

Abstract
Inflammatory reaction is a fundamental defense mechanism against threat towards normal integrity and physiology. On the other hand, chronic diseases such as obesity, type 2 diabetes, hypertension and atherosclerosis, have been causally linked to chronic, low-grade inflammation in various metabolic tissues. Recent cross-disciplinary research has led to identification of hypothalamic inflammatory changes that are triggered by overnutrition, orchestrated by hypothalamic immune system, and sustained through metabolic syndrome-associated pathophysiology. While continuing research is actively trying to underpin the identity and mechanisms of these inflammatory stimuli and actions involved in metabolic syndrome disorders and related diseases, proinflammatory IκB kinase-β (IKKβ), the downstream nuclear transcription factor NF-κB and some related molecules in the hypothalamus were discovered to be pathogenically significant. This article is to summarize recent progresses in the field of neuroendocrine research addressing the central integrative role of neuroinflammation in metabolic syndrome components ranging from obesity, glucose intolerance to cardiovascular dysfunctions.
Keywords: Inflammation, Type 2 diabetes, Obesity, IKKβ/NF-κB pathway, CNS, Hypothalamus
1. Introduction
While inflammatory responses in acute damages and disorders are necessary for homeostatic and defensive mechanisms of repair, regeneration and healing, chronic inflammatory signals in low-grade magnitude promote the development of a host of disorders ranging from cancer, diabetes, hypertension and cardiovascular disorders (CVDs). The etiologies of such chronic inflammatory factors are varied: in some cases, genetic inflammatory components work as a risk of developing chronic inflammatory diseases [1–3], and on the other hand, environmental factors such as smoke, stress, endocrine-disrupting pollutants and overnutrition (caloric excess) have been implicated as prominent triggering factors in the development and propagation of inflammation-related diseases [4,5]. Studies across the fields of immunology and endocrinology have established that caloric excess induces persistent inflammation in the circulation and peripheral metabolic tissues, disrupts metabolic homeostasis of the body, and thus contributes to a family of disorders including obesity, insulin resistance, glucose intolerance, hyperlipidemia and hypertension, collectively known as metabolic syndrome [6–14]. Unlike classical inflammatory responses, the root of such overnutrition-induced inflammation, descriptively termed “metabolic inflammation” [7,8,15–17] or “metaflammation” [9], is not necessarily pathogen borne but a result of metabolic homeostatic abnormalities. Interdisciplinary studies in neuroendocrinology and neuroimmunology have begun to elucidate the causal involvement of neuroinflammation in metabolic syndrome and related diseases, a phenomenon which did not receive research attention till recent years.
The central nervous system (CNS), in particular the hypothalamus, has been recognized to play a decisive regulatory role in maintaining metabolic homeostasis. Inflammation in the brain, which can be induced under chronic overnutrition, can disrupt neurohormone- and neurotransmitter-mediated central regulatory functions to propagate obesity and related disorders [6,8,15,16,18]. Lately, neuroinflammation has been recognized to have an expansive contribution towards health problems and diseases [19–30]. The proinflammatory axis comprising IκB kinase-β (IKKβ) and its downstream nuclear transcription factor NF-κB (IKKβ⧸NF-κB signaling) in the hypothalamus has been shown to be upregulated under overnutrition and promote chronic energy imbalance and changes in fat mass and body weight [19,21,22,24–27,30]. Interestingly, IKKβ/NF-κB-driven hypothalamic inflammation can employ parallel body weight-independent mechanisms to catapult to imbalance of glucose homeostasis, mediated by impaired insulin secretion, insulin resistance and glucose intolerance [21,25,27,31], or serve as an uncoupling point of obesity and hypertension [26]. Recently, non-neuronal cell types like astroglia and microglia have been shown to act as additional platforms of inducing hypothalamic inflammation in diet-induced obesity [32,33], thus broadening the scope of central inflammation-mediated metabolic dysfunctions. The current review describes the position of overnutrition-induced metabolic inflammation in the pathogenesis of a wide spectrum of diseases, emphasizing the underlying principal mediators and signaling pathways and the potential therapeutic implications.
2. Neuroinflammatory basis of obesity
Overweight and obesity are major risk factors for many diseases such as hyperglycemia, T2D, hypertension, atherosclerosis, less high-density lipoprotein cholesterol levels, and hypertriglyceridemia, and historically, when patients have three or more of these symptoms with or without obesity, the condition can be referred to as ‘metabolic syndrome’ [17,34–38]. Recent studies indicate that excessive body weight and obesity can also be a facilitator and predictor of neurodegenerative diseases [16,17], thus possibly further enlarging the definition of ‘metabolic syndrome’. Although pathogenic mechanisms encompassing this complex spectrum of disorders are yet to be fully understood, epidemiological, clinical and research studies have causally linked inflammatory factors and acute phase reactants such as C-reactive protein (CRP) [39–42], TNF-alpha (TNF-α) [40], IL-6 [42,43] and soluble adhesion molecules [44–47] to metabolic disorders. It has been widely accepted that there exists a chronic low-grade inflammation under conditions of overweight or obesity [6–8,15–17,41,48]. Evidences from experimental and clinical studies have unequivocally demonstrated that excess of caloric intake can evoke an inflammatory response, inscribing overfeeding to be a triggering factor of inflammation. The subsequent disease response is sustained via signaling pathways in metabolic tissues which involve local, intracellular actions of inflammatory molecules [6,9,11–13,49–53]. Recent studies unveiled that some of the intracellular pathways of inflammation in the hypothalamus have causative roles in weight gain and related disorders (reviewed in [7,8]). While the hypothalamus acts as the master regulator of energy balance by sensing metabolic cues and modulating the neurohormonal and neurotransmitter systems via endocrine signaling, trophic actions, complex neuronal plasticity and projections into the autonomic controlling centers of the brain [54–56], inflammation in the hypothalamus can affect many, if not all, of these critical regulatory machineries to provide a neuropathological basis for the development of metabolic diseases [8,17,57–63].
