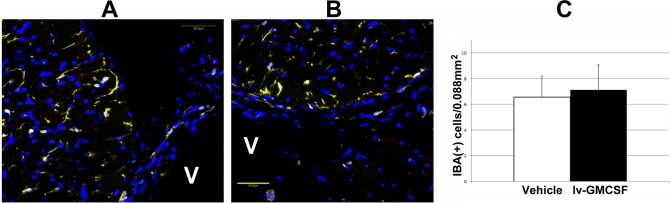
Brain microglial activity post–GM-CSF administration. IBA1(+) cells are shown in yellow. (A) Vehicle-treated CNS. (B) GM-CSF-treated CNS. There is little difference in appearance or distribution in either treatment group. V, ventricle. Scale bar: 50 μm. (C) The CNS-microglial quantification. Little difference in microglial numbers is seen in either treatment group 7 days after administration, although there is a slight trend toward more microglia in the GM-CSF treatment group.
GM-CSF Increases Postinduction ON Inflammation
Rodent NAION results in ON inflammation in the ON lamina and anterior ON.3 The GM-CSF-treated animals showed a trend towards increased numbers of inflammatory cells in infarcted lamina and uninfarcted ONs compared to vehicle (Fig. 2G, graph). IBA1 expression, which identifies inflammatory cells,39 was detectable on scattered protoplasmic cells in the anterior portion of the naïve (vehicle-uninduced) ON (Fig. 2B). Few extrinsic (ED1+) macrophages are identifiable in naïve tissue (Fig. 2D),5 or in the GM-CSF-treated uninduced lamina (Fig. 2A). At 7 days after rAION induction (4 days after treatment), microglial activity was upregulated in the laminae of vehicle- and GM-CSF-treated animals (Figs. 2B, 2E). This was seen as increased IBA1(+) cells in both treatment groups, although ONs of GM-CSF-treated animals typically showed a more intense inflammatory response, demonstrable by increased numbers of IBA1(+) cells, compared to the vehicle-treated animals (graph, Fig. 2G). The rAION-induced laminae of vehicle- and GM-CSF-treated animals showed an ED1(+) cellular infiltrate (Figs. 2B, 2E, in red, and graph, Fig. 2G, Lamina-rAION). The GM-CSF-treated animals had somewhat more ED1(+) cells in the anterior portion of the induced ON than did vehicle-treated animals (compare red cells in Figs. 2B vs. 2E, and comparative graph values in Fig. 2G). This trend is not significant. Many fewer ED1(+) cells were demonstrable in the posterior ON segment of the uninduced nerve of either treatment group (Fig. 2G, ON-uninduced). Similar to the results seen in the brain, GM-CSF did not significantly increase microglial activity in the uninduced ON (compare Figs. 2C, 2F). Thus, GM-CSF administered adjacent to the infarcted ON increases microglial activation, and slightly increases extrinsic macrophage recruitment, compared to vehicle treatment.
Figure 2.

Intraventricular GM-CSF increases inflammation in the infarcted ON. Confocal photos show inflammatory cells in representative sections of lamina (first 500 μm) and more distal (>2 mm) ON regions of the different treatment groups. (A–C) GM-CSF-treated animals. (D–F) Vehicle treated animals. (A, D) Uninduced lamina sections. Intrinsic microglial cells (IBA1[+], in green) are nonactivated with an extended/protoplasmic appearance). The majority of ED1(+) cells (in red) are present in vessels, with few ED1(+) cells present in the ON. (B, E) rAION-induced lamina sections. There is extensive microglial activation, and ED1(+) systemic macrophage invasion is seen in sections. (C, F) Uninduced distal ON sections. Few ED1(+) cells are present. The uninduced GM-CSF-treated ON (C) has microglial activity similar to that seen in the vehicle-treated (F) uninduced nerve. (G) Quantification of lamina/ON tissue sections from three individuals. The rAION-induced, GM-CSF-treated lamina shows a trend towards the greatest number of microglia and systemic macrophages, compared to the laminae from rAION-induced, vehicle-treated animals. This trend is continued in GM-CSF-treated tissues from uninduced eyes. Scale bars: 100 μm (B, E).
Intraventricular GM-CSF Does Not Improve Post-rAION RGC Survival
We compared RGC survival 30 days after rAION induction in GM-CSF- and vehicle- treated groups using nonbiased stereology (statistically robust cellular quantification). We quantified Brn3a immunostained RGCs in whole retinal flat mounts, compared to contralateral (uninduced) eyes from the same animals. This approach enabled comparison of the relative number of RGCs in the rAION-induced and uninduced eyes of both treatment groups. Results are summarized (in RGCs/mm2/retinal surface) for each of the individual groups in Figure 3.
