

Abstract
There is an urgent need to develop novel therapies for controlling chronic virus infections in immunocompromised patients. Disease associated with persistent γ-herpesvirus infection (EBV, human herpesvirus 8) is a significant problem in AIDS patients and transplant recipients, and clinical management of these conditions is difficult. Immune surveillance failure followed by γ-herpes-virus recrudescence can be modeled using murine γ-herpesvirus (MHV)-68 in mice lacking CD4+ T cells. In contrast with other chronic infections, no obvious defect in the functional capacity of the viral-specific CD8+ T cell response was detected. We show in this article that adoptive transfer of MHV-68–specific CD8+ T cells was ineffective at reducing the viral burden. Together, these indicate the potential presence of T cell extrinsic suppressive factors. Indeed, CD4-depleted mice infected with MHV-68 express increased levels of IL-10, a cytokine capable of suppressing the function of both APCs and T cells. CD4-depleted mice developed a population of CD8+ T cells capable of producing IL-10 that suppressed viral control. Although exhibiting cell surface markers indicative of activation, the IL-10–producing cells expressed increased levels of programmed death-1 but were not enriched in the MHV-68–specific compartment, nor were they uniformly CD44hi. Therapeutic administration of an IL-10R blocking Ab enhanced control of the recrudescent virus. These data implicate IL-10 as a promising target for the restoration of immune surveillance against chronic γ-herpesvirus infection in immunosuppressed individuals.
A immune response, instead persisting in the host with and EBV have the ability to escape the normally efficient variety of viruses such as hepatitis C virus (HCV), HIV, potentially dire consequences (1). Events leading to persistent infection remain poorly understood because T cell responses are generally competent at controlling intracellular pathogens such as viruses (2). One reason that these responses fail to control chronic infection is exhaustion of the antiviral T cell response (3–6). Chronic Ag stimulation of CD8+ T cells results in a gradual loss of functionality including the ability to proliferate, kill infected cells, and produce antiviral cytokines, but the degree of exhaustion varies according to the identify of the persisting pathogen and the inhibitory mechanisms involved in inhibiting the antiviral T cell response (6–11).
IL-10 has received particular attention for its correlation with persistent viral infections and for its ability to attenuate immune responses (12). Polymorphisms that cause increased IL-10 production have been associated with susceptibility to chronic HCV infection and severity of hepatitis B virus infection, whereas polymorphisms that reduce IL-10 expression associate with slower progression to AIDS (13–16). The effects of blocking the IL-10 pathway have been explored in several chronic virus infection models. Studies with lymphocytic choriomeningitis virus clone 13 and murine CMV indicated that Ab blockade or genetic removal of IL-10 signaling greatly improved the ability of mice to control infection through increased cytokine production and an increase in viral-specific T cells (17–20). IL-10 has also been implicated in persistent human infections, because CD4+ T cells from patients infected with HIV or HCV have enhanced responses on blockade of IL-10 signaling in vitro (21, 22). IL-10 affects a variety of APCs through alteration of MHC presentation and decreased inflammatory cytokine production (12). However, direct effects on CD8+ T cells have also been reported with Listeria monocytogenes infection (23). The source of IL-10 during chronic infections varies with the model studied because both myeloid and lymphocyte populations have been implicated in producing IL-10 (12).
Persistent infection of B lymphocytes with EBV is controlled mainly by CD8+ T cells, and patients are generally asymptomatic into advanced age (24). Conversely, immunosuppressed patients, such as those undergoing transplantation, can experience development of EBV-associated lymphoproliferative disease. In addition, the progression of HIV infection to AIDS can be accompanied by γ-herpesvirus–associated lymphomas (25). The development of AIDS is concomitant with declining CD4+ T cell numbers, and both human and mouse model data support the hypothesis that CD4 help is critical for controlling persistent viral infections (26, 27). However, the exact mechanisms by which CD4 T cell help promotes long-term immune surveillance and how immune surveillance of persistent infection breaks down in the absence of these cells remain unclear.
It is possible to dissect the interaction between CD4+ and CD8+ T cells in γ-herpesvirus infection through the use of the murine γ-herpesvirus (MHV) model, MHV-68. When mice lacking CD4+ T cells are infected with MHV-68, they control the initial viral burden with comparable kinetics to wild-type (WT) mice. However, control ultimately breaks down and the virus reactivates by day 40 postinfection (p.i.), whereas no detectable virus can be found in the lungs of WT animals. Interestingly, no decrease in Ag-specific CD8+ T cells or cytotoxicity has been reported for mice that lack CD4 help (28–30). In addition, therapeutic vaccination with recombinant vaccinia virus expressing an MHV-68 epitope did not reduce the viral burden despite dramatically expanding the Ag-specific population (31). Several β- and γ-herpesviruses such as human CMV and EBV have encoded IL-10 homologs in their genomes (32, 33). Although MHV-68 does not encode an IL-10 homolog, it does encode the M2 protein, which induces IL-10–dependent B cell proliferation, perhaps facilitating the establishment of virus latency in memory B cells (34). IL-10–deficient mice infected with MHV-68 have reduced peak lytic viral levels compared with WT controls; however, they also experience enhanced splenomegaly and lymphocytosis (35). In addition, MHV-68–induced IL-10 production has been shown to inhibit the ability of dendritic cells to fully activate T cell responses (36).
Our studies aimed to explore the role of known mechanisms of immune suppression on the MHV-68 CD8+ T cell response in CD4-depleted mice. Interestingly, CD4-depleted mice produced increased levels of IL-10 compared with intact animals and contained a population of CD8+ T cells that were capable of producing IL-10. Therapeutic blockade of IL-10R at late time points improved control over the viral replication in the lungs of CD4-depleted mice. These data indicate an important link between CD4+ T cell help and IL-10 production from a subset of CD8+ T cells, which is an important factor limiting long-term control of MHV-68 infection.
Materials and Methods
Mice and virus infections
MHV-68 virus (clone G2.4) was originally obtained from Dr. A.A. Nash (University of Edinburgh, U.K.). The virus was grown and titered using NIH3T3 cells as previously described (37). For MHV-68 infections, mice were given 400 PFU intranasally under anesthesia with either ketamine/xylazine or isoflurane. C57BL/6 and congenic B6-Ly5.2-CR mice were purchased from the National Cancer Institute (Bethesda, MD). IL-10 Thy1.1 reporter mice were obtained from Dr. Casey Weaver (University of Alabama, Birmingham, AL). IL-10R1−/− mice were obtained from Dr. Joonsoo Kang (University of Massachusetts Medical Center, Worcester, MA). MHC class II−/− mice were obtained from Taconic Laboratory. IL-10−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were bred and/or housed in the Dartmouth-Hitchcock Medical Center mouse facility. The Animal Care and Use Program of Dartmouth College approved all animal experiments.
