

Abstract
Many RNA viruses have evolved the ability to inhibit host cell transcription as a means to circumvent cellular defenses. For the study of these viruses, it is therefore important to have a quick and reliable way of measuring transcriptional activity in infected cells. Traditionally, transcription has been measured either by incorporation of radioactive nucleosides such as 3H-uridine followed by detection via autoradiography or scintillation counting, or incorporation of halogenated uridine analogs such as 5-bromouridine (BrU) followed by detection via immunostaining. The use of radioactive isotopes, however, requires specialized equipment and is not feasible in a number of laboratory settings, while the detection of BrU can be cumbersome and may suffer from low sensitivity.
The recently developed click chemistry, which involves a copper-catalyzed triazole formation from an azide and an alkyne, now provides a rapid and highly sensitive alternative to these two methods. Click chemistry is a two step process in which nascent RNA is first labeled by incorporation of the uridine analog 5-ethynyluridine (EU), followed by detection of the label with a fluorescent azide. These azides are available as several different fluorophores, allowing for a wide range of options for visualization.
This protocol describes a method to measure transcriptional suppression in cells infected with the Rift Valley fever virus (RVFV) strain MP-12 using click chemistry. Concurrently, expression of viral proteins in these cells is determined by classical intracellular immunostaining. Steps 1 through 4 detail a method to visualize transcriptional suppression via fluorescence microscopy, while steps 5 through 8 detail a method to quantify transcriptional suppression via flow cytometry. This protocol is easily adaptable for use with other viruses.
Keywords: Immunology, Issue 78, Virology, Chemistry, Infectious Diseases, Biochemistry, Genetics, Molecular Biology, Cellular Biology, Medicine, Biomedical Engineering, Arboviruses, Bunyaviridae, RNA, Nuclear, Transcription, Genetic, Rift Valley fever virus, NSs, transcription, click chemistry, MP-12, fluorescence microscopy, flow cytometry, virus, proteins, immunostaining, assay
Introduction
Traditionally, transcriptional activity has been measured through incorporation of either radioactive (3H-uridine)1 or halogenated (BrU)2 nucleosides into nascent RNA. These uridine analogs are taken up by cells, converted into uridine-triphosphate (UTP) through the nucleoside salvage pathway, and subsequently incorporated into newly synthesized RNA. 3H-uridine can be detected by autoradiography, which can be time-consuming, or scintillation counting, which requires specialized equipment. Furthermore, the use of radioactive isotopes may not be practical in a number of laboratory settings, including high containment biosafety laboratories. BrU can be detected through immunostaining with anti-BrU antibodies, which may require harsh permeabilization to ensure access of the antibody to the nucleus or partial denaturation of RNA, as RNA secondary structure might be a steric hindrance for antibody binding.
The protocol described here utilizes the recently developed click chemistry3 to measure transcriptional activity in cells infected with the RVFV strain MP-12. Click chemistry relies on a highly selective and efficient copper(I)-catalyzed cycloaddition reaction between an alkyne and an azide moiety4,5. In this protocol, nascent RNA in infected cells is first labeled through incorporation of the uridine analog EU. In a second step, the incorporated EU is detected through reaction with a fluorescent azide. The main advantages of this method are (1) that it does not require the use of radioactive isotopes and (2) that it does not require harsh permeabilization or denaturation of RNA. With a molecular weight of less than 50 Da, access of the fluorescent azide is not impeded by insufficient permeabilization or RNA secondary structure. Furthermore, researchers are not limited in their choice of primary antibodies when combining the detection of RNA with a classical immunostaining for viral proteins.
Two different methods are described in this protocol: one for visualizing transcription via fluorescence microscopy (steps 1-4), and one for quantifying transcription through flow cytometry (steps 5-8). Both of these methods combine labeling of nascent RNA with an intracellular immunostaining for viral proteins, which allows for a correlation between transcriptional activity and viral infection on a cell-by-cell basis.
The authors have successfully used the protocol described here to determine transcriptional activity in cells infected with the RVFV strain MP-126, cells infected with different MP-12 mutants7,8, 9, and cells infected with Toscana virus (TOSV)10. The protocol described here can be easily adapted for use with different viruses and has the potential to be modified to include immunofluorescence staining of specific viral or cellular proteins.