The mediobasal hypothalamus (MBH) plays a fundamental role in energy homeostasis, and the primary cell types governing this process are the orexigenic axis comprising the neuropeptide Y (NPY)/agouti-related peptide (AGRP) neurons and anorexigenic pro-opio-melanocortin (POMC) neurons. Thus, functionally, POMC and AGRP neurons reciprocally regulate energy homeostasis via negative and positive energy balance actions respectively and are also regulated by leptin in opposite manners [64–68]. In the event of metabolic inflammation, it seems that these first-order neurons get attacked readily, which negatively impact neuronal regulatory cascades such as leptin and insulin signaling, and also compromise the secretion of anorexigenic POMC-derived α-melanocyte stimulating hormone (α-MSH) and cocaine- and amphetamine-regulated transcript (CART), altogether resulting in increased appetite along with central leptin and insulin resistance to cause feeding and energy imbalance [30]. Mechanistically, these effects have been causally linked to overactivation of hypothalamic IKKβ/NF-κB pathway [30] and some relevant downstream mediators of leptin and insulin signaling inhibitors such as suppressor of cytokine signaling-3 (SOCS3) [69–71] and protein tyrosine phosphatase 1B [72]. Using experimental models of high-fat diet (HFD) feeding or intra-cerebroventricular lipid infusion in rodents to introduce overnutrition-induced hypothalamic inflammation, researchers have found that lipid excess activates hypothalamic IKKβ/NF-κB [19,22,25,30], consequently impairing hypothalamic leptin and insulin signaling to mediate weight gain and metabolic dysfunctions [30]. It has been shown that, mice with AGRP neuron-specific IKKβ ablation were partially protected from HFD-induced hyperphagia and obesity [30]. Ablation of IKKβ in POMC neurons in mice is insufficient to prevent HFD-induced obesity [26], but interestingly, it is able to reduce the cachectic effects resulting from illness-inducing classical inflammatory offenses by such as lipopolysaccharides (LPS) [73]. It was also reported that acute inflammation-induced Pomc gene activation results in food intake suppression, physical inactivity and cachectic changes via a pathway that is independent of leptin and STAT3 signaling in POMC neurons [74], thus indicating alternative hypothalamic pathway(s) in conveying cachectic inflammation to affect POMC cells [75]. Altogether, future research is much needed to depict the divergent roles and pathways of hypothalamic inflammation in obesogenic weight gain vs. cachectic weight loss outcomes.
In addition, molecules which interact with IKKβ/NF-κB signaling cascade, such as myeloid differentiation primary response gene 88 (MyD88) [19] or c-Jun N-terminal kinase 1 (JNK1) [28,76–80], also play significant roles in the development of obesity, insulin resistance and dyslipidemia. Notably, these inflammatory pathways that mediate insulin insensitivity are closely linked to an intracellular endoplasmic reticulum (ER) stress process [81]. ER stress has been known to activate NF-κB via signaling cross talk between IKKβ/NF-κB pathway and unfolded protein response (UPR) elements via PKR-like ER kinase, inositol requiring enyzyme-1, and activating transcription factor-6 [82–84]. Under overnutritional condition, there is a positive feedback between hypothalamic IKKβ/NF-κB activation and induction of neuronal ER stress [27,30]. In fact, mice with genetic ablation of ER stress activator X-box binding protein-1 have been shown to be susceptible to central leptin resistance and diet-induced weight gain [85]. In an attempt to validate the possible therapeutic potential of targeting these inflammatory mediators, researchers found that brain-specific ablation of IKKβ [30] or MyD88 [19], chemical chaperone-mediated lowering of hypothalamic ER stress [85], MBH-specific inhibition of autophagy defect [21], and whole-body knockdown of NF-κB subunit p50 [86], can all similarly improve leptin sensitization and alleviate diet-induced weight gain and obesity. Furthermore, it was reported that JNK1 knockout in the brain but not in other tissues [28,87], just like whole-body knockdown of JNK1 [76], provided anti-obesity effect in mice. Consistently, brain-specific SOCS3 knockout mice displayed anti-obesity effects with improved central leptin sensitivity when animals were subjected to HFD feeding [88].
However, it is yet to be fully understood, what are the inducers of hypothalamic IKKβ/NF-κB signaling activation in the context of obesity and related metabolic diseases. Studies on Toll-like receptors (TLRs) of the innate immune system revealed that TLR2 [89,90] or TLR4 [22,91] knockdown in mice could significantly reduce HFD-induced inflammation and protect against dietary obesity. Also, inflammasomes, which are known as macromolecular innate immune cell sensors, have been recognized to increase metabolic stress, insulin resistance and obesity [92–96]. Studies have also tackled Nod-like receptor 3 (NLRP3) inflammasome components, which can activate IKKβ/NF-κB pathway through inflammatory IL-1β and IL-18 release. When NLRP3 was ablated in HFD-fed mice, it led to improved glucose tolerance and insulin sensitivity and prevented obesity-induced activation of adipose tissue interferon-γ expression [95]. Collectively, these data suggested a potential role of inflammasomes in mediating IKKβ/NF-κB-dependent metabolic inflammation, and that molecular intervention in inflammasome-mediated pathways could improve obesity-associated inflammation and metabolic dangers.
It is also important to point out that glial cells, such as microglia and astrocytes, are involved in overnutrition-induced central inflammation. Research has demonstrated that early postnatal nutritional overload through HFD feeding leads to excessive production of IL-6 in activated microglia and consequent weight gain [97]. In concurrence with the microglial participation, the astrocytes have been found to be activated by various stimuli such as proinflammatory cytokines [98] and saturated fatty acids [99] – which further trigger the release of inflammatory cytokines like TNF-α and IL-6 that can contribute to central metabolic inflammation. Also in rodents, HFD feeding has been reported to result in reactive gliosis in the mediobasal hypothalamus and weight gain [33]. Therefore, central metabolic inflammation is evidently a consummation of both neuronal and non-neuronal contributions and their possible cross-talks. In sum, central metabolic inflammation exhibits a functionally integrative point of overnutrition-induced obesity. Mechanistically, this central inflammatory state is sustained by hypothalamic stress and inflammation that is critically mediated by IKKβ/NF-κB and its related signaling components (Fig. 1). Based on promising intervention results in animals, therapeutic strategies for human patients may expect to follow by targeting the key elements of inflammation in the neuronal and glial cells in order to counteract obesity and related co-morbidities.
Fig. 1.
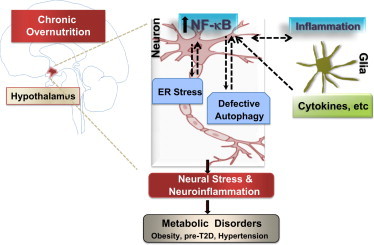
Role of hypothalamic neuroinflammation in metabolic disorders. Overnutrition activates pro-inflammatory IKK-β/NF-κB in the hypothalamic neurons, which occurs through intracellular neuronal stressors like ER stress, defective autophagy machinery as well as glia-neuron inflammatory cross-talk, collectively leading to hypothalamic metabolic deregulations that cause a spectrum of metabolic disorders.