CD4 depletion
For depletion of CD4+ T cells, mice were treated with 500 μg anti-CD4 (GK1.5) on days −1 and 0 relative to infection, followed by 250 μg on day 3 p.i. and twice weekly thereafter. Control mice were either untreated or given rat IgG (Jackson ImmunoResearch, West Grove, PA).
Tissue preparation
Single-cell suspensions of spleens were prepared by passing through cell strainers. Lungs were injected with 1 ml MEM containing 417.5 μg/ml Liberase CI and 200 μg/ml DNase I (Roche, Indianapolis, IN). Tissues were minced with scissors and then incubated for 30 min at 37°C and passed through cell strainers. Suspensions were resuspended in 80% isotonic Percoll and subsequently overlaid with 40% isotonic Percoll. Samples were then centrifuged at 400 × g for 25 min. Cells at the 40/80 interface were collected, washed, and counted.
Tetramer, Ab staining, and flow cytometric analysis
MHC/peptide tetramers for the MHV-68 epitopes, ORF6487–495/Db and ORF61524–531/Kb, conjugated to APC were obtained from the National Institutes of Health Tetramer Core facility (Emory University, Atlanta, GA). Cells were incubated with anti-CD16/32 Ab for 10 min on ice. Cells were washed and then stained for 1 h in the dark with tetramers as previously described (38), then stained with cell surface markers or the appropriate isotype controls for 20 min on ice. Samples were analyzed using FACSCalibur, FACSCanto, or Accuri Flow cytometers. Data were then analyzed using either FlowJo or Accuri software.
Intracellular cytokine staining
Splenocytes were incubated with 1 μg/ml of the appropriate peptide plus 10 μg/ml brefeldin A in complete medium at 37°C for 5 h. Cells were then stained with cell surface markers, fixed in 1% formaldehyde, and rendered permeable with 0.5% saponin solution before staining with Abs against IFN-γ, TNF-α, or granzyme B, or the appropriate isotype control. The percentage of cytokine-producing cells was established by subtracting from the no-peptide controls.
Ab blockade of inhibitory cytokines and receptors
Anti–glucocorticoid-induced TNFR-related protein mAb (clone DTA-1) was provided by Dr. Mary Jo Turk (Dartmouth Medical School, Lebanon, NH). Mice were injected with 250 μg per injection on days 33 and 39 p.i. Anti–programmed death-1 (PD-1) (clone RMP1-14) was provided by Dr. Hideo Yagita (Juntendo University, Tokyo, Japan). Mice were injected with 250 μg of the mAb given twice per week starting on day 33 p.i. For anti–CTLA-4, clone 4F10 was purchased from the American Type Culture Collection (Manassas, VA). Mice were given 200 μg twice per week starting on day 33 p.i. IL-10R Ab (clone 1B1.3a) was purchased from BioExcel (West Lebanon, NH). Mice were administered 250 μg every 3 d. Anti–Gr-1 Ab (clone RB6-8C5) was given every day for 5 d. Mice were given 250 μg per injection. All injections were performed via the peritoneal route.
IL-10 ELISA
Serum was collected from mice infected at least 42 d p.i. by cardiac puncture. Supernatants were collected from splenocytes in culture for 48 h. The eBioscience mouse ready-SET-Go kit was used according to the manufacturer’s protocol (eBioscience, San Diego, CA).
CFSE proliferation assay
Splenocytes from Thy1.1–IL-10 reporter mice were incubated with 0.5 μg CFSE at 1 × 107 cells/ml for 10 min, washed, and then added to a 96-well plate coated with anti-CD3. Cells were incubated for 72 h at 37°C, and CD8+ T cells were analyzed for dye dilution by flow cytometry.
In vivo cytotoxicity assay
Naive C57BL/6 splenocytes were incubated with 1 μg/ml of the ORF6 or ORF61 peptide, or no peptide for 5 h. Each population was then labeled with 2.5, 0.25, or 0.025 μM CFSE (Molecular Probes, Eugene, OR). Cells were then mixed at a 1:1:1 ratio, and 2 × 107 total cells were injected i.v. Six hours later, mice were sacrificed and spleen cell suspensions were incubated with 20 μg/ml 7-aminoactinomycin D (7-AAD; Sigma-Aldrich, St. Louis, MO) for 15 min at room temperature in the dark. Cells were then analyzed by flow cytometry, and specific lysis was calculated using the following formula: Specific killing = 100 − {[(% peptide pulsed infected/% unpulsed infected)/(% peptide pulsed uninfected/% unpulsed uninfected)] × 100}.
Generation of ORF61-specific CD8+ T cell line
Splenocytes were isolated from mice infected with MHV-68 for 60 d. A total of 4 × 106 cells were placed in a volume of 2 ml with the ORF61 peptide (1 ng/ml) and IL-2 (10 U/ml). After 6 d, the cells were cultured with irradiated naive spleen cells with ORF61 peptide (1 ng/ml) and IL-2 (10 U/ml) in a small cell culture flask. Media was changed every second day, adding fresh IL-2 at 10 U/ml, and cultures were restimulated weekly with peptide and IL-2 as described earlier, until p79 tetramer positive reached >99% purity.
Bone marrow chimera generation
To generate mixed bone marrow chimeras, we lethally irradiated B6-Ly5.2 recipient mice with a split dose totaling 1000 rad and subsequently injected them i.v. with a total of 106 bone marrow cells. For generation of the CD8α−/−:WT, CD8α−/−:IL10−/−, and CD8α−/−:IL-10R1−/− mixed chimeras, bone marrow from CD8α−/− mice was mixed at a 80:20 ratio with the additional population. Mice were then left to reconstitute for 50 d before infection.
Plaque assay
Lung tissue was disrupted and 10-fold serial dilutions were incubated on 3T3 monolayers for 1 h at 37°C. Two milliliters of a 1:1 mixture of carboxymethyl-cellulose and DMEM was added and cultured for 5 d at 37°C. Media was then aspirated and monolayers were fixed with methanol at room temperature for 20 min and then were stained for 180 min with 8% Giemsa stain (Sigma-Aldrich). Plaques were counted microscopically.
Quantitative PCR for viral transcripts
Latent viral DNA was quantified by quantitative fluorescent PCR for the ORF50 gene as previously described (38).