Figure 3.
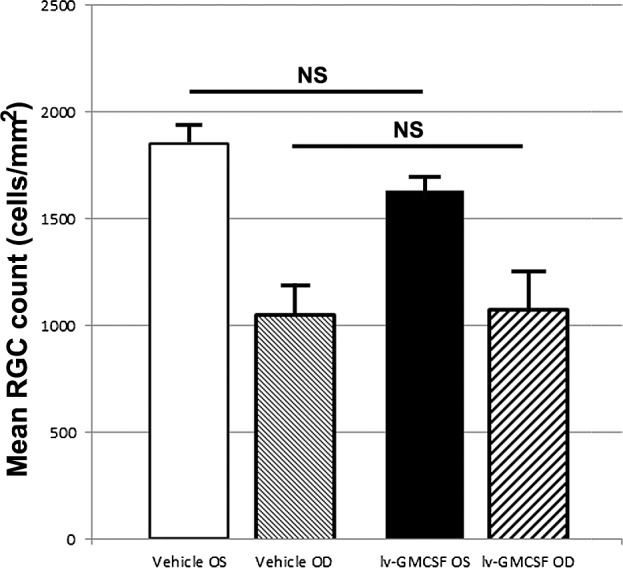
Stereological analysis of vehicle and GM-CSF-treated animals. Average cell counts per mm2 retinal area is shown for each condition. The rAION induction resulted in a 42.9% RGC loss in vehicle-treated animals versus a 33.9% RGC loss in GM-CSF-treated, animals when calculated against their contralateral control eyes. The difference in overall RGC numbers in the treated eyes between the two treatment groups is nonsignificant (P = 0.91, 2-tailed t-test, n = 9 animals/treatment group).
There was a trend toward fewer RGCs in the retinae of uninduced eyes of GM-CSF-treated animals, than in the uninduced eyes of vehicle controls (mean 1633 vs. 1854 cells/mm2). This difference was nonsignificant (P > 0.058, 2-tailed t-test). Interestingly, while a trend was seen toward increased RGC preservation in rAION-induced, GM-CSF-treated eyes, when induced eyes were compared to their contralateral (uninduced) retinae (33.9% RGC loss in GM-CSF versus 42.9% loss in vehicle-treated controls), the total numbers of RGCs in both rAION-induced treatment arms were nearly identical (1079 in GM-CSF-treated animals versus 1057 in vehicle-treated animals, P > 0.91, 2-tailed t-test). Thus, intraventricularly administered GM-CSF was not neuroprotective when considered for overall RGC survival.
ON-RhoA Activation After ON Infarct
Because of the difficulty in generating sufficient affected anterior ON tissue for Western analysis, we used rhotekin-affinity immunolabeling of infarcted ON. Anti-GST labeling of the bound GST-rhotekin protein construct enabled localization of active RhoA, and relative ON-RhoA signal intensity in vehicle- and GM-CSF-treated animals can be compared directly. This is shown in Figure 4.
Figure 4.

RhoA activation is upregulated following rAION. Rhotekin immunostaining and densitometric assay of: (A) Vehicle treated, uninduced lamina. Dotted lines indicate the regions used in the densitometric assay. (B). Vehicle-treated, rAION-induced lamina. The arrow indicates focal areas of increased active RhoA. (C) Uninduced ON section. (D) GM-CSF-treated, rAION induced lamina. Arrowheads surround an area of increased diffuse rhotekin immunostaining. (E) Densitometric assay of rhotekin immunostaining, using Image J. Identical-sized areas in each section were used for analysis. Two sections were averaged for each laminar analysis. Rhotekin binding at 7 days was greatest in vehicle-treated, rAION-induced lamina. Rhotekin binding was less in GM-CSF-treated, rAION induced lamina, and minimal in uninduced (naïve) ON. Scale bar: 100 μm (D).
We compared laminar regions from GM-CSF- and vehicle-treated animals (Figs. 4A–D) 1 week after induction (4 days after treatment). Lamina cross-sections were discernible by the lima bean shape of the ON. Densitometric analysis of rhotekin signal (indicative of relative RhoA activity) in the different ON regions was performed using the ImageJ package (available in the public domain at http://rsbweb.nih.gov/ij/download.html, Fig. 4E).