3. Neuroinflammatory basis of diabetic glucose disorders
The CNS can regulate whole-body glucose balance, and the hypothalamus governs the peripheral and central glucose homeostasis via multiple complex neural networks through neuropeptide and neurotransmitter actions [68,100–102]. For example, central insulin and leptin signaling have been shown to be required for maintaining glucose homeostasis at least via suppressing hepatic glucose production and promoting peripheral glucose uptake (reviewed in [6,7,15,17]). Several studies have echoed the regulatory action of hypothalamic arcuate nucleus-mediated leptin signaling on glucose metabolism independent of its actions on feeding or body weight [101,103–105]. Leptin deficiency can impair glucose metabolism irrespective of being obese [105] or lean [104], and leptin treatment in some models effectively improves these glucose disorders without requiring changes of food intake or body weight [103,104]. It has also been appreciated that glutamatergic or GABAergic neurotransmission of the hypothalamus can acutely mediate hypothalamic control of glucose homeostasis [106,107]. From disease perspective, while literature over time has convincingly established the role of overnutrition-induced weight gain and peripheral metabolic inflammation on development of glucose intolerance, insulin resistance, insulin insensitivity and leptin resistance [6,11,14,81], recent findings revealed that systemic glucose homeostasis can be impaired by overnutrition-induced pathological activation of central immune system through body weight-independent mechanisms [68,100–102]. Prolonged metabolic stress through overnutrition, in part through subjecting hypothalamic immune cells to a chronic state of overactivation, disrupts the normal physiological functioning of central insulin and leptin signaling that are needed for the control of systemic glucose homeostasis (reviewed in [6,7,15,17]). While molecular mechanisms of central dysregulation in glucose disorders largely remain to be investigated, a body of literature over the past decade has recognized IKKβ/NF-κB and JNK pathways to be the targets of metabolic insults for the development of peripheral insulin resistance and glucose intolerance. For example, overnutrition-induced increases of intracellular stimuli, such as ligands of CRP [39], TNF-alpha [40], IL-6 [42], TLRs [108], were all shown to activate IKKβ/NF-κB to cause insulin resistance and T2D. Also, intracellular stresses including reactive oxygen species (ROS) and ER stress are widely demonstrated to exalt deleterious effects on glucose balance [30,41,48]. By targeting hypothalamic inflammation, existing experimental data has provided initial evidence to support a direct link between diet-induced activation of pro-inflammatory factors in the hypothalamus and development of T2D/pre-T2D that can occur in an obesity-independent manner [27]. These findings suggest that while inhibition of hypothalamic inflammation leads to improvement of systemic glucose metabolism that is secondary to the anti-obesity effect [21,27,30,109], this therapy can simultaneously introduce a direct neural action from inhibiting hypothalamic inflammation in improving central control of glucose homeostasis. Further research is much needed to decipher the central mechanistic pathways that can primarily link hypothalamic inflammation to the development of glucose disorders.
Regarding superoxides, we need to keep in mind that, while excessive production of ROS is classically associated with oxidative stress, pathology of insulin resistance, T2D, muscular dystrophies and aging [110–113], recent evidences showed that ROS can facilitate beneficial actions depending on the amount and source of its generation [110,114–118]. ROS accumulation has been observed in glucose-utilizing POMC neurons even when the body is in a state of positive energy balance, which can solicit a satiety-promoting action of POMC neuron-generated ROS upon glucose triggering [114]. In the long run, sustained elevated ROS levels could be detrimental for the POMC neuronal functioning and therefore promote weight gain. By contrast, during a state of negative energy balance, the overactive NPY/AGRP neurons which utilize free fatty acids but not glucose as their primary substrate, do not accrue high levels of ROS [115]. Such decrement of free radicals even during increased substrate utilization is reported to be mediated by a feed-forward buffering action of ROS scavenger uncoupling protein 2 (UCP 2) [115]. On the other hand, uncontrolled ROS production in NPY/AGRP neurons impairs the neuronal firing of these cells [115]. Evidently, the beneficial or deleterious actions of ROS are determined by the source of its fuels and the amount of its accumulation at a given time in a particular cellular group. In this line, other studies have also reported that ROS, when generated within a boundary of low concentrations in the plasma membrane or the endomembrane can provide a protective action via normal cellular functioning and intracellular signaling which could be beneficial for reversal of insulin resistance [110,116,117]. These findings give a new perspective of the protective actions of ROS which was classically believed to be an intra-cellular stressor.
Taken together, these experimental evidences have in general pointed to a causal link between the hypothalamic inflammatory mediators and T2D/pre-T2D, and inhibitors of inflammatory molecules can be considered as prospective therapeutic options for developing novel anti-T2D treatments. Indeed under experimental conditions of using pharmacological tools, biochemical modulators or genetic manipulations, brain specific annihilation of metabolic inflammation via IKKβ/NF-κB inhibition and ER stress suppression, have both been shown to improve insulin action and glucose metabolism [27,30]. Recently, some epidemiological evidence interestingly demonstrated the lower mortality among mild overweight or small-degree (grade 1) obesity, while higher levels of obesity were associated with a significant risk of death [119], further highlighting the value of developing weight control-independent solutions for treating T2D and related deleterious complications.
4. Neuroinflammatory basis of hypertension and cardiovascular disease
Hypertension is an integral component of obesity-related metabolic disorders and is a predictor of CVDs such as atherosclerosis and stroke [17,120]. While cardiac output, vascular compliance, blood volume and endocrine balance are major determinants of blood pressure, vasoconstriction and sodium retention are considered as primary contributors of hypertension and both processes are closely regulated by the CNS. Comparatively, obesity-related hypertension is governed by even more complex interactions among large numbers of genetic and environmental factors. Earlier studies implicated adipose tissue regulation of cardiovascular–renal functioning and leptin-mediated sympatho-excitation to be causally important for obesity-induced hypertension [121]. Further studies recognized the contribution of inflammation in some of the peripheral components such as smooth muscles, endothelial cells and vascular macrophages towards the development of obesity-related hypertension [122–124]. Ample evidence now points towards the critical involvement of neuroendocrine modulators such as melanocortin system [125,126] and the sympathetic nervous system (SNS) [26,27] in obesity-associated hypertension. Although, the mechanism through which obesity directly induces hypertension is still an emerging area of investigation, an induction of neuroinflammatory condition has been observed in obesity-related hypertension. Of note, recent research has identified hypothalamic metabolic inflammation as a vital pathogenic link between obesity and blood pressure increase [26,27]. A salient observation of this study was that obesity-induced activation of the hypothalamic inflammatory system, and more specifically, the IKKβ/NF-κΒ pathway in POMC neurons, could account for the effects of chronic HFD in causing hypertension, and therapeutically, inhibition of IKKβ/NF-κΒ pathway in POMC neurons [26] or pharmacological inhibition of brain ER stress that lies upstream of NF-κB activation [27], can acutely alleviate sympathetic upregulation and hypertensive outcomes prior to the chronic effects of obesity reduction. Thus, the convergent point of hypothalamic metabolic inflammation and SNS overactivation-mediated hypertension represents a novel platform for antihypertensive drug targeting. It is even more meaningful, since central NF-κB pathway gains further support for anti-hypertensive drug targeting in general, in addition to obesity-related hypertension, as the inflammatory machinery has been found to be involved with several other forms of hypertension such as essential hypertension, spontaneous hypertension, and angiotensin II-induced hypertension [127–129], albeit the downstream mechanisms of NF-κB could be different among these types of hypertension.