Protocol
Analysis of transcription by fluorescence microscopy
1. Infect Cells with MP-12 and Label Nascent RNA with EU
Place 12-mm round coverslips into 12-well tissue culture plate, one coverslip per well.
Note: If cells have trouble adhering to coverslips, coat with poly-L-lysine (MW ≥ 70,000) before placing in wells. To this aim, submerge coverslips in a sterile 0.1 mg/ml solution in H2O of poly-L-lysine for 5 min. Rinse coverslips with sterile H2O and air-dry for at least 2 hr.
To seed 293 cells onto coverslips, add 5 x 104 cells in 1 ml growth medium (DMEM containing 10% fetal bovine serum, 100 U/ml Penicillin and 100 mg/ml Streptomycin) per well. Incubate in a humidified incubator at 37 °C and 5% CO2 O/N.
Infect cells with MP-12 at a multiplicity of infection (m.o.i.) of 3. Include at least two uninfected wells: one of the two wells will serve as the uninfected control, the other will be treated with actinomycin D (ActD) at step 1.5 as a control for transcriptional suppression. Remove growth medium and dilute the virus stock so that the total volume added to each well is 200 ml. Incubate for 1 hr in a humidified incubator at 37 °C and 5% CO2.
Note: Instead of infecting cells, cells can also be transfected with plasmid DNA or in vitro synthesized RNA encoding a specific viral protein. To this aim, transfect cells with an appropriate chemical transfection reagent according to the manufacturer's instructions and proceed to step 1.5 at 16 hr post transfection. It is advisable to optimize the transfection conditions and incubation time before RNA labeling and fixation.
Remove inoculum and add 1 ml fresh growth medium per well. Incubate at 37 °C for 15 hr.
Note: If adapting this protocol for use with another virus, it is advisable to perform a time course experiment to determine the optimal time point for RNA labeling and fixation. Set up this time course so that the infection times are staggered and all samples can be labeled, fixed, and stained at the same time.
At 15 hr post infection (h.p.i.), replace growth medium with medium containing 1 mM EU. To one of the mock infected wells, also add 5 μg/ml ActD. This will serve as the control for transcriptional suppression, as ActD inhibits DNA-dependent RNA synthesis. Return cells to the incubator.
At 16 h.p.i., remove the medium and wash the coverslips once with 1 ml PBS (KCl 0.20 g/L, KH2PO4 0.20 g/L, NaCl 8.00 g/L, Na2HPO4 1.15 g/L, pH 7.4) per well. Fix cells by adding 1 ml of 4% paraformaldehyde (PFA) per well and incubating for 30 min at RT.
Note: To prepare 4% PFA fixation solution, dissolve 10 g paraformaldehyde in 150 ml H2O heated to 60 °C on a heated magnetic stirrer. Add 500 μl of 1M NaOH and continue to stir until solution becomes clear. Filter PFA solution through a paper filter to remove particulates and add 62.5 ml phosphate buffer (77 mM NaH2PO4, 287 mM Na2HPO4, pH 7.4). Add H2O up to a total volume of 250 ml. Freeze at -20 °C in single-use aliquots.
Wash once with 1 ml PBS per well. No incubation time is required for this and all subsequent wash steps. Proceed immediately to step 2.
Note: It is important to immediately proceed with the click reaction, as the RNA degenerates if kept at this stage for prolonged periods of time; a loss of the RNA fluorescence signal can already be observed if cells are kept for 1 hr at 4 °C. Similarly, if performing a time course experiment, be sure to label, fix, and stain all samples at the same time.
2. Click Reaction to Detect Labeled RNA
Permeabilize cells by adding 1 ml 0.2% Triton X-100 in PBS. Incubate for 10 min at RT.
Note: All solutions used in steps 2.1 to 2.3 need to be nuclease free.
Wash three times with 1 ml PBS per well.
Remove coverslips from 12-well plate and transfer into humid chamber. Cover each coverslip with 50 - 100 μl click staining solution (100 mM Tris pH 8.5, 1 mM CuSO4, 20 μM fluorescent azide [excitation/emission maximum: 590/617 nm], 100 mM ascorbic acid) each. Incubate for 1 hr at RT in the dark.
Note: To assemble the humid chamber, line the bottom of a cell culture or petri dish with a 10 cm diameter with plastic paraffin film. Line the inside of the edge of the dish with damp paper towels. Place the coverslips onto the plastic paraffin film.