Rodent NAION results in RhoA activation in the ON of vehicle- and GM-CSF-treated animals, compared to the uninduced ONs. Granulocyte-macrophage colony-stimulating factor administration reduced RhoA activity, compared with vehicle-treated animals. Uninduced ON showed minimal RhoA activity as demonstrable by Rhotekin binding (Fig. 4). The rAION induction resulted in upregulation of active intraneural RhoA in the area of primary infarct (Figs. 4B, 4D). Administration of GM-CSF reduced RhoA activity in the lamina of rAION-induced animals (Fig. 4D), although no improvement in axonal regeneration of GM-CSF-treated animals was detected by GAP43 immunostaining (data not shown).
ON Infarction Results in Postinfarct Demyelination
Postinfarct demyelination is a well-documented occurrence following CNS ischemia.40 Early in vivo electrophysiological analyses did not demonstrate functional ON demyelination. However, numerous variables can cloud interpretation of ON function changes based on VEP analysis,41 and more recent analyses have suggested that ON demyelination or myelin damage resulting in functional alterations may occur.42,43 We evaluated ON function directly after ON infarct using ex vivo–based CAPs from isolated ONs. We analyzed numerous naïve controls, as well as vehicle- and GM-CSF-treated rAION induced ONs 1 month after induction (Fig. 5). Considerable CAP variations can occur between naïve ONs (data not shown).4 We minimized potential intra-animal artifacts by comparing intraneural responses in the same animals (Fig. 5A, naïve [no treatment]; Fig. 5C, vehicle-treated; and Fig. 5D, GM-CSF-treated) and using identical induction parameters designed to ensure consistent results without damage (see Methods). Figure 5A reveals that rAION induction generates postinfarct demyelination 1 month after rAION compared to the contralateral (naïve) ON. This was demonstrable by a diminution of signal amplitude and delayed transmission speed. Although the largest diameter fibers in vehicle-treated animals typically showed the most sensitivity and loss following infarct (compare the Fig. 5A-1A axon fiber response in vehicle-treated uninduced with that of the induced ON, Fig. 5A), this was not consistent. Interestingly, demyelination was prominent in the 1C (small diameter) component in vehicle-treated animals (compare arrows in naïve and rAION-induced ONs, Fig. 5C). Granulocyte-macrophage colony-stimulating factor–treated animals showed a selective loss of the small fiber component (Fig. 5D-rAION induced). The small (1C) fibers in the uninduced ONs from GM-CSF-treated animals showed a slight, but nonsignificant decrease in the transmission speed (Fig. 5B table: latency in the ONs from naive eyes 2.5 ± 0.5 vs. 3.6 ± 1.2 msec in ONs from uninduced, GM-CSF-treated animals). This suggests subthreshold changes may occur in GM-CSF-treated animals, even in uninduced eyes.
Figure 5.

Rodent NAION results in postinfarct demyelination/myelin dysfunction. Ex vivo electrophysiological analysis using CAPs. (A) Comparison between contralateral naïve and rAION induced ONs (same animal comparisons). The ON-CAPs reveal three myelinated axonal components: Large (1A), medium (1B), and small (1C) diameter fibers. At 1 month post-rAION in untreated (rAION only, no intraventricular treatment), there is a considerable decrease in the amplitudes of all three components, while 1B and 1C fibers also show reduced transmission speed, consistent with postinfarct demyelination. (B) Quantification of CAP parameters in representative animals. The last column on the right indicates the number of animals tested per group. Vehicle-treated animals show a reduction in amplitude and delayed transmission speed in all three fiber sizes in the induced eyes 1 month after rAION. The GM-CSF-treated animals also reveal decreased amplitude and reduced transmission speed of the largest fibers, loss of amplitude in the mid-size fibers, and a complete dropout of the smallest (1C) myelinated fiber component. (C, D) Qualitative comparison of ONs from (C) vehicle- and (D) GM-CSF-treated animals. There is decreased 1A and 1B fiber amplitude, and 1C fiber transmission speed delay in the vehicle-treated animal. There is total loss of the 1C fiber component in the rAION-induced, GM-CSF-treated ON. The 1A peak is increased in amplitude, while the 1B peak is reduced in amplitude. Scale bars: 1.5 mV and 0.75 msec (A), 3 mV and 1.5 msec (C, D).