Uncontrolled high blood pressure is a predisposing risk factor of stroke. A growing number of recent investigations have established a critical role of brain inflammation in pathogenesis of ischemia and stroke [130–132] in an obesity-dependent or obesity-independent manner. As a prophylactic or therapeutic option, several anti-inflammatory agents have been proven successful in treating stroke [133–135], and of interest, inhibition of brain IKKβ/NF-κB also provides a striking protection against ischemia [136–139]. In addition, anti-inflammatory treatment with IL-1 receptor blocker in a clinical trial showed improvement in neurological impairments in stroke patients [140]. Thus a “primed” inflammatory environment in the brain can promote the risk of ischemia and stroke, and conversely, curtailing neuroinflammation could be therapeutically effective. However, an important caveat for use of anti-inflammatory drugs in stroke patients is that, stroke is often accompanied with the risk of adaptive immune response of lymphoid organs, hence druggable choices should be focused on selective anti-neuroinflammatory factors without spillovers on systemic immune functioning [141].
5. From bench to bedside: perspectives and challenges
Although basic experimental research on animal models are the foundation of designing therapeutic strategies and drug discovery, these models that are subjected to high fat/calorie feeding, intra-brain proinflammatory challenges or gene manipulations cannot unerringly clone the different pathological manifestations in patients across diverse race, feeding habits, behavioral practices and environmental cues. As a result, our biggest challenge remains in recapitulating the complex and sundry human disease conditions. While a ‘top–down’ approach could be attempting to replicate the disease symptoms and pathological causes, the reverse ‘bottom–up’ strategy of screening human populations for cellular and genetic alterations that mirror observed aberrations in experimental models will provide meaningful information. Recent report by Thaler et al. [33] depicting the association between hypothalamic neuronal and glial injury with obesity both in humans and rodent models, as well as the discovered role of lipid sensor GPR120 in the control of energy balance in humans and rodents [142] aptly recounts this strategy. Another key aspect to be taken into account is the paradoxical actions of several classical toxic and degenerative elements in health and pathophysiological state. As we have discussed earlier, overt concentrations of ROS contribute to inflammation-mediated metabolic deregulations, but subtle presence in confined regions convene a protective action [114,115,118]. Similar considerations need to be given to certain pro-inflammatory cytokines which have anorexic effects, as well as some cytokines which have anti-inflammatory actions. Of interest, slight weight gain has been shown to delay aging and mortality [119], which should increase our caution in order to develop appropriate anti-obesity strategies. Largely the challenge towards meaningful translation of basic research findings to therapeutic options for patients remains in identifying the balance between the right environment, concentration and duration of the insults in experimental animal models. Regardless, chronic inflammation in the body, in particular in the CNS, represents a bold mechanistic player for a spectrum of metabolic syndrome-related diseases, and is clearly worth being a target for combating these human diseases.
6. Concluding remarks
Convincing evidences from research during the past decade have established the contribution of neuroinflammation and in particular hypothalamic inflammation in the pathogenesis of obesity, insulin resistance, T2D/pre-T2D and CVD. The effects of metabolic inflammation due to chronic overnutrition is not just circumscribed to the peripheral metabolic tissues as was believed earlier, but is etiologically important for the CNS and especially the hypothalamus which drives central dysregulation of metabolic homeostasis. Hypothalamic neuroinflammation exerts its enervating actions via neurohormonal as well as autonomic regulations of nutrient sensing and energy balance, leading to a spectrum of metabolic syndrome disorders that mediate T2D and CVDs. The IKKβ/NF-κB pathway in the hypothalamus has been recognized as the prime machinery directing overnutrition-induced metabolic dysfunctions, and in this process, several forms of intracellular organelle changes in hypothalamic cells including ER stress, oxidative stress and autophagic defect, all of which are known to foster metabolic impairment in concert with IKKβ/NF-κB activation, have been proven to contribute towards disease manifestations (Fig. 1). In sum, current findings have substantially advanced our knowledge in the field of overnutrition-induced central inflammation and the mechanisms of metabolic dysfunctions. Also, anti-inflammatory therapies for chronic diseases have been successful to some extent. However, high risks to activate compensatory or counteracting mechanisms in some cases pose a challenge to strategize appropriate therapeutic options. Therefore, continuing research in understanding the detailed characteristics of neuroinflammatory mechanisms will be indispensable for harnessing the promising empiric findings to cogent therapeutic options, so that effective and selective therapies can be developed to treat patients without blunting other important aspects of health.
Conflict of Interest
None declared.