Statistical analysis
The p values were calculated using either Student t test or one-way ANOVA with a Bonferroni posttest. A p value <0.05 was considered significant.
Results
MHV-68–infected mice maintain functional capability in the absence of T cell help
Previous work indicates that WT mice infected with MHV-68 are able to maintain control over the persistent infection. Depletion of CD4+ T cells does not alter the ability of the host to control the initial lytic infection. However, control is ultimately lost and the virus reactivates to high levels in the lungs within 6 wk p.i. (Fig. 1A) (28, 29). In contrast with chronic infections with other viruses such as lymphocytic choriomeningitis virus clone 13, we did not detect a decrease in the number of Ag-specific CD8+ T cells in either the spleen (Supplemental Fig. 1A) or the lung (Fig. 1B); in fact, the CD8+ T cell response was increased in CD4-depleted mice, consistent with previously published results (30). The proportion of virus-specific CD8+ T cells producing IFN-γ reflected the pattern seen by tetramer staining (Fig. 1C, 1D); however, a smaller proportion of cells coproduced IFN-γ and TNF-α, consistent with previous studies (28). The amount of IFN-γ produced by p56-specific cells was reduced in the CD4-depleted group; however, the p79-specific population was unaffected (Fig. 1C, 1D, Supplemental Fig. 1B, 1C). These changes did not impact on the in vivo cytolytic ability of the CD8+ T cells, which were unaffected in all cases (Fig. 1E). Overall, no consistent defects were apparent that would account for the loss of T cell-mediated control of MHV-68 in CD4-deficient mice.
FIGURE 1.
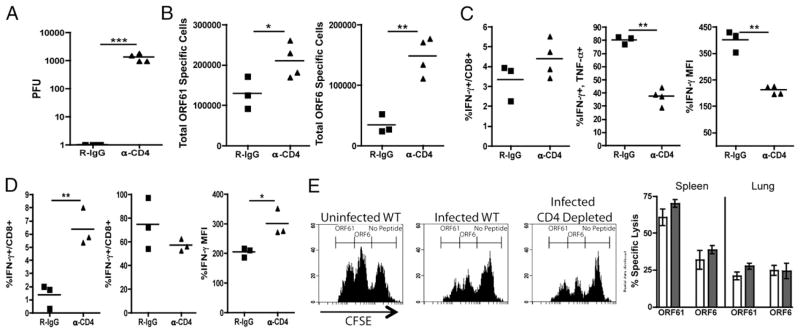
Virus recrudescence in the absence of CD4 help. B6 mice were infected with 400 PFU MHV-68 intranasally and were either given rat IgG or CD4-depleting Ab. A, Lung viral titers were measured by plaque assay on day 40 p.i. B, Total number of ORF6- and ORF61-specific CD8+ T cells in the lung assessed by tetramer staining at 42 d p.i. C and D, Cytokine production by MHV-68–specific CD8+ T cells in the lung in response to restimulation with ORF61 (C) or ORF6 (D) as analyzed by intracellular cytokine staining. E, Cytotoxicity assessed by an in vivo cytotoxicity assay at day 57 p.i. A mixture of ORF61, ORF6, or no peptide-pulsed targets was labeled with CFSE and injected i.v. Six hours later, targets in the spleen and lungs were analyzed. Representative histograms are from the spleen. White bars represent Rat-IgG–treated mice; gray bars represent CD4-depleted mice. Error bars indicate SD. Each dot represents data from individual mice. Experiment was repeated three times with a minimum of three mice per group. *p < 0.05, **p < 0.01, ***p < 0.001.
Adoptive CD8+ T cell therapy is inhibited in MHV-68–infected helpless mice during viral reactivation
Postexposure vaccination with vaccinia virus expressing an MHV-68 epitope successfully induces robust expansion of Ag-specific CD8+ T cells; however, no effect can be seen on the virus load in the lungs of treated animals (31). Because these cells are primed in a CD4-deficient environment, they may carry an inherent defect in their ability to respond optimally to vaccination. To circumvent this issue, we generated an MHV-68–specific CD8+ T cell line from virus-infected WT mice. In brief, WT mice were infected with MHV-68, and splenocytes were isolated and cultured in vitro with the ORF61 peptide until >99% of the CD8+ T cells were specific for the epitope (Supplemental Fig. 2). A total of 1 × 106 CD8+ T cells were then adoptively transferred into MHV-68–infected hosts either at the time of infection or 60 d p.i., when reactivation is readily apparent. Transfer of cells during the primary response was able to reduce the viral burden in the lungs by >40-fold, whereas transfer of an identical number of cells during reactivation had no effect on the viral load (Fig. 2A). This suggests that transferred CD8+ T cells were being actively inhibited from controlling the virus in CD4-deficient mice. If the number of Ag-specific cells was increased to 5 × 106 or 1 × 107, the CD8+ T cells were able to overcome the putative suppressive environment and reduce the viral load (Fig. 2B). Taken together, the data indicate that despite having no obvious defect in the Ag-specific CD8+ T cell response, a suppressive environment likely exists in the absence of CD4 cell help that is capable of dampening the antiviral activity of both endogenous and adoptively transferred CD8+ T cells.
FIGURE 2.
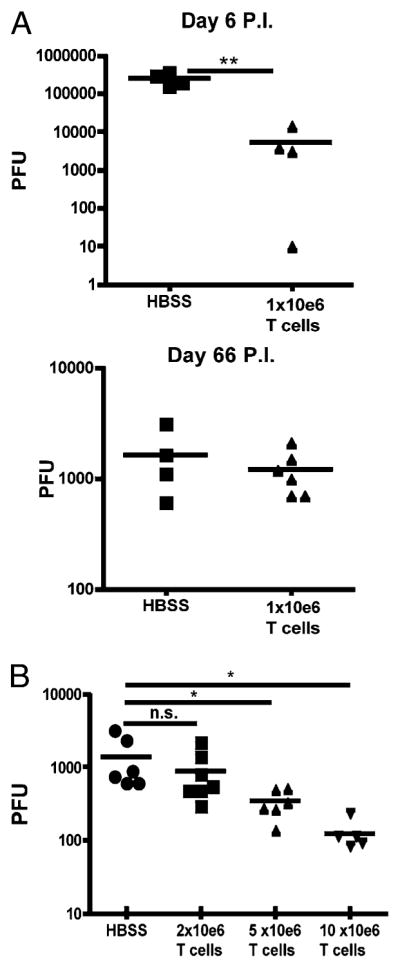
Transfer of an ORF61-specific CD8+ T cell line in MHV-68–infected CD4-depleted mice. WT mice were infected with MHV-68 and splenocytes were isolated during maintenance of the memory population (day 60 p.i.) and cultured in vitro with the ORF61 peptide until >99% of the CD8+ T cells were specific for the epitope. A, A total of 1 × 106 CD8+ T cells were adoptively transferred into MHV-68–infected hosts either at the time of infection (top) or into CD4-depleted hosts 60 d p.i. (bottom), and viral titers were determined by plaque assay 6 d later. B, Various numbers of ORF61-specific CD8+ T cells were transferred into helpless mice at day 60 p.i., and viral titers were determined 6 d later. Each dot represents data from individual mice. Experiments were repeated at least two times with a minimum of four mice per group. *p < 0.05, **p < 0.01. n.s., not significant.