Note: Be careful not to let the coverslips dry out during this or any other step in the protocol. It is best to add the staining solution to each coverslip directly after it has been placed in the humid chamber.
Note: Prepare the click staining solution fresh immediately prior to this step. Stock solutions of 1.5 M Tris pH 8.5, 100 mM CuSO4, and 500 mM ascorbic acid may be stored at 4 °C. However, discard any ascorbic acid stock solution that has turned yellow.
Note: Be sure to select a fluorophore that matches the filters on your fluorescent microscope.
Remove coverslips from humid chamber and place into a fresh 12-well plate and wash three times with 1 ml PBS per well.
Note: If no detection of viral proteins is desired, coverslips can be mounted and imaged after this step (proceed to step 3.3).
3. Immunofluorescence to Detect Expression of Viral Proteins
Add 1 ml of blocking buffer (3% (w/v) bovine serum albumin (BSA) in PBS) to each well and incubate for 30 min at RT in the dark.
Remove coverslips from 12-well plate, transfer into humid chamber and cover with 50 - 100 μl of primary antibody (anti-RVFV, a kind gift from Dr. R.B. Tesh, UTMB) diluted in blocking buffer (1:500). Incubate for 1 hr at RT in the dark.
Note: When adapting this protocol for use with a different virus, determine the optimal dilution for your primary antibody first.
Note: Alternatively, this step can be performed O/N at 4 °C in the dark.
Remove coverslips from humid chamber and place back into 12-well plate and wash three times with 1 ml PBS per well.
Remove coverslips from 12-well place, place into humid chamber and cover with 50 - 100 μl of secondary antibody (anti-mouse IgG conjugated to fluorescent dye [excitation/emission maximum: 495/519 nm]) diluted in blocking buffer (1:500). Incubate for 1 hr at RT in the dark.
Note: Be sure to select a secondary antibody that can recognize your primary antibody and matches the filters on your fluorescent microscope.
Note: If desired, 200 ng/ml DAPI can be added to the secondary antibody dilution to visualize nuclei.
Remove coverslips from humid chamber and place back into 12-well plate and wash three times with 1 ml PBS per well.
Mount coverslips by placing one drop (ca. 15 μl) of Fluoromount-G onto a microscope slide. Dip the coverslip into H2O, remove excess H2O by touching the side against a paper towel, and place cell-side down into the drop of Fluoromount-G. Air dry for 5 min.
Note: If imaging on an inverted microscope, allow Fluoromount-G to solidify at 4 °C O/N or affix the coverslips to the microscope slide by sealing the edge with transparent nail polish.
Note: Slides can be imaged immediately or stored at 4 °C in the dark for up to a week.
4. Acquisition of Data
Image using a fluorescent microscope.
Note: Be sure to select appropriate fluorophores which can be visualized using your microscope. For reference, the following are the fluorophores and filter sets used to acquire the images shown in this publication.
DAPI : Excitation filter: BP 330 - 385 Dichromatic mirror: DM 400 Emission filter: LP 420
Fluorophore with an excitation/emission maximum of 495/519: Excitation filter: BP 460 - 490 Dichromatic mirror: DM 505 Emission filter: LP 510
Fluorophore with an excitation/emission maximum of 590/617: Excitation filter: BP 545 - 580 Dichromatic mirror: DM 600 Emission filter: LP 610
Analysis of transcription by flow cytometry
5. Infect Cells with MP-12 and Label Nascent RNA with EU
Seed 293 cells into 6-well tissue culture plate in growth medium (DMEM containing 10% fetal bovine serum, 100 U/ml Penicillin, 100 μg/ml Streptomycin). Incubate in a humidified incubator at 37 °C and 5% CO2 until cells have reached 80 - 100% confluency.
Infect cells with MP-12 at an m.o.i. of 3. Include at least two uninfected wells: one of the two wells will serve as the uninfected control, the other will be treated with ActD at step 5.4. Remove growth medium and dilute the virus stock so that the total volume added to each well is 400 μl. Incubate for 1 hr in a humidified incubator at 37 °C and 5% CO2.
Remove inoculum and add 2 ml fresh growth medium per well. Incubate at 37 °C for 12 hr.