ON Infarct Results in Postinfarct Myelin Damage and Demyelination
In contrast with uninduced ONs, rAION induction results in ON-axonal loss, which has been reported previously.33 Results from isolated ON-CAPs following rAION suggested that an ON infarct reduces myelin integrity in addition to simple axonal loss. We evaluated ON ultrastructure in the naïve and treatment groups 30 days after induction. The ONs were postfixed in 4FIG and examined by TEM at ×6500. The ON axons from the uninduced eye of vehicle-treated animals (Fig. 6A) and ONs from the uninduced eye of GM-CSF-treated animals (Fig. 6C) revealed tightly packed myelinated axons of varying diameter. We measured the circumferential lengths of the different axon fiber sizes (large, medium, and small) for 10 axons of each fiber size (Fig. 6E, white bars) for each treatment group. The total amount of myelin damage in length (defined as either areas of myelin swelling or loss of myelin lamination with lucency; see Fig. 6F) was measured for each axon and averaged, yielding the mean values for each group. Myelin lucency was taken to suggest myelin damage and focal dissolution. We discounted nonspecific changes in myelin, such as simple unwinding, which could be due to delay in perfusion-fixation, although we always compared rAION and uninduced ONs from the same animal to minimize this possibility. All axons used for measurement had intact axoplasm, defined as having intact mitochondria and neurofilaments. Results are shown in Figure 6.
Figure 6.

Rodent NAION results in long-term focal myelin damage. (A) Uninduced (naïve) ON. Intact axons of varying calibers are packed together, with normal myelin. Few disruptions are discernible. (B) Vehicle-treated, rAION-induced. While axoplasm is intact with mitochondria and neurofilaments, focal regions of myelin damage and swelling are apparent (areas indicated by white arrows). (C) GM-CSF-treated, uninduced. Axonal structure is similar to that seen in (A). (D) GM-CSF-treated, rAION-induced. Similar to (B), there are focal areas of myelin degeneration and swelling, indicated by white arrows. (E) Myelin damage quantification in naïve, vehicle-rAION and GM-CSF-rAION–induced animals (n = 10 axons of each caliber for each group). The method of determining axonal size by circumference and myelin damage for each axon is shown in (F). In (D), three axonal fiber sizes (L, large; M, medium; S, small) are revealed by circumferential measurement. Hatched areas within each larger bar represent mean myelin damage. Few naïve (white bars) axons of any size show myelin damage. By contrast, vehicle-treated rAION-induced (gray bars) axons and GM-CSF-treated rAION-induced (black bars) axons of all sizes show significant levels of myelin damage (***P < 0.01; 2-tailed t-test) compared to that seen in their naïve equivalent fiber size. The GM-CSF-treated ONs show a nonstatistically significant trend towards more myelin damage. Scale bars: 500 nm (A, F).
The majority of myelin sheaths from naïve- and GM-CSF-uninduced ONs were intact (compare Figs. 6A, naïve-OS and 6C, GM-CSF-OS). Intact axons frequently had visible mitochondria (arrowheads) and always had intact neurofilaments in parallel (small arrow). Large axons from naïve ONs had a mean myelin damage score of 5.4% ± 8.3% (±SD). Medium and small axons from naïve ONs also showed minimal myelin damage (8% ± 10.5% and 4.6% ± 9.2%, respectively). This suggests that GM-CSF-associated inflammation in the ON results in myelin compromise and dysfunction of otherwise surviving axons.
The rAION-induced vehicle-treated ONs 30 days after induction revealed that large, medium, and small fibers had 31% ± 15.8%, 43.7% ± 10.2%, and 35.9% ± 18% myelin damage, respectively. The rAION-induced GM-CSF-treated ONs also showed postinfarct demyelination and focal myelin damage, which was slightly greater for the largest and smallest fiber components, compared to vehicle-treated animals (compare hatched areas in the gray and black bars in the graph in Fig. 6E). Thus, rAION induction results in long-term (30 days) axonal damage with intact axoplasm.
Discussion
Granulocyte-macrophage colony-stimulating factor–inflammatory modulation has been proposed as a neuroprotective strategy following CNS trauma and ischemia, but evidence is conflicting. Some previous reports suggested that locally as well as systemically administered GM-CSF neuroprotect following spinal cord trauma, and reduces scarring.23,27 Other studies have suggested that while exogenously administered GM-CSF can be neuroprotective, it can reduce axonal regeneration and increase glial response by stimulating extrinsic macrophages to generate extracellular matrix molecules.44,45 These differences may stem from model differences that primarily affect either the neuron cell body or axonal damage, since GM-CSF can be protective when directly administered to neuron soma.46 Since the rAION lesion selectively damages the ON, we administered GM-CSF into the CSF to enable it to circulate to the axons. The introduction into CSF circulation enables axon-cytokine exposure at higher concentration than if administered intravenously. We found that GM-CSF administered in this manner does not neuroprotect; rather, it upregulated the postinfarct inflammation detected at the lamina and actually may increase postinfarct damage.