Increased levels of IL-10 are produced in MHV-68–infected helpless mice
We determined whether known suppressive mechanisms associated with other chronic infections were involved in inhibiting the CD8+ T cell response in CD4-depleted MHV-68–infected mice. To do so, we treated CD4+ T cell-depleted mice with Abs against PD-1, glucocorticoid-induced TNFR-related protein, or CTLA-4 during late time points when virus had reactivated; however, no therapeutic value was observed (Supplemental Fig. 3A). In addition, we detected a dramatic increase in the number of CD11b+, Gr-1+ cells in the lungs and spleen of MHV-68–infected mice in the absence of CD4+ T cell help (Supplemental Fig. 3B). Myeloid cells with this phenotype are known to have suppressive capabilities in a variety of tumor models and to be induced by various infections (39–41). However, depletion of this population using anti–Gr-1 Ab had no effect on viral load in the lungs of CD4+ T cell-depleted mice (Supplemental Fig. 3C).
Interestingly, we observed an increase in both serum levels of IL-10 from helpless mice compared with WT mice, as well as from splenocytes cultured for 48 h ex vivo (Fig. 3A, 3B). To test whether IL-10 was contributing to the loss of long-term control of the virus in CD4-deficient animals, we used mice genetically deficient in the IL-10R1 chain. MHV-68–infected helpless mice lacking IL-10R1 had lower levels of virus reactivation in the lungs compared with WT mice (Fig. 3C), implicating a role for IL-10 in virus reactivation. To confirm that IL-10 was suppressing the immune response against MHV-68 in helpless mice, we generated virus-specific CD8+ T cell lines from WT or IL-10R1−/− mice, and adoptively transferred identical numbers into CD4-deficient mice undergoing virus reactivation. Transfer of 2 × 106 cells from either population had no effect. However, although transfer of 4 × 106 WT cells had no effect on viral control, 4 × 106 IL-10R1−/− cells were able to reduce the viral burden (Fig. 3D).
FIGURE 3.
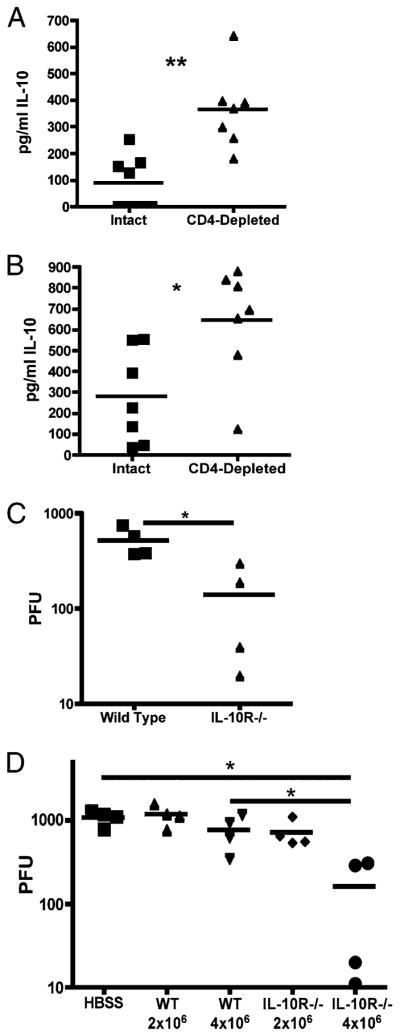
MHV-68–infected CD4-depleted mice have increased levels of IL-10 contributing to suppression of CD8+ T cell responses. A, Serum from intact and CD4-depleted mice infected for 42 d was analyzed for IL-10 by ELISA. B, Splenocytes were isolated from intact and CD4-depleted mice infected for 42 d and cultured at 37°C for 48 h with the ORF61 peptide before supernatants were analyzed by ELISA for IL-10. C, WT and IL-10R1−/− mice were CD4 depleted and infected with MHV-68 for 60 d. Lungs were then analyzed for viral titer by plaque assay. D, CD4-depleted MHV-68–infected mice were infected for 60 d. Then either 0, 2 × 106, or 4 × 106 ORF61-specific WT or IL-10R1−/− CD8+ T cells were adoptively transferred. Six days later, the lungs were analyzed for viral titers by plaque assay. Each dot represents data from individual mice. Experiments were repeated at least two times with a minimum of four mice per group. *p < 0.05, **p < 0.01.
CD4-depleted mice develop a population of CD8+ T cells capable of producing IL-10
We next determined which cell populations were producing increased amounts of IL-10 in the absence of CD4 help on infection with MHV-68. To do so, we made use of Thy1.1–IL-10 reporter mice, which were genetically modified to include the Thy1.1 gene under control of the IL-10 promoter (42). Therefore, when the promoter of IL-10 is active, the cell expresses Thy1.1 on the cell surface. We examined intact and CD4-depleted mice infected with MHV-68 for 60 d for Thy1.1+ populations. No increases in Thy1.1 expression were found between the groups in CD11b+, CD11c+, Gr-1+, or CD19+ populations (Supplemental Fig. 4). A small population of CD8− cells produced IL-10 in the intact but not CD4-depleted group, and further examination revealed these to be CD4 T cells (data not shown). Interestingly, we found a population of CD8+ T cells in both the spleen and lung that expressed the Thy1.1 marker only in the absence of CD4+ T cell help (Fig. 4A). The MHV-68–specific CD8+ T cell populations ORF6 and ORF61 were not enriched for Thy1.1+ cells in either the spleen or lung (Fig. 4B). To determine whether the Thy1.1+ cells were, in fact, capable of producing IL-10, we sorted CD8+, CD11c− cells into Thy1.1− and Thy1.1+ populations and cultured them for 24 h in either media alone or with agonistic anti-CD3 Ab. Although the Thy1.1− cells failed to produce IL-10, Thy1.1+ CD8+ T cells produced IL-10 on stimulation (Fig. 4C). Anti-CD3 stimulation failed to induce proliferation, as well as robust IFN-γ and TNF-α production from the Thy1.1+ cells compared with the Thy1.1− cells (Fig. 4D, 4E). We next analyzed the CD8+ Thy1.1+ population using a broad array of cell surface differentiation markers. Thy1.1+ cells were found to be CD127low, CD43hi, and had an increased percentage of CD62Llo cells, characteristic of an activated phenotype (Fig. 5). The Thy1.1+ CD8+ cells also expressed increased levels of CD44 compared with the naive CD44lo population; however, expression was decreased compared with Ag-experienced CD44hi CD8+ T cells (Fig. 5).