Note: If adapting this protocol for use with another virus, it is advisable to perform a time course experiment to determine the optimal time point for labeling and harvesting. Set up this time course so that the infection times are staggered and all samples can be labeled, harvested, and stained at the same time.
At 12 h.p.i., replace growth medium with medium containing 0.5 mM EU. To one of the mock infected wells, also add 5 μg/ml ActD. This will serve as the control for transcriptional suppression, as ActD inhibits DNA-dependent RNA synthesis. Return cells to the incubator.
At 13 h.p.i., wash cells once with PBS and harvest by trypsinization of cells. Wash harvested cells three times with PBS containing 1 mM EDTA (PBS-EDTA).
Note: During this and subsequent steps, do not centrifuge cells faster than 500 - 1,000 x g to prevent cell breakage. Centrifuge cells for 1 - 2 min to sediment.
Note: If a surface immunostaining is planned following the click reaction, harvest using a rubber policeman or disposable cell scraper instead.
Fix cells by resuspending them in 1 ml of 4% PFA and incubate for 30 min at RT. Refer to step 1.6 for instructions on how to prepare 4% PFA.
Wash once with 1 ml PBS-EDTA per well. Proceed immediately to step 6.
Note: It is important to immediately proceed with the click reaction, as the RNA degenerates if kept at this stage for prolonged periods of time. Similarly, if performing a time course experiment, be sure to label, fix and stain all samples at the same time.
6. Click Reaction to Detect Labeled RNA
Permeabilize cells by resuspending in 0.5 ml of 0.2% Triton X-100 in PBS. Incubate for 10 min at RT.
Note: All solutions used in steps 6.1 to 6.3 need to be nuclease free.
Note: If cells do not sediment properly during this step, increase the centrifugation time to 5 min. Sedimentation behavior will improve once cells are suspended in FACS buffer (step 6.4).
Wash once with 1 ml PBS.
Note: Do not use EDTA in this step as this will chelate the Cu2+ ions necessary for the click reaction.
Resuspend cells in 200 μl click staining solution (100 mM Tris pH 8.5, 1 mM CuSO4, 200nM azide labeled with fluorescent dye [excitation/emission maximum: 650/665 nm], 100 mM ascorbic acid). Incubate for 1 hr at RT in the dark.
Note: Prepare the click staining solution fresh immediately prior to this step. Stock solutions of 1.5 M Tris pH 8.5, 100 mM CuSO4, and 500 mM ascorbic acid may be stored at 4 °C. However, discard any ascorbic acid stock solution that has turned yellow.
Note: If cells clump together during this step, resuspend by pipetting up and down several times.
Note: Be sure to select a fluorophore that can be detected by your flow cytometer.
Note: Also prepare one sample of infected cells which are not subjected to the click staining procedure; these will be needed for calibration of the flow cytometer. Either use half of the infected cells or include an additional infected well at step 5.2. These control cells will be immunostained at step 7.1.
Wash twice with FACS buffer (PBS containing 1 mM EDTA and 0.5% (w/v) BSA).
Note: If no immunostaining of viral proteins is desired, cells can be analyzed immediately (proceed to step 8).
7. Immunofluorescence to Detect Expression of Viral Proteins
Resuspend cells in 200 μl of primary antibody (anti-RVFV) diluted in FACS buffer (1:10,000) and incubate for 1 hr at RT in the dark.
Note: Alternatively, this step can be performed O/N at 4 °C in the dark.
Note: Also prepare one sample of uninfected cells which were subjected to the click staining procedure but will not be immunostained, these will be needed for calibration of the flow cytometer. Either use half of the mock infected cells or include an additional uninfected well at step 5.2.
Note: When adapting this protocol for use with a different virus, determine the optimal dilution for your primary antibody first.
Wash cells twice in FACS buffer.
Resuspend cells in 200 μl of secondary antibody (anti-mouse IgG conjugated to fluorescent dye [excitation/emission maximum: 495/519 nm]) diluted in FACS buffer (1:1,000) and incubate for 1 hr at RT in the dark.
Wash cells once in FACS buffer.
Note: At this point, cells can be stored O/N at 4 °C in the dark.
8. Acquisition of Data
Resuspend cells in 500 μl FACS buffer, transfer into 5 ml polystyrene tubes and analyze using a flow cytometer. Use MP-12 infected cells (no click reaction, anti-RVFV staining only) and mock infected cells (click reaction only) to calibrate the instrument.