Acknowledgments
The authors sincerely thank Cai lab members for related research. D.C. is supported by NIH R01 DK078750, R01 AG031774, and American Diabetes Association Grant 1-12-BS-20. D.C. is a recipient of Irma T. Hirschl Scholarship.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Reference
- 1.Glass C.K., Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nature Reviews Immunology. 2006;6(1):44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- 2.Chawla A., Repa J.J., Evans R.M., Mangelsdorf D.J. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294(5548):1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 3.Heap G.A., van Heel D.A. The genetics of chronic inflammatory diseases. Human Molecular Genetics. 2009;18(R1):R101–R106. doi: 10.1093/hmg/ddp001. [DOI] [PubMed] [Google Scholar]
- 4.Richter C.A., Birnbaum L.S., Farabollini F., Newbold R.R., Rubin B.S., Talsness C.E., Vandenbergh J.G., Walser-Kuntz D.R., vom Saal F.S. In vivo effects of bisphenol A in laboratory rodent studies. Reproductive Toxicology. 2007;24(2):199–224. doi: 10.1016/j.reprotox.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz M.W., Porte D., Jr. Diabetes, obesity, and the brain. Science. 2005;307(5708):375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 6.Cai D. NFkappaB-mediated metabolic inflammation in peripheral tissues versus central nervous system. Cell Cycle. 2009;8(16):2542–2548. doi: 10.4161/cc.8.16.9386. [DOI] [PubMed] [Google Scholar]
- 7.Cai D., Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Annals of the New York Academy of Sciences. 2011;1243:E1–39. doi: 10.1111/j.1749-6632.2011.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai D., Liu T. Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging (Albany, NY) 2012;4(2):98–115. doi: 10.18632/aging.100431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregor M.F., Hotamisligil G.S. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 10.Lehrke M., Lazar M.A. Inflamed about obesity. Nature Medicine. 2004;10(2):126–127. doi: 10.1038/nm0204-126. [DOI] [PubMed] [Google Scholar]
- 11.Lumeng C.N., Saltiel A.R. Inflammatory links between obesity and metabolic disease. Journal of Clinical Investigation. 2011;121(6):2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk S., Saberi M., Olefsky J.M. Insulin sensitivity: modulation by nutrients and inflammation. Journal of Clinical Investigation. 2008;118(9):2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoelson S.E., Goldfine A.B. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nature Medicine. 2009;15(4):373–374. doi: 10.1038/nm0409-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odegaard J.I., Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai D. One step from prediabetes to diabetes: hypothalamic inflammation? Endocrinology. 2012;153(3):1010–1013. doi: 10.1210/en.2011-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends in Endocrinology & Metabolism. 2013;24(1):40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai D. Neuroinflammation in overnutrition-induced diseases. Vitamins & Hormones. 2013;91:195–218. doi: 10.1016/B978-0-12-407766-9.00008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thaler J.P., Schwartz M.W. Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151(9):4109–4115. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinridders A., Schenten D., Konner A.C., Belgardt B.F., Mauer J., Okamura T., Wunderlich F.T., Medzhitov R., Bruning J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cellular Metabolism. 2009;10(4):249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Tang Y., Cai D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nature Cell Biology. 2012;14(10):999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng Q., Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. Journal of Biological Chemistry. 2011;286(37):32324–32332. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milanski M., Degasperi G., Coope A., Morari J., Denis R., Cintra D.E., Tsukumo D.M., Anhe G., Amaral M.E., Takahashi H.K. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. Journal of Neuroscience. 2009;29(2):359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milanski M., Arruda A.P., Coope A., Ignacio-Souza L.M., Nunez C.E., Roman E.A., Romanatto T., Pascoal L.B., Caricilli A.M., Torsoni M.A. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes. 2012;61(6):1455–1462. doi: 10.2337/db11-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh I., Thaler J.P., Ogimoto K., Wisse B.E., Morton G.J., Schwartz M.W. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. American Journal of Physiology – Endocrinology and Metabolism. 2010;299(1):E47–E53. doi: 10.1152/ajpendo.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Posey K.A., Clegg D.J., Printz R.L., Byun J., Morton G.J., Vivekanandan-Giri A., Pennathur S., Baskin D.G., Heinecke J.W., Woods S.C. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. American Journal of Physiology – Endocrinology and Metabolism. 2009;296(5):E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purkayastha S., Zhang G., Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nature Medicine. 2011;17(7):883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purkayastha S., Zhang H., Zhang G., Ahmed Z., Wang Y., Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(7):2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabio G., Cavanagh-Kyros J., Barrett T., Jung D.Y., Ko H.J., Ong H., Morel C., Mora A., Reilly J., Kim J.K. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes & Development. 2010;24(3):256–264. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., Li B., Liu G., Cai D. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497(7448):211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X., Zhang G., Zhang H., Karin M., Bai H., Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arruda A.P., Milanski M., Coope A., Torsoni A.S., Ropelle E., Carvalho D.P., Carvalheira J.B., Velloso L.A. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011;152(4):1314–1326. doi: 10.1210/en.2010-0659. [DOI] [PubMed] [Google Scholar]
- 32.Horvath T.L., Sarman B., Garcia-Caceres C., Enriori P.J., Sotonyi P., Shanabrough M., Borok E., Argente J., Chowen J.A., Perez-Tilve D. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14875–14880. doi: 10.1073/pnas.1004282107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thaler J.P., Yi C.X., Schur E.A., Guyenet S.J., Hwang B.H., Dietrich M.O., Zhao X., Sarruf D.A., Izgur V., Maravilla K.R. Obesity is associated with hypothalamic injury in rodents and humans. Journal of Clinical Investigation. 2012;122(1):153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carantoni M., Zuliani G., Volpato S., Palmieri E., Mezzetti A., Vergnani L., Fellin R. Relationships between fasting plasma insulin, anthropometrics, and metabolic parameters in a very old healthy population. Associazione Medica Sabin. Metabolism. 1998;47(5):535–540. doi: 10.1016/s0026-0495(98)90236-0. [DOI] [PubMed] [Google Scholar]
- 35.Ferrannini E., Haffner S.M., Mitchell B.D., Stern M.P. Hyperinsulinaemia: the key feature of a cardiovascular and metabolic syndrome. Diabetologia. 1991;34(6):416–422. doi: 10.1007/BF00403180. [DOI] [PubMed] [Google Scholar]
- 36.Gray R.S., Fabsitz R.R., Cowan L.D., Lee E.T., Howard B.V., Savage P.J. Risk factor clustering in the insulin resistance syndrome. The Strong Heart Study. American Journal of Epidemiology. 1998;148(9):869–878. doi: 10.1093/oxfordjournals.aje.a009712. [DOI] [PubMed] [Google Scholar]