FIGURE 4.

CD8+ T cells are capable of producing IL-10 in the absence of CD4 help. Intact and CD4-depleted IL-10 Thy1.1 reporter mice were infected with MHV-68 for at least 42 d. A, Representative plots of intact and CD4-depleted mice expressing Thy1.1 in the CD8+ T cell compartment. B, Thy1.1 expression was analyzed for the bulk CD8+ T cell compartment, as well as for the ORF6- and ORF61-specific populations. C, IL-10 Thy1.1 reporter mice were depleted of CD4+ T cells for 42 d, and CD8+ CD11c− cells were isolated by flow sorting into Thy1.1+ and Thy1.1− populations. Cells were then stimulated with immobilized anti-CD3 Ab for 48 h, and supernatant IL-10 levels were determined by ELISA. D, Splenocytes from MHV-68–infected CD4-depleted mice were CFSE labeled, then stimulated with anti-CD3 for 72 h, and proliferation of Thy1.1+ and Thy1.1− populations was measured by CFSE dilution. E, Splenocytes were stimulated with anti-CD3 for 24 h, and production of IFN-γ and TNF-α was determined for the Thy1.1+ and Thy1.1− populations. Error bars indicate SD. Each dot represents data from individual mice. Experiments were repeated at least two times with a minimum of four mice per group. **p < 0.01, ***p < 0.001. n.s., not significant.
FIGURE 5.

Thy1.1+ CD8+ T cells express cell surface markers of activation. IL-10 Thy1.1 reporter mice were CD4 depleted for 60 d, and the expression of cell surface markers was analyzed by flow cytometry for CD8+ Thy1.1+ and Thy1.1− cells. Representative histograms and statistical analysis are shown. Solid histograms represent Thy1.1+; open histograms represent Thy1.1−. Error bars indicate SD. Experiments were repeated at least two times with a minimum of four mice per group. **p < 0.01, ***p < 0.001.
Two populations of regulatory CD8+ T cells have recently been described in mice. One population expresses Foxp3 and suppresses via TGF-β (43). Another population capable of suppressing graft rejection through IL-10 has been reported to be separated from conventional CD8+ T cells by coexpression of CD122+ and PD-1 (44). When we analyzed the CD8+ population, we found that although the Thy1.1+ cells did not express any detectable Foxp3 (data not shown), they were, in fact, CD122+ and expressed increased levels of PD-1, indicating a phenotype similar to a previously described regulatory CD8+ T cell population (Fig. 5).
To determine whether this CD8+ population of IL-10–producing cells directly contributed to suppression of the MHV-68–specific immune response, we isolated the ability to either produce or respond to IL-10 to the CD8+ T cell compartment using mixed bone marrow chimeric mice. We reconstituted lethally irradiated mice with CD8α−/− bone marrow mixed with either WT, IL-10−/−, or IL-10R1−/− bone marrow at a 4:1 ratio. In this manner, we generated mice where the CD8+ compartment contained T cells that were exclusively either WT-, IL-10–, or IL-10R1–deficient, respectively, but the large majority of all other cells expressed these molecules. Chimeric mice were then CD4 depleted and infected with MHV-68 for at least 60 d; then the viral load was determined in the lungs. IL-10−/− bone marrow chimeric mice had significantly lower levels of virus compared with WT mice in the absence of CD4+ T cell help, indicating that IL-10 produced by CD8+ cells contributes to suppression of the MHV-68 immune response (Fig. 6A). IL-10R1−/− bone marrow chimeric mice also had lower viral load compared with WT chimeras (Fig. 6A). The decrease was slightly less robust than that seen with the IL-10−/− chimeras, suggesting that both direct and indirect effects on the Ag-specific CD8+ T cell response were potentially responsible for improved viral control. Notably, two IL-10−/− chimera and three IL-10R1−/− mice had no detectable lytic virus in the lungs, whereas all WT chimera mice had high levels of reactivation.
FIGURE 6.

IL-10 produced by CD8+ cells suppresses MHV-68 immune responses at least partly through a direct mechanism. A, WT mice were lethally irradiated and reconstituted with a mixture of bone marrow cells. Eighty percent CD8α−/− bone marrow cells were combined with 20% WT, IL-10−/−, or IL-10R1−/− cells, and injected into irradiated mice and left for 50 d. The chimeric mice were then CD4 depleted and infected for 60 d. Viral titers in the lungs were then analyzed by plaque assay. B, Helpless MHV-68 infected mice were either left untreated or were treated with anti–IL-10R Ab on days 0 and 7, days 21–42, or for the duration of the experiment. Viral titers were then analyzed by plaque assay at day 45 p.i., and latent virus was measured by QT-PCR (C). Helpless mice were infected with MHV-68 for 60 d. Mice were then given anti–IL-10R twice weekly for 2 wk, and the viral titer in the lung was determined by plaque assay (D). The effects of IL-10R blockade were determined by measuring production of TNF-α by ORF61-specific CD8+ T cells, and by expression of the costimulation marker CD80 by CD11c+ cells (E). Solid histograms represent rat IgG-treated mice; open histograms represent anti–IL-10R–treated mice. Each dot represents data from individual mice. Experiments were repeated at least two times with a minimum of four mice per group. *p < 0.05, **p < 0.01.