Note: Be sure to select appropriate fluorophores which can be detected by your instrument. For reference, the following are the laser lines and filter sets used to acquire the data shown in this publication.
Fluorophore with an excitation/emission spectrum of 495/519: Laser: 488 nm Filter: 530/30
Fluorophore with an excitation/emission spectrum of 650/665: Laser: 640 nm Filter: 670/20
Representative Results
The NSs protein of RVFV (family Bunyaviridae, genus Phlebovirus) 11 inhibits host cell general transcription through two distinct mechanisms: (i) by sequestering the p44 subunit of the basal transcription factor TFIIH11 and (ii) by promoting the proteasomal degradation of the p62 subunit of TFIIH 6. The RVFV MP-12 vaccine strain 13 has been used for the experiments described in this protocol since MP-12 strain can be handled in a biosafety level 2 laboratory and is excluded from the select agent rule in the U.S., which applies to other wild-type RVFV strains. The NSs protein of MP-12 strain is fully functional and retains the ability to suppress host transcription6. The virus rMP12-C13type, which carries a deletion encompassing 69% of the NSs gene 14, has been included as a control virus lacking all NSs functions, including the ability to suppress host cell transcription.
Figure 1 shows images acquired by fluorescence microscopy. In mock infected cells (Figures 1a-1d), nuclei appear bright red due to ongoing transcription. Since cells have only been labeled with EU for 1 hr, followed by immediate fixation, most RNAs remain in the nucleus. In contrast, when cells have been treated with ActD to pharmacologically inhibit transcription (Figures 1e-1h), the red fluorescence of nuclei is markedly reduced. This fluorescence is similarly reduced when cells are infected with MP-12 (Figures 1n-1q), but not with the NSs deletion mutant rMP12-C13type (Figures 1i-1m). Figure 2 also shows images acquired by fluorescence microscopy, but here cells have been transfected with in vitro synthesized mRNA instead of infection with live virus. When cells were transfected with mRNA encoding GFP (Figures 2g-2i), no change in transcriptional activity compared to untransfected cells (Figures 2a-2c), as visualized by red fluorescence in the nuclei, can be detected. Only transfection with mRNA encoding the RVFV virulence factor NSs (Figures 2k-2m) results in decreased red fluorescence.
Figure 3A depicts quantitative data obtained by flow cytometry. The data is represented as scatter plots with fluorescent signal for EU incorporation into nascent RNA plotted on the y-axis and that for anti-RVFV staining plotted on the x-axis. The quadrant gates were set so that the majority of mock infected cells was situated in the upper left quadrant, the majority of ActD treated cells was situated in the lower left quadrant, and the majority of infected cells was situated in the right half of the plot. When cells were infected with MP-12, 81.5% of total cells, or 93% of anti-RVFV positive cells, showed reduced transcriptional activity. In contrast, when cells were infected with rMP12-C13type, 91.6% of total cells, or 97% of anti-RVFV positive cells, showed transcriptional activity which was comparable to that of mock infected cells. Figure 3B depicts the same data as Figure 3A in the form of a histogram for fluorescence RNA labeling with EU incorporation.
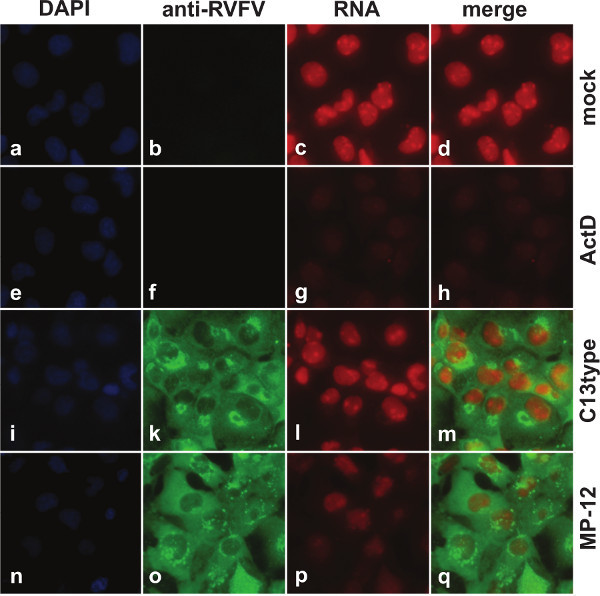 Figure 1. Fluorescence microscopy of cells infected with MP-12 and rMP12-C13type. 293 cells were mock infected or infected with either MP-12 or the NSs deletion mutant rMP-12-C13type. Between 15 and 16 h.p.i., cells were labeled with 1 mM EU. As a control for transcriptional suppression, mock infected cells were treated with 5 mg/ml ActD concurrently with the EU treatment. Nuclei are visualized through DAPI staining (blue), expression of viral proteins is visualized by staining with anti-RVFV antibodies (green), and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 590/617 nm) (red).