- 37.Reaven G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Hernandez H., Simental-Mendia L.E., Rodriguez-Ramirez G., Reyes-Romero M.A. Obesity and inflammation: epidemiology, risk factors, and markers of inflammation. International Journal of Endocrinology. 2013;2013:678159. doi: 10.1155/2013/678159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anty R., Bekri S., Luciani N., Saint-Paul M.C., Dahman M., Iannelli A., Amor I.B., Staccini-Myx A., Huet P.M., Gugenheim J. The inflammatory C-reactive protein is increased in both liver and adipose tissue in severely obese patients independently from metabolic syndrome, Type 2 diabetes, and NASH. American Journal of Gastroenterology. 2006;101(8):1824–1833. doi: 10.1111/j.1572-0241.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 40.Hotamisligil G.S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 41.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 42.Mohamed-Ali V., Goodrick S., Rawesh A., Katz D.R., Miles J.M., Yudkin J.S., Klein S., Coppack S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. Journal of Clinical Endocrinology and Metabolism. 1997;82(12):4196–4200. doi: 10.1210/jcem.82.12.4450. [DOI] [PubMed] [Google Scholar]
- 43.Heinrich P.C., Castell J.V., Andus T. Interleukin-6 and the acute phase response. Biochemical Journal. 1990;265(3):621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingelsson E., Hulthe J., Lind L. Inflammatory markers in relation to insulin resistance and the metabolic syndrome. European Journal of Clinical Investigation. 2008;38(7):502–509. doi: 10.1111/j.1365-2362.2008.01962.x. [DOI] [PubMed] [Google Scholar]
- 45.Haught W.H., Mansour M., Rothlein R., Kishimoto T.K., Mainolfi E.A., Hendricks J.B., Hendricks C., Mehta J.L. Alterations in circulating intercellular adhesion molecule-1 and L-selectin: further evidence for chronic inflammation in ischemic heart disease. American Heart Journal. 1996;132(1 Pt 1):1–8. doi: 10.1016/s0002-8703(96)90383-x. [DOI] [PubMed] [Google Scholar]
- 46.Koh K.K., Han S.H., Quon M.J. Inflammatory markers and the metabolic syndrome: insights from therapeutic interventions. Journal of the American College of Cardiology. 2005;46(11):1978–1985. doi: 10.1016/j.jacc.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 47.Kressel G., Trunz B., Bub A., Hulsmann O., Wolters M., Lichtinghagen R., Stichtenoth D.O., Hahn A. Systemic and vascular markers of inflammation in relation to metabolic syndrome and insulin resistance in adults with elevated atherosclerosis risk. Atherosclerosis. 2009;202(1):263–271. doi: 10.1016/j.atherosclerosis.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 48.Hotamisligil G.S. Inflammation and endoplasmic reticulum stress in obesity and diabetes. International Journal of Obesity (London) 2008;32(Suppl. 7):S52–S54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai D., Frantz J.D., Tawa N.E., Jr., Melendez P.A., Oh B.C., Lidov H.G., Hasselgren P.O., Frontera W.R., Lee J., Glass D.J. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119(2):285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 50.Cai D., Yuan M., Frantz D.F., Melendez P.A., Hansen L., Lee J., Shoelson S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nature Medicine. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donath M.Y., Shoelson S.E. Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 52.Ferrante A.W., Jr. Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. Journal of Internal Medicine. 2007;262(4):408–414. doi: 10.1111/j.1365-2796.2007.01852.x. [DOI] [PubMed] [Google Scholar]
- 53.Glass C.K., Olefsky J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cellular Metabolism. 2012;15(5):635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz M.W., Woods S.C., Porte D., Jr., Seeley R.J., Baskin D.G. Central nervous system control of food intake. Nature. 2000;404(6778):661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 55.Zelzer E., Levy Y., Kahana C., Shilo B.Z., Rubinstein M., Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO Journal. 1998;17(17):5085–5094. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blouet C., Schwartz G.J. Hypothalamic nutrient sensing in the control of energy homeostasis. Behavioural Brain Research. 2010;209(1):1–12. doi: 10.1016/j.bbr.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 57.Coll A.P., Farooqi I.S., O’Rahilly S. The hormonal control of food intake. Cell. 2007;129(2):251–262. doi: 10.1016/j.cell.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 59.Flier J.S. Neuroscience. Regulating energy balance: the substrate strikes back. Science. 2006;312(5775):861–864. doi: 10.1126/science.1127971. [DOI] [PubMed] [Google Scholar]
- 60.Lam T.K., Schwartz G.J., Rossetti L. Hypothalamic sensing of fatty acids. Nature Neuroscience. 2005;8(5):579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 61.Morton G.J., Cummings D.E., Baskin D.G., Barsh G.S., Schwartz M.W. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 62.Sandoval D., Cota D., Seeley R.J. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annual Review of Physiology. 2008;70:513–535. doi: 10.1146/annurev.physiol.70.120806.095256. [DOI] [PubMed] [Google Scholar]
- 63.Marino J.S., Xu Y., Hill J.W. Central insulin and leptin-mediated autonomic control of glucose homeostasis. Trends in Endocrinology & Metabolism. 2011;22(7):275–285. doi: 10.1016/j.tem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barsh G.S., Schwartz M.W. Genetic approaches to studying energy balance: perception and integration. Nature Reviews Genetics. 2002;3(8):589–600. doi: 10.1038/nrg862. [DOI] [PubMed] [Google Scholar]
- 65.Saper C.B., Chou T.C., Elmquist J.K. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36(2):199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz M.W., Woods S.C., Seeley R.J., Barsh G.S., Baskin D.G., Leibel R.L. Is the energy homeostasis system inherently biased toward weight gain? Diabetes. 2003;52(2):232–238. doi: 10.2337/diabetes.52.2.232. [DOI] [PubMed] [Google Scholar]
- 67.Zigman J.M., Elmquist J.K. Minireview: from anorexia to obesity--the yin and yang of body weight control. Endocrinology. 2003;144(9):3749–3756. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]
- 68.Elmquist J.K., Coppari R., Balthasar N., Ichinose M., Lowell B.B. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. Journal of Comparative Neurology. 2005;493(1):63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 69.Howard J.K., Flier J.S. Attenuation of leptin and insulin signaling by SOCS proteins. Trends in Endocrinology & Metabolism. 2006;17(9):365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 70.Howard J.K., Cave B.J., Oksanen L.J., Tzameli I., Bjorbaek C., Flier J.S. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nature Medicine. 2004;10(7):734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 71.Myers M.G., Cowley M.A., Munzberg H. Mechanisms of leptin action and leptin resistance. Annual Review of Physiology. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 72.Zabolotny J.M., Kim Y.B., Welsh L.A., Kershaw E.E., Neel B.G., Kahn B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. Journal of Biological Chemistry. 2008;283(21):14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jang P.G., Namkoong C., Kang G.M., Hur M.W., Kim S.W., Kim G.H., Kang Y., Jeon M.J., Kim E.H., Lee M.S. NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. Journal of Biological Chemistry. 2010;285(13):9706–9715. doi: 10.1074/jbc.M109.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi X., Wang X., Li Q., Su M., Chew E., Wong E.T., Lacza Z., Radda G.K., Tergaonkar V., Han W. Nuclear factor kappaB (NF-kappaB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia. 2013;56(4):925–936. doi: 10.1007/s00125-013-2831-2. [DOI] [PubMed] [Google Scholar]