Blockade of IL-10R during reactivation is able to reduce the viral burden in MHV-68–infected helpless mice
Although genetic disruption of the IL-10R1 gene was able to reduce the virus in reactivating “helpless” mice, this could be a result of altered kinetics during the primary response, because IL-10−/− mice infected with MHV-68 have been reported to have lower peak viral loads than WT mice (35). We therefore took a complementary approach to address the role of IL-10 in immune surveillance, by administering a blocking Ab at various times throughout the course of infection. Interestingly, blockade of IL-10R during the primary lytic infection (days 0–7), during reactivation (day 21+), or throughout the duration of the experiment all resulted in a reduction in viral load (Fig. 6B). Although IL-10R blockade improved control of the lytic virus, no effect was observed on the latent viral loads (Fig. 6C). For IL-10 blockade to be a clinically relevant therapy, it must be capable of restoring immune surveillance at late stages after virus reactivation has occurred. Therefore, CD4-depleted MHV-68 infected mice were left for 60 d, then given twice weekly injections of the anti–IL-10R Ab for 2 wk and virus titers were determined. Blockade of the IL-10R at these times resulted in a reduction of viral load compared with rat Ig-treated controls (Fig. 6D). Similar therapeutic efficacy was observed when class II−/− mice infected with MHV-68 were administered IL-10R blocking Abs (data not shown). This confirms the therapeutic value of IL-10 blockade even at late stages after virus reactivation. When the Ag-specific CD8+ T cell response was analyzed via bronchoalveolar lavage, enhanced effector responses were observed, as more TNF-α was produced on restimulation (Fig. 6E). A small but statistically significant increase in IFN-γ was also observed (data not shown). In addition, CD11c+ cells from the lung expressed increased levels of the costimulatory marker CD80 (Fig. 6E). Thus, disruption of IL-10 signaling allows for enhanced immune surveillance in immunocompromised MHV-68–infected mice.
Discussion
Previous studies have identified CD4+ T cells as being critical for the long-term control of MHV-68 infection (29). Our understanding of how the immune response against MHV-68 is suppressed in the absence of CD4 T cell help remains unclear because Ag-specific CD8+ T cell numbers, proliferative capability, and cytolytic activity of CD8+ T cells appear to be largely normal in helpless mice (Fig. 1, Supplemental Fig. 1) (28, 30, 31). A recent study indicated that class II−/− mice infected with MHV-68 express increased levels of PD-1 on Ag-specific CD8+ T cells, and that blockade of the PD-1 pathway improves viral control (10). Conversely, in our experiments, administration of PD-1 blocking Abs to CD4-depleted MHV-68–infected mice did not improve viral control. The exact reason for this discrepancy is unclear but could be a result of differences between using MHC class II−/− mice in the previous study versus depleting GK1.5 anti-CD4 Ab in this study. Our study aimed to explore various mechanisms to enhance control of MHV-68 in the immunocompromised state of a CD4-deficient host. We generated an ORF61-specific CD8+ T cell line in vitro and adoptively transferred the cells into MHV-68–infected mice that lack CD4+ T cell help. Although the T cell line was highly effective at reducing the viral load during the initial lytic infection, an identical number of cells was unable to provide therapeutic value if transferred at day 60 p.i. when MHV-68 has reactivated to high levels in the lungs (Fig. 2). This indicates that in addition to any potential CD8+ T cell intrinsic defects, a suppressive environment exists that is capable of inhibiting antiviral activity by the T cell line. Therefore, it was particularly relevant when we detected increased IL-10 secretion in helpless mice undergoing virus reactivation, because IL-10 has been implicated in the suppression of immune responses against a variety of acute and persistent infections (12, 45, 46). In contrast with other models, we observed an increased production of IL-10 only under conditions of CD4+ T cell depletion (Fig. 4), specifically by a population of CD8+ T cells (8, 17, 18).
The IL-10–producing CD8+ T cells expressed markers indicating Ag experience based on expression of surface markers. Interestingly, they were not enriched in the Ag-specific population recognizing the ORF6 or ORF61 epitopes, although we cannot rule out that the IL-10–producing CD8 T cells are derived from CD8 T cells responding to other viral epitopes. These cells expressed intermediate levels of the marker CD44 and had increased levels of PD-1 and CD43 (Figs. 4, 5), a phenotype that differed from the tetramer-positive population, suggesting these cells may be in a different stage of activation. IL-10 produced by CD8+ T cells contributed to suppression of the MHV-68 response as bone marrow chimeras where only CD8+ cells lacked the IL-10 gene had significantly lower viral loads than WT chimeras. In addition, mAb blockade of IL-10R at any stage of the infection enhanced control of the lytic virus, indicating an important role for IL-10 in subduing the T cell response throughout the infection.
During acute infection, MHV-68 induces expression of IL-10 from the dendritic cell compartment (36), which facilitates colonization of the host by the virus. In addition, IL-10 induction by the MHV-68 M2 protein facilitates induction of the latent state by promoting B cell proliferation and survival (34). Our results are distinct as we observe increased levels of IL-10 during chronic reactivation of MHV-68 only in the absence of CD4+ T cell help from a source distinct from that seen during the initial infection. IL-10 production appears to be responsible, in part, for the breakdown of immune surveillance, allowing virus reactivation to occur. In other experiments, we have observed Thy1.1 expression from CD8+ T cells after infection of CD4-depleted IL-10 reporter mice with vaccinia virus, a virus that does not persist in the absence of CD4 help. Therefore, induction of the IL-10–producing CD8+ T regulatory population appears to be linked to the depletion of CD4+ T cells rather than virus persistence per se. One of the major roles associated with CD4+ T cell help is to appropriately “license” dendritic cells via CD40L–CD40 interactions (47, 48). These fully licensed dendritic cells then are capable of appropriately priming the CD8+ T cell response. In the absence of T cell help, inhibitory molecules such as TRAIL are upregulated, limiting the memory capabilities of the CD8+ T cell population (49). As such, one hypothesis is that IL-10 production preferentially occurs in CD8+ T cells primed by unlicensed dendritic cells, which may transmit different signals to the CD8 T cell, for example, costimulatory signals or cytokines, compared with licensed APCs.
Recently, suppressive CD8+ T cells have been identified in a number of infectious diseases. Similar to CD4+ regulatory T cells, CD8+ T cells can suppress through several mechanisms including IL-10, CTLA-4, and TGF-β (43). In mice, two distinct types of regulatory CD8+ T cells have been identified: CD25+ Foxp3+ cells that suppress via CTLA-4 and TGF-β, and CD122+ PD-1+ cells that suppress via IL-10 (43, 44, 50). Although the IL-10–producing cells identified in our experiments did not express Foxp3 (data not shown), they did express increased levels of PD-1 and CD122 (Fig. 5). With respect to human infection, regulatory CD8+ T cells have been identified in patients with HCV (51–53), HIV (54–56), and HSV-2 (57). Interestingly, HIV-specific CD8+ T cell production of IL-10 is correlated with declining CD4+ T cell numbers, increased HIV plasma RNA levels, and decreased cytolysis of infected cells (55, 56).