Figure 1. Fluorescence microscopy of cells infected with MP-12 and rMP12-C13type. 293 cells were mock infected or infected with either MP-12 or the NSs deletion mutant rMP-12-C13type. Between 15 and 16 h.p.i., cells were labeled with 1 mM EU. As a control for transcriptional suppression, mock infected cells were treated with 5 mg/ml ActD concurrently with the EU treatment. Nuclei are visualized through DAPI staining (blue), expression of viral proteins is visualized by staining with anti-RVFV antibodies (green), and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 590/617 nm) (red).
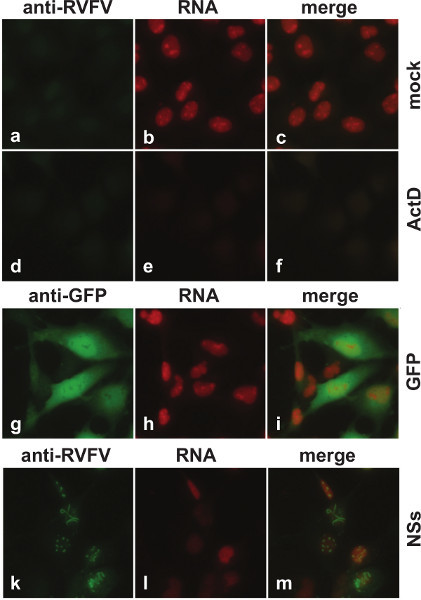 Figure 2. Fluorescence microscopy of cells transfected with in vitro synthesized RNA for either GFP or MP-12 NSs. 293 cells were mock transfected, or transfected with in vitro synthesized RNA coding for either GFP or MP-12 NSs. Between 15 and 16 hr posttransfection, cells were labeled with 1 mM EU. As a control for transcriptional suppression, mock transfected cells were treated with 5 mg/ml ActD concurrently with the EU treatment. The expression of GFP is visualized by staining with an anti-GFP antibody (g-i), the expression of NSs is visualized by staining with anti-RVFV antibodies (a-f, k-m) followed by anti-mouse IgG conjugated to fluorescent dye (excitation/emission maximum: 495/519 nm) (green), and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 590/617 nm) (red).
Figure 2. Fluorescence microscopy of cells transfected with in vitro synthesized RNA for either GFP or MP-12 NSs. 293 cells were mock transfected, or transfected with in vitro synthesized RNA coding for either GFP or MP-12 NSs. Between 15 and 16 hr posttransfection, cells were labeled with 1 mM EU. As a control for transcriptional suppression, mock transfected cells were treated with 5 mg/ml ActD concurrently with the EU treatment. The expression of GFP is visualized by staining with an anti-GFP antibody (g-i), the expression of NSs is visualized by staining with anti-RVFV antibodies (a-f, k-m) followed by anti-mouse IgG conjugated to fluorescent dye (excitation/emission maximum: 495/519 nm) (green), and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 590/617 nm) (red).