- 75.Clarke I.J. Whatever way weight goes, inflammation shows. Endocrinology. 2010;151(3):846–848. doi: 10.1210/en.2009-1470. [DOI] [PubMed] [Google Scholar]
- 76.Hirosumi J., Tuncman G., Chang L., Gorgun C.Z., Uysal K.T., Maeda K., Karin M., Hotamisligil G.S. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 77.Sabio G., Das M., Mora A., Zhang Z., Jun J.Y., Ko H.J., Barrett T., Kim J.K., Davis R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322(5907):1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sabio G., Cavanagh-Kyros J., Ko H.J., Jung D.Y., Gray S., Jun J.Y., Barrett T., Mora A., Kim J.K., Davis R.J. Prevention of steatosis by hepatic JNK1. Cellular Metabolism. 2009;10(6):491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Solinas G., Vilcu C., Neels J.G., Bandyopadhyay G.K., Luo J.L., Naugler W., Grivennikov S., Wynshaw-Boris A., Scadeng M., Olefsky J.M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cellular Metabolism. 2007;6(5):386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 80.Vallerie S.N., Furuhashi M., Fucho R., Hotamisligil G.S. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS One. 2008;3(9):e3151. doi: 10.1371/journal.pone.0003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng J., Lu P.D., Zhang Y., Scheuner D., Kaufman R.J., Sonenberg N., Harding H.P., Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and Cellular Biology. 2004;24(23):10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu P., Han Z., Couvillon A.D., Kaufman R.J., Exton J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Molecular and Cellular Biology. 2006;26(8):3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamazaki H., Hiramatsu N., Hayakawa K., Tagawa Y., Okamura M., Ogata R., Huang T., Nakajima S., Yao J., Paton A.W. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. Journal of Immunology. 2009;183(2):1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ozcan L., Ergin A.S., Lu A., Chung J., Sarkar S., Nie D., Myers M.G., Jr., Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cellular Metabolism. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 86.Gao Z., Yin J., Zhang J., He Q., McGuinness O.P., Ye J. Inactivation of NF-kappaB p50 leads to insulin sensitization in liver through post-translational inhibition of p70S6K. Journal of Biological Chemistry. 2009;284(27):18368–18376. doi: 10.1074/jbc.M109.007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Belgardt B.F., Mauer J., Wunderlich F.T., Ernst M.B., Pal M., Spohn G., Bronneke H.S., Brodesser S., Hampel B., Schauss A.C. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):6028–6033. doi: 10.1073/pnas.1001796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mori H., Hanada R., Hanada T., Aki D., Mashima R., Nishinakamura H., Torisu T., Chien K.R., Yasukawa H., Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature Medicine. 2004;10(7):739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 89.Caricilli A.M., Nascimento P.H., Pauli J.R., Tsukumo D.M., Velloso L.A., Carvalheira J.B., Saad M.J. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. Journal of Endocrinology. 2008;199(3):399–406. doi: 10.1677/JOE-08-0354. [DOI] [PubMed] [Google Scholar]
- 90.Himes R.W., Smith C.W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB Journal. 2010;24(3):731–739. doi: 10.1096/fj.09-141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsukumo D.M., Carvalho-Filho M.A., Carvalheira J.B., Prada P.O., Hirabara S.M., Schenka A.A., Araujo E.P., Vassallo J., Curi R., Velloso L.A. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 92.Church L.D., Cook G.P., McDermott M.F. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nature Clinical Practice Rheumatology. 2008;4(1):34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- 93.Lamkanfi M., Kanneganti T.D. The inflammasome: a remote control for metabolic syndrome. Cell Research. 2012;22(7):1095–1098. doi: 10.1038/cr.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schroder K., Zhou R., Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 95.Vandanmagsar B., Youm Y.H., Ravussin A., Galgani J.E., Stadler K., Mynatt R.L., Ravussin E., Stephens J.M., Dixit V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stienstra R., van Diepen J.A., Tack C.J., Zaki M.H., van d.V., Perera D., Neale G.A., Hooiveld G.J., Hijmans A., Vroegrijk I. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tapia-Gonzalez S., Garcia-Segura L.M., Tena-Sempere M., Frago L.M., Castellano J.M., Fuente-Martin E., Garcia-Caceres C., Argente J., Chowen J.A. Activation of microglia in specific hypothalamic nuclei and the cerebellum of adult rats exposed to neonatal overnutrition. Journal of Neuroendocrinology. 2011;23(4):365–370. doi: 10.1111/j.1365-2826.2011.02113.x. [DOI] [PubMed] [Google Scholar]
- 98.Belanger M., Allaman I., Magistretti P.J. Differential effects of pro- and anti-inflammatory cytokines alone or in combinations on the metabolic profile of astrocytes. Journal of Neurochemistry. 2011;116(4):564–576. doi: 10.1111/j.1471-4159.2010.07135.x. [DOI] [PubMed] [Google Scholar]
- 99.Gupta S., Knight A.G., Gupta S., Keller J.N., Bruce-Keller A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. Journal of Neurochemistry. 2012;120(6):1060–1071. doi: 10.1111/j.1471-4159.2012.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herman M.A., Kahn B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. Journal of Clinical Investigation. 2006;116(7):1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morton G.J. Hypothalamic leptin regulation of energy homeostasis and glucose metabolism. Journal of Physiology. 2007;583(Pt 2):437–443. doi: 10.1113/jphysiol.2007.135590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Plum L., Belgardt B.F., Bruning J.C. Central insulin action in energy and glucose homeostasis. Journal of Clinical Investigation. 2006;116(7):1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwartz M.W., Baskin D.G., Bukowski T.R., Kuijper J.L., Foster D., Lasser G., Prunkard D.E., Porte D., Jr., Woods S.C., Seeley R.J. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45(4):531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 104.Shimomura I., Hammer R.E., Ikemoto S., Brown M.S., Goldstein J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 105.Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J.M. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 106.Chan O., Paranjape S., Czyzyk D., Horblitt A., Zhu W., Ding Y., Fan X., M. Seashore, R. Sherwin. Increased GABAergic output in the ventromedial hypothalamus contributes to impaired hypoglycemic counterregulation in diabetic rats. Diabetes. 2011;60(5):1582–1589. doi: 10.2337/db10-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tong Q., Ye C., McCrimmon R.J., Dhillon H., Choi B., Kramer M.D., Yu J., Yang Z., Christiansen L.M., Lee C.E. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cellular Metabolism. 2007;5(5):383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shi H., Kokoeva M.V., Inouye K., Tzameli I., Yin H., Flier J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. Journal of Clinical Investigation. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H., Zhang G., Gonzalez F.J., Park S.M., Cai D. Hypoxia-inducible factor directs POMC gene to mediate hypothalamic glucose sensing and energy balance regulation. PLoS Biology. 2011;9(7):e1001112. doi: 10.1371/journal.pbio.1001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends in Pharmacological Sciences. 2011;32(2):82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 111.Murphy M.E., Kehrer J.P. Activities of antioxidant enzymes in muscle, liver and lung of chickens with inherited muscular dystrophy. Biochemical and Biophysical Research Communications. 1986;134(2):550–556. doi: 10.1016/s0006-291x(86)80455-7. [DOI] [PubMed] [Google Scholar]