IBA-1(+) microglia typically are scattered sparsely within the naïve anterior ON. Immediately after rAION, there is BBB disruption, with immediate activation of intrinsic microglia. Rodent NAION induces microglial activation, as well as extrinsic macrophage invasion, easily identifiable 7 days after induction (seen in Fig. 2). Colony-stimulating factor–administered GM-CSF administration apparently increased postinfarct extrinsic macrophage recruitment, but this was a nonsignificant trend. The major effect of GM-CSF was microglial activation, which was seen primarily in infarct-affected tissue. There was little effect in total microglial numbers or activation state seen in brain periventricular regions in GM-CSF- versus vehicle-treated animals. A similar result was seen in uninduced ONs. Thus, GM-CSF fails to stimulate a vigorous inflammatory response in uninfarcted or unstressed tissues.
Intraventricularly administered GM-CSF did not increase retinal ganglion cell survival, as measured by stereology 30 days after induction. Indeed, there was a trend toward total RGC loss in GM-CSF-treated, uninduced eyes, compared to vehicle-treated animals (from 1854 ± 289 vs. 1633 ± 150 RGCs/mm3), but the total number of surviving RGCs after induction was almost identical (1057 vs. 1079/mm3, r = 0.91). Thus, locally administered GM-CSF does not improve postinfarct RGC survival, despite its effect on ON inflammation, and microglial activation and recruitment.
Interestingly, intraventricular GM-CSF resulted in reduced levels of active RhoA in the infarcted ON, as measured by rhotekin-affinity immunostaining. This may be associated with the ability of GM-CSF to increase microglial activity and early myelin elimination in the area of the primary lesion, or it may be related to extrinsic macrophage activation. Activated microglia are more able to eliminate degenerate myelin than extrinsic macrophages.47 We did not assay for RhoA-related changes in axonal regeneration. Costaining the ON for GAP43 (a marker of axonal regeneration) revealed minimal GAP43 expression in the primary lesion of either vehicle- or GM-CSF-treated animals, suggesting that GM-CSF administered in this fashion did not improve axonal regrowth.
An important, clinically relevant finding (to NAION-affected individuals) is that sudden anterior ON ischemia results in postinfarct demyelination and/or focal damage. Postinfarct axonal demyelination has been identified previously in other regions of the CNS.40,48 Demyelination also is associated with spinal cord trauma49 and optic nerve transection,50 but was not suspected until recently as an element in NAION.42,43,51 Direct changes in myelination-function are demonstrable by ON-CAP analysis, and also are seen in ON transection.50 In addition to the loss of amplitude in rAION-induced eyes, there was increased latency (peak = Tmax) in all fiber types (large, medium, and small) of vehicle-treated rAION-affected eyes (compare naïve and vehicle-rAION induced in Fig. 5B). The CAPs from GM-CSF-treated ON infarcted nerves also showed highly variable amplitudes in the different fiber types, and loss of the smallest (Fig. 1C) fiber responses. There was increased latency in the medium size (Fig. 1B) fibers compared to naïve ONs (compare naïve with rAION-GM-CSF values). These results suggest that, in addition to myelin damage that increases conduction time, the smallest (Fig. 1C) fibers are likely more sensitive to inflammation-associated damage. The TEM and electrophysiological findings suggest that increased inflammation associated with ON infarct and subsequent GM-CSF administration reduces the quality of overall ON transmission, as well as reducing the total axonal number, and that GM-CSF does not reduce ON demyelination or damage.
The TEM analysis confirmed demyelination and myelin damage as focal swelling in axons with intact axoplasm (intact mitochondria and neurofilaments) >1 month after induction. A minimal amount of myelin damage was present even in control axons, but the degree of myelin damage 1 month after rAION induction was dramatically increased, in vehicle- and GM-CSF-treated animals, surrounding or within areas of generalized axonal loss (Figs. 6B, 6D, arrows). Granulocyte-macrophage colony-stimulating factor–treated animals trended towards more myelin damage (compare graph in Fig. 6E, GM-CSF versus vehicle-treated animals), but this was not significant. Regardless of the treatment, ON infarction leads to postinfarct myelin damage and demyelination with functional consequences. Demyelination/myelin damage may increase axonal noise-to-signal ratios, result in mistiming, and have an important role in loss of function.49 Recovery from myelin damage also may contribute to later visual recovery.