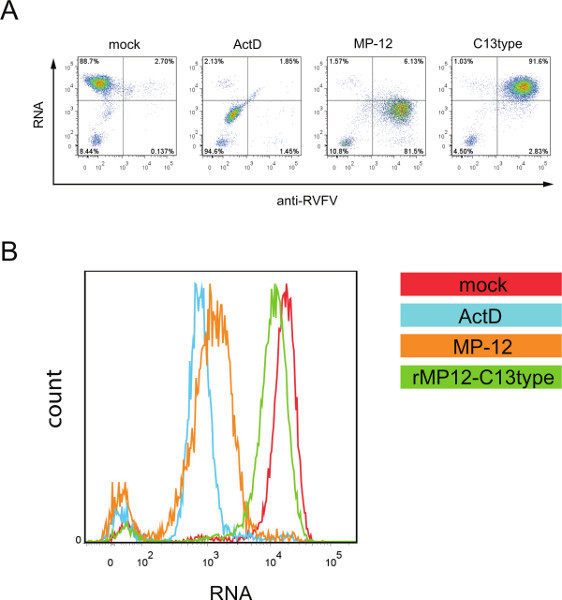 Figure 3. (A) Flow cytometric analysis of cells infected with MP-12 or rMP12-C13type. Cells were mock infected, or infected with either MP-12 or rMP12-C13type. Between 12 and 13 h.p.i., cells were labeled with 0.5 mM EU. As a control for transcriptional suppression, mock transfected cells were treated with 5 μg/ml ActD concurrently with the EU treatment. Expression of viral proteins is visualized by staining with anti-RVFV antibodies followed by a secondary antibody labeled with fluorescent dye (excitation/emission maximum: 495/519 nm) (anti-RVFV) and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 650/665 nm) (RNA). The data is represented as a scatter plot. (B) Representation of the same data shown in A as a histogram. Fluorescence intensity for EU incorporation is plotted on the x-axis, cell counts are plotted on the y-axis. Click here to view larger figure.
Figure 3. (A) Flow cytometric analysis of cells infected with MP-12 or rMP12-C13type. Cells were mock infected, or infected with either MP-12 or rMP12-C13type. Between 12 and 13 h.p.i., cells were labeled with 0.5 mM EU. As a control for transcriptional suppression, mock transfected cells were treated with 5 μg/ml ActD concurrently with the EU treatment. Expression of viral proteins is visualized by staining with anti-RVFV antibodies followed by a secondary antibody labeled with fluorescent dye (excitation/emission maximum: 495/519 nm) (anti-RVFV) and RNA is visualized by detection of the incorporated EU with azide labeled with fluorescent dye (excitation/emission maximum: 650/665 nm) (RNA). The data is represented as a scatter plot. (B) Representation of the same data shown in A as a histogram. Fluorescence intensity for EU incorporation is plotted on the x-axis, cell counts are plotted on the y-axis. Click here to view larger figure.
Discussion
The provided protocol describes a method to measure the effect of viral infection on host cell transcription via incorporation of the uridine analog EU into nascent RNA. This method has several advantages over previous methods: it is fast, sensitive, and it does not rely on the use of radioactive isotopes. Furthermore, the method can be adapted to yield qualitative data through fluorescence microscopy or quantitative data via flow cytometry using essentially the same reagents. When combined with immunofluorescence staining for viral antigens, this method also allows the researcher to differentiate infected from uninfected cells.
It is important to note that this method also labels newly synthesized viral RNA at the same rate as it labels host transcripts, a drawback which is shared with other methods relying on 3H-uridine or BrU incorporation. At least in the case of RVFV MP-12 infection, however, we have found that the amount of host transcripts by far outweighs the amount of viral transcripts, and thus the influence of viral RNA on total RNA measured by this assay is negligible. When adapting this protocol for use with another virus, researchers may wish to identify viral transcripts either through their localization, through treatment of infected cells with ActD, or though fluorescent in-situ hybridization (FISH)15. If the virus replicates in the cytoplasm, such as MP-12, then viral RNA can be easily distinguished from cellular RNA, which is still mostly contained in the nucleus after 1 hr of labeling. Infected cells can also be treated with ActD to suppress host cell transcription while leaving viral RNA synthesis unaffected. ActD functions by binding to double-stranded DNA16 and therefore only inhibits DNA-dependent transcription but has no effect on viral RNA-dependent RNA polymerases.
When performing this protocol, it is critical to avoid contamination with nucleases between the permeabilization and the click labeling reaction, as EU labeled RNA remains sensitive to degradation by RNases 3. Furthermore, researchers should proceed immediately to the click labeling reaction, as prolonged incubation times between fixation and click labeling reaction, even at 4 °C, lead to degradation of RNA and ultimately result in a loss of the RNA fluorescence signal. This is of special concern when performing time-course experiments; researchers should design their experiments so that all cells can be labeled, fixed, and stained at the same time. Fortunately, the RNA fluorescence signal remains stable after the click labeling reaction, so the protocol can easily be stopped after this step.