- 112.Andersen J.K. Oxidative stress in neurodegeneration: cause or consequence? Nature Medicine. 2004;10(Suppl.):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 113.Anderson E.J., Lustig M.E., Boyle K.E., Woodlief T.L., Kane D.A., Lin C.T., Price J.W., III, Kang L., Rabinovitch P.S., Szeto H.H. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Diano S., Liu Z.W., Jeong J.K., Dietrich M.O., Ruan H.B., Kim E., Suyama S., Kelly K., Gyengesi E., Arbiser J.L. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nature Medicine. 2011;17(9):1121–1127. doi: 10.1038/nm.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Andrews Z.B., Liu Z.W., Walllingford N., Erion D.M., Borok E., Friedman J.M., Tschop M.H., Shanabrough M., Cline G., Shulman G.I. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454(7206):846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barbieri E., Sestili P. Reactive oxygen species in skeletal muscle signaling. Journal of Signal Transduction. 2012;2012:982794. doi: 10.1155/2012/982794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B.J., Skiba B., Ooms L.M. Reactive oxygen species enhance insulin sensitivity. Cellular Metabolism. 2009;10(4):260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Horvath T.L., Andrews Z.B., Diano S. Fuel utilization by hypothalamic neurons: roles for ROS. Trends in Endocrinology & Metabolism. 2009;20(2):78–87. doi: 10.1016/j.tem.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 119.Flegal K.M., Carroll M.D., Kit B.K., Ogden C.L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307(5):491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- 120.Donnan G.A., Fisher M., Macleod M., Davis S.M. Stroke. Lancet. 2008;371(9624):1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- 121.Sharma A.M., Engeli S., Pischon T. New developments in mechanisms of obesity-induced hypertension: role of adipose tissue. Current Hypertension Reports. 2001;3(2):152–156. doi: 10.1007/s11906-001-0030-x. [DOI] [PubMed] [Google Scholar]
- 122.Duan S.Z., Usher M.G., Mortensen R.M. PPARs: the vasculature, inflammation and hypertension. Current Opinion in Nephrology and Hypertension. 2009;18(2):128–133. doi: 10.1097/MNH.0b013e328325803b. [DOI] [PubMed] [Google Scholar]
- 123.Harrison D.G., Guzik T.J., Goronzy J., Weyand C. Is hypertension an immunologic disease? Current Cardiology Reports. 2008;10(6):464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 124.Savoia C., Schiffrin E.L. Inflammation in hypertension. Current Opinion in Nephrology and Hypertension. 2006;15(2):152–158. doi: 10.1097/01.mnh.0000203189.57513.76. [DOI] [PubMed] [Google Scholar]
- 125.Rahmouni K., Haynes W.G., Mark A.L. Cardiovascular and sympathetic effects of leptin. Current Hypertension Reports. 2002;4(2):119–125. doi: 10.1007/s11906-002-0036-z. [DOI] [PubMed] [Google Scholar]
- 126.Rahmouni K., Correia M.L., Haynes W.G., Mark A.L. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45(1):9–14. doi: 10.1161/01.HYP.0000151325.83008.b4. [DOI] [PubMed] [Google Scholar]
- 127.Kang Y.M., Ma Y., Zheng J.P., Elks C., Sriramula S., Yang Z.M., Francis J. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovascular Research. 2009;82(3):503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qadri F., Hauser W., Johren O., Dominiak P. Kinin B1 and B2 receptor mRNA expression in the hypothalamus of spontaneously hypertensive rats. Canadian Journal of Physiology and Pharmacology. 2002;80(4):258–263. doi: 10.1139/y02-051. [DOI] [PubMed] [Google Scholar]
- 129.Wu K.I., Schmid-Schonbein G.W. Nuclear factor kappa B and matrix metalloproteinase induced receptor cleavage in the spontaneously hypertensive rat. Hypertension. 2011;57(2):261–268. doi: 10.1161/HYPERTENSIONAHA.110.158709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iadecola C., Anrather J. The immunology of stroke: from mechanisms to translation. Nature Medicine. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jin R., Yang G., Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells 1. Journal of Leukocyte Biology. 2010;87(5):779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Drake C., Boutin H., Jones M.S., Denes A., McColl B.W., Selvarajah J.R., Hulme S., Georgiou R.F., Hinz R., Gerhard A. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain, Behavior, and Immunity. 2011;25(6):1113–1122. doi: 10.1016/j.bbi.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo Y., Xu X., Li Q., Li Z., Du F. Anti-inflammation effects of picroside 2 in cerebral ischemic injury rats. Behavioral and Brain Functions. 2010;6:43. doi: 10.1186/1744-9081-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liao S.L., Kao T.K., Chen W.Y., Lin Y.S., Chen S.Y., Raung S.L., Wu C.W., Lu H.C., Chen C.J. Tetramethylpyrazine reduces ischemic brain injury in rats. Neuroscience Letters. 2004;372(1–2):40–45. doi: 10.1016/j.neulet.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 135.Lu C.Z., Xiao B.G. Neuroprotection of G-CSF in cerebral ischemia. Frontiers in Bioscience. 2007;12:2869–2875. doi: 10.2741/2278. [DOI] [PubMed] [Google Scholar]
- 136.Cheng C.C., Chen Y.H., Chang W.L., Yang S.P., Chang D.M., Lai J.H., Ho L.J. Phytoestrogen bavachin mediates anti-inflammation targeting Ikappa B kinase-I kappaB alpha-NF-kappaB signaling pathway in chondrocytes in vitro. European Journal of Pharmacology. 2010;636(1-3):181–188. doi: 10.1016/j.ejphar.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 137.Herrmann O., Baumann B., de L.R., Muhammad S., Zhang W., Kleesiek J., Malfertheiner M., Kohrmann M., Potrovita I., Maegele I. IKK mediates ischemia-induced neuronal death. Nature Medicine. 2005;11(12):1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- 138.Jiang T., Gao L., Guo J., Lu J., Wang Y., Zhang Y. Suppressing inflammation by inhibiting NF-kappaB pathway contributes to the neuroprotection ofik Angiotensin-(1-7) in rats with permanent cerebral ischemia. British Journal of Pharmacology. 2012:20. doi: 10.1111/j.1476-5381.2012.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Y., Zhang X.J., Yang C.H., Fan H.G. Oxymatrine protects rat brains against permanent focal ischemia and downregulates NF-kappaB expression. Brain Res. 2009;1268:174–180. doi: 10.1016/j.brainres.2009.02.069. [DOI] [PubMed] [Google Scholar]
- 140.Emsley H.C., Smith C.J., Georgiou R.F., Vail A., Hopkins S.J., Rothwell N.J., Tyrrell P.J.A. randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. Journal of Neurology, Neurosurgery, and Psychiatry. 2005;76(10):1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meisel C., Schwab J.M., Prass K., Meisel A., Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nature Reviews Neuroscience. 2005;6(10):775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- 142.Ichimura A., Hirasawa A., Poulain-Godefroy O., Bonnefond A., Hara T., Yengo L., Kimura I., Leloire A., Liu N., Iida K. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483(7389):350–354. doi: 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]