While previous studies have suggested that modulating macrophage-associated inflammation can be neuroprotective and axon-regenerative following ON damage, we did not observe this effect. There are a number of caveats to the current study. We did not measure GM-CSF levels following direct intraventricular administration, but this approach generates considerably higher levels of circulating CSF peptide than those given by IV administration.52 Intraventricularly administered proteins may remain in the CSF compartment for many hours, if not days, while intravenously administered peptides may be cleared rapidly.53–56 A high concentration of GM-CSF administered intraventricularly may generate such a strong nonspecific response that macrophage activation may flood out any selective neuroprotective response. Additionally, while we used GM-CSF to recruit and increase extrinsic macrophage activity, the primary effect we observed in this study was an increase in intrinsic microglial activity, which may be less neuroprotective than extrinsic macrophages.12 Finally, extrinsic macrophage and intrinsic microglial activity can be either neurodegenerative (the M1 response) or neuroprotective (the M2 response). Our current results suggested that increasing macrophage activity by a relatively nonspecific cytokine such as GM-CSF is unlikely to provide any benefit to recovery. A successful approach to inflammatory-mediated ON repair and recovery likely will require selective modulation of the specific inflammatory components, which can stimulate postischemic remyelination and axonal regeneration.
Acknowledgments
The authors thank John Strong and Johanna Sotiris of the UMB core ultrastructural imaging facility for careful ultrastructural work, and Zara Mehrabyan (UMB Department of Ophthalmology) for the ImageJ-RhoA quantification and her suggestions for manuscript revision.
Supported by National Institutes of Health Grants EY-015304 and EY-019529 (SLB).
Disclosure: B.J. Slater, None; F.L. Vilson, None; Y. Guo, None; D. Weinreich, None; S. Hwang, None; S.L. Bernstein, None
References
- 1. Kerr NM, Chew SS, Danesh-Meyer HV. Non-arteritic anterior ischaemic optic neuropathy: a review and update. J Clin Neurosci. 2009; 16: 994–1000 [DOI] [PubMed] [Google Scholar]
- 2. IONDT study group Ischemic Optic Neuropathy Decompression Trial: twenty-four-month update. Arch Ophthalmol. 2000; 118: 793–798 [PubMed] [Google Scholar]
- 3. Zhang C, Guo Y, Miller NR, Bernstein SL. Optic nerve infarction and postischemic inflammation in the rodent model of anterior ischemic optic neuropathy (rAION). Brain Res. 2009; 1264: 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicholson JD, Puche AC, Guo Y, Weinreich D, Slater BJ, Bernstein SL. PGJ2 provides prolonged CNS stroke protection by reducing white matter edema. PLoS One. 2012; 7: e50021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salgado C, Vilson F, Miller NR, Bernstein SL. Cellular inflammation in nonarteritic anterior ischemic optic neuropathy and its primate model. Arch Ophthalmol. 2011; 129: 1583–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009; 40: 1849–1857 [DOI] [PubMed] [Google Scholar]
- 7. Henning EC, Ruetzler CA, Gaudinski MR. Feridex preloading permits tracking of CNS-resident macrophages after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009; 29: 1229–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldenberg-Cohen N, Kramer M, Bahar I, Monselise Y, Weinberger D. Elevated plasma levels of interleukin 8 in patients with acute anterior ischemic optic neuropathy. Br J Ophth. 2004; 88: 1538–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldenberg-Cohen N, Guo Y, Margolis FL, Miller NR, Cohen Y, Bernstein SL. Oligodendrocyte dysfunction following induction of experimental anterior optic nerve ischemia. Invest Ophthalmol Vis Sci. 2005; 46: 2716–2725 [DOI] [PubMed] [Google Scholar]
- 10. Pangratz-Fuehrer S, Kaur K, Ousman SS, Steinman L, Liao YJ. Functional rescue of experimental ischemic optic neuropathy with αB-crystallin. Eye (Lond). 2011; 6: 809–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiernan JA. Axonal and vascular changes following injury to the rat's optic nerve. J Anat. 1985; 141: 139–154 [PMC free article] [PubMed] [Google Scholar]