A study of EBV-infected patients revealed that CD8+ regulatory cells could be expanded from immunocompromised patients (58). Interestingly, the same population could not be expanded from EBV-infected healthy control patients, indicating the potential for preferential induction of regulatory CD8+ T cells in an immuno-compromised state. The expanded regulatory CD8+ T cells produced both IFN-γ and IL-10, and required cell-to-cell contact to suppress CD4+ T cell proliferation. This is again reminiscent of the MHV-68 system because our data indicate production of IL-10 in an immunocompromised host. The IL-10 pathway is likely a key factor in the host–virus equilibrium in several herpesvirus infections, because many of these viruses, such as human CMV, EBV, and veterinary γ-herpesviruses, encode IL-10 homologs in their genomes (32, 33) and induce IL-10 production by the host. Therefore, disruption of the IL-10 pathway may provide a novel therapeutic for enhanced control of human γ-herpesvirus–induced disease in immunocompromised patients. In addition, it adds to our understanding of the host–virus equilibrium in persistent virus infections in general, and in particular the functions of CD4+ T cell help and the biology of IL-10 in these infections.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AI069943, CA103642, and T32AI07363.
Abbreviations used in this article
- HCV
hepatitis C virus
- MHV
murine γ-herpes-virus
- PD-1
programmed death-1
- p.i
postinfection
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Verucchi G, Calza L, Manfredi R, Chiodo F. Human immunodeficiency virus and hepatitis C virus coinfection: epidemiology, natural history, therapeutic options and clinical management. Infection. 2004;32:33–46. doi: 10.1007/s15010-004-3063-7. [DOI] [PubMed] [Google Scholar]
- 2.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. 2005;79:10514–10527. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–879. doi: 10.1038/ni1241. [DOI] [PubMed] [Google Scholar]
- 5.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 6.Fuse S, Molloy MJ, Usherwood EJ. Immune responses against persistent viral infections: possible avenues for immunotherapeutic interventions. Crit Rev Immunol. 2008;28:159–183. doi: 10.1615/critrevimmunol.v28.i2.40. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha SJ, West EE, Araki K, Smith KA, Ahmed R. Manipulating both the inhibitory and stimulatory immune system towards the success of therapeutic vaccination against chronic viral infections. Immunol Rev. 2008;223:317–333. doi: 10.1111/j.1600-065X.2008.00638.x. [DOI] [PubMed] [Google Scholar]
- 10.Dias P, Giannoni F, Lee LN, Han D, Yoon S, Yagita H, Azuma M, Sarawar SR. CD4 T-cell help programs a change in CD8 T-cell function enabling effective long-term control of murine gammaherpesvirus 68: role of PD-1-PD-L1 interactions. J Virol. 2010;84:8241–8249. doi: 10.1128/JVI.00784-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 13.Miyazoe S, Hamasaki K, Nakata K, Kajiya Y, Kitajima K, Nakao K, Daikoku M, Yatsuhashi H, Koga M, Yano M, Eguchi K. Influence of interleukin-10 gene promoter polymorphisms on disease progression in patients chronically infected with hepatitis B virus. Am J Gastroenterol. 2002;97:2086–2092. doi: 10.1111/j.1572-0241.2002.05926.x. [DOI] [PubMed] [Google Scholar]
- 14.Persico M, Capasso M, Persico E, Masarone M, Renzo A, Spano D, Bruno S, Iolascon A. Interleukin-10 - 1082 GG polymorphism influences the occurrence and the clinical characteristics of hepatitis C virus infection. J Hepatol. 2006;45:779–785. doi: 10.1016/j.jhep.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 15.Knapp S, Hennig BJ, Frodsham AJ, Zhang L, Hellier S, Wright M, Goldin R, Hill AV, Thomas HC, Thursz MR. Interleukin-10 promoter polymorphisms and the outcome of hepatitis C virus infection. Immunogenetics. 2003;55:362–369. doi: 10.1007/s00251-003-0594-5. [DOI] [PubMed] [Google Scholar]
- 16.Shin HD, Winkler C, Stephens JC, Bream J, Young H, Goedert JJ, O’Brien TR, Vlahov D, Buchbinder S, Giorgi J, et al. Genetic restriction of HIV-1 pathogenesis to AIDS by promoter alleles of IL10. Proc Natl Acad Sci USA. 2000;97:14467–14472. doi: 10.1073/pnas.97.26.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maris CH, Chappell CP, Jacob J. Interleukin-10 plays an early role in generating virus-specific T cell anergy. BMC Immunol. 2007;8:8. doi: 10.1186/1471-2172-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphreys IR, de Trez C, Kinkade A, Benedict CA, Croft M, Ware CF. Cytomegalovirus exploits IL-10-mediated immune regulation in the salivary glands. J Exp Med. 2007;204:1217–1225. doi: 10.1084/jem.20062424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rigopoulou EI, Abbott WG, Haigh P, Naoumov NV. Blocking of interleukin-10 receptor—a novel approach to stimulate T-helper cell type 1 responses to hepatitis C virus. Clin Immunol. 2005;117:57–64. doi: 10.1016/j.clim.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Clerici M, Wynn TA, Berzofsky JA, Blatt SP, Hendrix CW, Sher A, Coffman RL, Shearer GM. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest. 1994;93:768–775. doi: 10.1172/JCI117031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas PS, Pedicord V, Ploss A, Menet E, Leiner I, Pamer EG. Pathogen-specific CD8 T cell responses are directly inhibited by IL-10. J Immunol. 2007;179:4520–4528. doi: 10.4049/jimmunol.179.7.4520. [DOI] [PubMed] [Google Scholar]
- 24.Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu Rev Immunol. 1997;15:405–431. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- 25.Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. 2007;60:1365–1372. doi: 10.1136/jcp.2007.051953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Far M, Halwani R, Said E, Trautmann L, Doroudchi M, Janbazian L, Fonseca S, van Grevenynghe J, Yassine-Diab B, Sékaly RP, Haddad EK. T-cell exhaustion in HIV infection. Curr HIV/AIDS Rep. 2008;5:13–19. doi: 10.1007/s11904-008-0003-7. [DOI] [PubMed] [Google Scholar]