Finally, researchers need to keep in mind that the cytopathic effect (CPE) caused by many viruses negatively affects cellular RNA synthesis even in the absence of any specific transcriptional suppression. It is therefore important to measure transcriptional activity at a time post infection when cells are still viable. If a mutant virus is available which lacks the ability to suppress transcription, such as rMP-12-C13type (Figures 1 and 3), this can be a great tool in determining the optimal time point for measuring transcription.
When planning a large number of experiments, researchers might want to consider direct labeling of their primary antibody with a fluorochrome17, thereby eliminating the need for a secondary antibody and reducing the time required for the detection of viral antigens.
In summary, this protocol provides a rapid and reliable way to measure the effect of viral infection on host cell transcription. It can easily be adapted for use with different viruses and has the potential to be modified to include immunostainings for specific viral or cellular proteins.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank R.B. Tesh for providing the mouse polyclonal anti-RVFV antiserum and M. Griffin of the UTMB Flow Cytometry Core Facility for help with flow cytometry. This work was supported by 5 U54 AI057156 through the Western Regional Center of Excellence, NIH grant R01 AI08764301, and funding from the Sealy Center for Vaccine Development at UTMB.B.K. was supported by the James W. McLaughlin Fellowship Fund at UTMB.O.L. was supported by a Maurice R. Hilleman Early-Stage Career Investigator Award.
References
- Fakan S. Structural support for RNA synthesis in the cell nucleus. Methods Achiev. Exp. Pathol. 1986;12:105–140. [PubMed] [Google Scholar]
- Jensen PO, Larsen J, Christiansen J, Larsen JK. Flow cytometric measurement of RNA synthesis using bromouridine labelling and bromodeoxyuridine antibodies. Cytometry. 1993;14:455–458. doi: 10.1002/cyto.990140416. [DOI] [PubMed] [Google Scholar]
- Jao CY, Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Tornoe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Kalveram B, Lihoradova O, Ikegami T. NSs protein of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 2011;85:6234–6243. doi: 10.1128/JVI.02255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalveram B, et al. Rift Valley fever virus NSs inhibits host transcription independently of the degradation of dsRNA-dependent protein kinase PKR. Virology. 2012. [DOI] [PMC free article] [PubMed]
- Head JA, Kalveram B, Ikegami T. Functional analysis of Rift Valley fever virus NSs encoding a partial truncation. PLoS One. 2012;7:e45730. doi: 10.1371/journal.pone.0045730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lihoradova OA, et al. Characterization of Rift Valley Fever Virus MP-12 Strain Encoding NSs of Punta Toro Virus or Sandfly Fever Sicilian Virus. PLoS Negl Trop Dis. 2013;7:e2181. doi: 10.1371/journal.pntd.0002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalveram B, Ikegami T. Toscana Virus NSs Protein Promotes Degradation of Double-Stranded RNA-Dependent Protein Kinase. J. Virol. 2013. [DOI] [PMC free article] [PubMed]
- Schmaljohn C, Nichol S. In: In Fields Virology. Knipe DM, et al., editors. Lippincott Williams and Wilkins; 2007. pp. 1741–1789. [Google Scholar]
- Le May N, et al. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116:541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- Caplen H, Peters CJ, Bishop DH. Mutagen-directed attenuation of Rift Valley fever virus as a method for vaccine development. J. Gen. Virol. 1985;66:2271–2277. doi: 10.1099/0022-1317-66-10-2271. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol. 2006;80:2933–2940. doi: 10.1128/JVI.80.6.2933-2940.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyboh K, Ajamian L, Mouland AJ. Detection of viral RNA by fluorescence in situ hybridization (FISH) J. Vis. Exp. 2012. p. e4002. [DOI] [PMC free article] [PubMed]
- Reich E, Goldberg IH. Actinomycin and nucleic acid function. Prog. Nucleic Acid Res. Mol. Biol. 1964;3:183–234. doi: 10.1016/s0079-6603(08)60742-4. [DOI] [PubMed] [Google Scholar]
- Holmes KL, Lantz LM, Russ W. Conjugation of fluorochromes to monoclonal antibodies. Curr. Protoc. Cytom. 2001;Chapter 4(Unit 4 2) doi: 10.1002/0471142956.cy0402s00. [DOI] [PubMed] [Google Scholar]