- 12. Yin Y, Cui Q, Li Y, et al. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003; 23: 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mi S, Lee X, Shao Z, Thill G. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004; 7: 221–228 [DOI] [PubMed] [Google Scholar]
- 14. Alblas J, Ulfman L, Hordijk P, Koenderman L. Activation of RhoA and ROCK are essential for detachment of migrating leukocytes. Mol Biol Cell. 2001; 12: 2137–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jordan J, Segura T, Brea D, Galindo MF, Castillo J. Inflammation as therapeutic objective in stroke. Curr Pharm Des. 2008; 14: 3549–3564 [DOI] [PubMed] [Google Scholar]
- 16. Busch SA, Horn KP, Silver DJ, Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009; 29: 9967–9976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Setzu A, Lathia JD, Zhao C, et al. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia. 2006; 54: 297–303 [DOI] [PubMed] [Google Scholar]
- 18. Kotter MR, Setzu A, Sim FJ, van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 2001; 35: 204–212 [DOI] [PubMed] [Google Scholar]
- 19. Yin Y, Henzl MT, Lorber B, et al. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006; 9: 843–852 [DOI] [PubMed] [Google Scholar]
- 20. Hartung HP, Jung S, Stoll G, et al. Inflammatory mediators in demyelinating disorders of the CNS and PNS. J Neuroimmunol. 1992; 40: 197–210 [DOI] [PubMed] [Google Scholar]
- 21. Khajah M, Millen B, Cara DC, Waterhouse C, McCafferty DM. Granulocyte-macrophage colony-stimulating factor (GM-CSF): a chemoattractive agent for murine leukocytes in vivo. J Leukoc Biol. 2011; 89: 945–953 [DOI] [PubMed] [Google Scholar]
- 22. Bouhy D, Malgrange B, Multon S, et al. Delayed GM-CSF treatment stimulates axonal regeneration and functional recovery in paraplegic rats via an increased BDNF expression by endogenous macrophages. FASEB J. 2007; 20: 1239–1248 [DOI] [PubMed] [Google Scholar]
- 23. Huang X, Kim JM, Kong TH, et al. GM-CSF inhibits glial scar formation and shows long-term protective effect after spinal cord injury. J Neurol Sci. 2009; 277: 87–97 [DOI] [PubMed] [Google Scholar]
- 24. Sugiyama Y, Yagita Y, Oyama N, et al. Granulocyte colony-stimulating factor enhances arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke. Stroke. 2011; 42: 770–775 [DOI] [PubMed] [Google Scholar]
- 25. Love R. GM-CSF induced arteriogenesis: a potential treatment for stroke? Lancet Neurol. 2003; 2: 458 [DOI] [PubMed] [Google Scholar]
- 26. Schabitz WR, Kruger C, Pitzer C, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood Flow Metab. 2008; 28: 29–43 [DOI] [PubMed] [Google Scholar]
- 27. Hayashi K, Ohta S, Kawakami Y, Toda M. Activation of dendritic-like cells and neural stem/progenitor cells in injured spinal cord by GM-CSF. Neurosci Res. 2009; 64: 96–103 [DOI] [PubMed] [Google Scholar]
- 28. Kruger C, Laage R, Pitzer C, Schabitz WR, Schneider A. The hematopoietic factor GM-CSF (granulocyte-macrophage colony-stimulating factor) promotes neuronal differentiation of adult neural stem cells in vitro. BMC Neurosci. 2007; 8: 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evans RD, Weston DA, McLaughlin M, Brown AM. A non-linear regression analysis method for quantitative resolution of the stimulus-evoked compound action potential from rodent optic nerve. J Neurosci Methods. 2010; 188: 174–178 [DOI] [PubMed] [Google Scholar]
- 30. Sautter J, Schwartz M, Duvdevani R, Sabel BA. GM1 ganglioside treatment reduces visual deficits after graded crush of the rat optic nerve. Brain Res. 1991; 565: 23–33 [DOI] [PubMed] [Google Scholar]
- 31. Malliri A, ten Klooster JP, Olivio C, Collard JG. Determination of the activity of Rho-like GTPases in cells. Methods Mol Biol. 2002; 189: 99–109 [DOI] [PubMed] [Google Scholar]
- 32. Reid T, Furuyashiki T, Ishizaki T, Watanabe G, et al. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996; 271: 13556–13560 [DOI] [PubMed] [Google Scholar]
- 33. Bernstein SL, Guo Y, Kelman SE, Flower RW, Johnson MA. Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2003; 44: 4153–4162 [DOI] [PubMed] [Google Scholar]