- 27.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 28.Belz GT, Liu H, Andreansky S, Doherty PC, Stevenson PG. Absence of a functional defect in CD8+ T cells during primary murine gammaherpesvirus-68 infection of I-A(b−/−) mice. J Gen Virol. 2003;84:337–341. doi: 10.1099/vir.0.18821-0. [DOI] [PubMed] [Google Scholar]
- 29.Cardin RD, Brooks JW, Sarawar SR, Doherty PC. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J Exp Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevenson PG, Belz GT, Altman JD, Doherty PC. Virus-specific CD8(+) T cell numbers are maintained during gamma-herpesvirus reactivation in CD4-deficient mice. Proc Natl Acad Sci USA. 1998;95:15565–15570. doi: 10.1073/pnas.95.26.15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belz GT, Stevenson PG, Castrucci MR, Altman JD, Doherty PC. Postexposure vaccination massively increases the prevalence of gamma-herpesvirus-specific CD8+ T cells but confers minimal survival advantage on CD4-deficient mice. Proc Natl Acad Sci USA. 2000;97:2725–2730. doi: 10.1073/pnas.040575197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholas J. Human gammaherpesvirus cytokines and chemokine receptors. J Interferon Cytokine Res. 2005;25:373–383. doi: 10.1089/jir.2005.25.373. [DOI] [PubMed] [Google Scholar]
- 33.Slobedman B, Barry PA, Spencer JV, Avdic S, Abendroth A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J Virol. 2009;83:9618–9629. doi: 10.1128/JVI.01098-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siegel AM, Herskowitz JH, Speck SH. The MHV68 M2 protein drives IL-10 dependent B cell proliferation and differentiation. PLoS Pathog. 2008;4:e1000039. doi: 10.1371/journal.ppat.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peacock JW, Bost KL. Murine gammaherpesvirus-68-induced interleukin-10 increases viral burden, but limits virus-induced splenomegaly and leukocytosis. Immunology. 2001;104:109–117. doi: 10.1046/j.0019-2805.2001.01286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flaño E, Kayhan B, Woodland DL, Blackman MA. Infection of dendritic cells by a gamma2-herpesvirus induces functional modulation. J Immunol. 2005;175:3225–3234. doi: 10.4049/jimmunol.175.5.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J Gen Virol. 1992;73:2347–2356. doi: 10.1099/0022-1317-73-9-2347. [DOI] [PubMed] [Google Scholar]
- 38.Usherwood EJ, Ward KA, Blackman MA, Stewart JP, Woodland DL. Latent antigen vaccination in a model gammaherpesvirus infection. J Virol. 2001;75:8283–8288. doi: 10.1128/JVI.75.17.8283-8288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serafini P, De Santo C, Marigo I, Cingarlini S, Dolcetti L, Gallina G, Zanovello P, Bronte V. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 43.Dinesh RK, Skaggs BJ, La Cava A, Hahn BH, Singh RP. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun Rev. 2010;9:560–568. doi: 10.1016/j.autrev.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai H, Wan N, Zhang S, Moore Y, Wan F, Dai Z. Cutting edge: programmed death-1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J Immunol. 2010;185:803–807. doi: 10.4049/jimmunol.1000661. [DOI] [PubMed] [Google Scholar]
- 45.Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007;15:143–146. doi: 10.1016/j.tim.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 46.Filippi CM, von Herrath MG. IL-10 and the resolution of infections. J Pathol. 2008;214:224–230. doi: 10.1002/path.2272. [DOI] [PubMed] [Google Scholar]
- 47.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 48.Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–1148. doi: 10.1038/ni1129. [DOI] [PubMed] [Google Scholar]
- 49.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 50.Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, Santarlasci V, Manetti R, Vanini V, Romagnani P, et al. Human CD8+CD25 + thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood. 2003;102:4107–4114. doi: 10.1182/blood-2003-04-1320. [DOI] [PubMed] [Google Scholar]
- 51.Billerbeck E, Blum HE, Thimme R. Parallel expansion of human virus-specific FoxP3- effector memory and de novo-generated FoxP3+ regulatory CD8+ T cells upon antigen recognition in vitro. J Immunol. 2007;179:1039–1048. doi: 10.4049/jimmunol.179.2.1039. [DOI] [PubMed] [Google Scholar]
- 52.Accapezzato D, Francavilla V, Paroli M, Casciaro M, Chircu LV, Cividini A, Abrignani S, Mondelli MU, Barnaba V. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J Clin Invest. 2004;113:963–972. doi: 10.1172/JCI20515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alatrakchi N, Graham CS, van der Vliet HJ, Sherman KE, Exley MA, Koziel MJ. Hepatitis C virus (HCV)-specific CD8+ cells produce transforming growth factor beta that can suppress HCV-specific T-cell responses. J Virol. 2007;81:5882–5892. doi: 10.1128/JVI.02202-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elrefaei M, Baker CA, Jones NG, Bangsberg DR, Cao H. Presence of suppressor HIV-specific CD8+ T cells is associated with increased PD-1 expression on effector CD8+ T cells. J Immunol. 2008;180:7757–7763. doi: 10.4049/jimmunol.180.11.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elrefaei M, Barugahare B, Ssali F, Mugyenyi P, Cao H. HIV-specific IL-10-positive CD8+ T cells are increased in advanced disease and are associated with decreased HIV-specific cytolysis. J Immunol. 2006;176:1274–1280. doi: 10.4049/jimmunol.176.2.1274. [DOI] [PubMed] [Google Scholar]
- 56.Elrefaei M, Ventura FL, Baker CA, Clark R, Bangsberg DR, Cao H. HIV-specific IL-10-positive CD8+ T cells suppress cytolysis and IL-2 production by CD8+ T cells. J Immunol. 2007;178:3265–3271. doi: 10.4049/jimmunol.178.5.3265. [DOI] [PubMed] [Google Scholar]
- 57.Nordström I, Nurkkala M, Collins LV, Eriksson K. CD8+ T-cells suppress antigen-specific and allogeneic CD4+ T-cell responses to herpes simplex virus type 2-infected human dendritic cells. Viral Immunol. 2005;18:616–626. doi: 10.1089/vim.2005.18.616. [DOI] [PubMed] [Google Scholar]
- 58.Popescu I, Macedo C, Abu-Elmagd K, Shapiro R, Hua Y, Thomson AW, Morelli AE, Storkus WJ, Metes D. EBV-specific CD8+ T cell reactivation in transplant patients results in expansion of CD8+ type-1 regulatory T cells. Am J Transplant. 2007;7:1215–1223. doi: 10.1111/j.1600-6143.2007.01740.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.