- 34. Paxinos G, Watson C. The Rat Brain, a Stereotaxic Atlas. 4th ed. New York, NY: Academic Press; 2001. [Google Scholar]
- 35. Fischer D, Petkova V, Thanos S, Benowitz LI. Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci. 2004; 24: 8726–8740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009; 50: 3860–3868 [DOI] [PubMed] [Google Scholar]
- 37. Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005; 130: 813–831 [DOI] [PubMed] [Google Scholar]
- 38. Duvdevani R, Lavie V, Segel L, Schwartz M. A new method for expressing axonal size: rat optic nerve analysis. J Electron Microsc. (Tokyo). 1993; 42: 412–414 [PubMed] [Google Scholar]
- 39. Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001; 32: 1208–1215 [DOI] [PubMed] [Google Scholar]
- 40. Frost SB, Barbay S, Mumert ML, Stowe AM, Nudo RJ. An animal model of capsular infarct: endothelin-1 injections in the rat. Behav Brain Res. 2006; 169: 206–211 [DOI] [PubMed] [Google Scholar]
- 41. Holder GE. Electrophysiological assessment of optic nerve disease. Eye. 2004; 18: 1133–1143 [DOI] [PubMed] [Google Scholar]
- 42. Parisi V, Gallinaro G, Ziccardi L, Coppola G. Electrophysiological assessment of visual function in patients with non-arteritic ischaemic optic neuropathy. Eur J Neurol. 2008; 15: 839–845 [DOI] [PubMed] [Google Scholar]
- 43. Janaky M, Fulop Z, Palffy A, Benedek K, Benedek G. Electrophysiological findings in patients with nonarteritic anterior ischemic optic neuropathy. Clin Neurophysiol. 2006; 117: 1158–1166 [DOI] [PubMed] [Google Scholar]
- 44. Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp Neurol. 1997; 148: 587–603 [DOI] [PubMed] [Google Scholar]
- 45. Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008; 28: 9330–9341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schallenberg M, Charalambous P, Thanos S. GM-CSF regulates the ERK1/2 pathways and protects injured retinal ganglion cells from induced death. Exp Eye Res. 2009; 89: 665–677 [DOI] [PubMed] [Google Scholar]
- 47. Durafourt BA, Moore CS, Zammit DA, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012; 60: 717–727 [DOI] [PubMed] [Google Scholar]
- 48. Kovari E, Gold G, Herrmann FR, et al. Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke. 2004; 35: 410–414 [DOI] [PubMed] [Google Scholar]
- 49. McDonald JW, Belegu V. Demyelination and remyelination after spinal cord injury. J Neurotrauma. 2006; 23: 345–359 [DOI] [PubMed] [Google Scholar]
- 50. Sugioka M, Sawai H, Adachi E, Fukuda Y. Changes of compound action potentials in retrograde axonal degeneration of rat optic nerve. Exp Neurol. 1995; 132: 262–270 [DOI] [PubMed] [Google Scholar]
- 51. Argyropoulou MI, Zikou AK, Tzovara I, et al. Non-arteritic anterior ischaemic optic neuropathy: evaluation of the brain and optic pathway by conventional MRI and magnetisation transfer imaging. Eur Radiol. 2007; 17: 1669–1674 [DOI] [PubMed] [Google Scholar]
- 52. Collins JM, Riccardi R, Trown P, O'Neill D, Poplack DG. Plasma and cerebrospinal fluid pharmacokinetics of recombinant interferon alpha A in monkeys: comparison of intravenous, intramuscular, and intraventricular delivery. Cancer Drug Deliv. 1985; 2: 247–253 [DOI] [PubMed] [Google Scholar]
- 53. Jones PM, Robinson IC. Differential clearance of neurophysin and neurohypophysial peptides from the cerebrospinal fluid in conscious guinea pigs. Neuroendocrinology. 1982; 34: 297–302 [DOI] [PubMed] [Google Scholar]
- 54. Zhao ML, Brosnan CF, Lee SC. 15-deoxy-delta (12, 14)-PGJ2 inhibits astrocyte IL-1 signaling: inhibition of NF-kappaB and MAP kinase pathways and suppression of cytokine and chemokine expression. J Neuroimmunol. 2004; 153: 132–142 [DOI] [PubMed] [Google Scholar]
- 55. Li W, Mase M, Inui T, et al. Pharmacokinetics of recombinant human lipocalin-type prostaglandin D synthase/beta-trace in canine. Neurosci Res. 2008; 61: 289–293 [DOI] [PubMed] [Google Scholar]
- 56. Imberti R, Cusato M, Accetta G, et al. Pharmacokinetics of colistin in cerebrospinal fluid after intraventricular administration of colistin methanesulfonate. Antimicrob Agents Chemother. 2012; 56: 4416–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]