

Abstract
Studies in human and animals have demonstrated that nutritionally induced low birth-weight followed by rapid postnatal growth increases the risk of metabolic syndrome and cardiovascular disease. Although the mechanisms underlying such nutritional programming are not clearly defined, increased oxidative-stress leading to accelerated cellular aging has been proposed to play an important role. Using an established rodent model of low birth-weight and catch-up growth, we show here that post-weaning dietary supplementation with coenzyme Q10, a key component of the electron transport chain and a potent antioxidant rescued many of the detrimental effects of nutritional programming on cardiac aging. This included a reduction in nitrosative and oxidative-stress, telomere shortening, DNA damage, cellular senescence and apoptosis. These findings demonstrate the potential for postnatal antioxidant intervention to reverse deleterious phenotypes of developmental programming and therefore provide insight into a potential translatable therapy to prevent cardiovascular disease in at risk humans.
Abbreviations: CoQ, coenzyme Q; CVD, cardiovascular disease; OGG-1, 8 oxoguanine DNA glycosylase 1; NTHL1, Nthl endonuclease III like-1; NEIL1, nei endonuclease VIII-like 1; BER, base excision repair; NOX, nicotinamide adenine dinucleotide diphosphate oxidase; XO, xanthine oxidase; ROS, reactive oxidative species; RNS, reactive nitrogen species; RIS, reactive inflammatory species; 3-NT, 3-nitrotyrosine; 4-HNE, 4-hydroxynonenal; O2, superoxide anion; OH-, hydroxy radicals; MnSOD, manganese superoxide dismutase; CuZnSOD, copper-zinc superoxide dismutase; PRDX, peroxidiredoxin; GR, glutathione reductase; GPx, glutathione peroxidase; Bax, Bcl2-associated protein; nppa, natriuretic peptide A; nppb, natriuretic peptide B; actin, alpha-1; acta1, sarco endoplasmic reticulum Ca(2+) ATPase; serca2, single strand breaks, SSBs; ETC, electron transport chain; DIG, dioxygenin; PGFE, pulsed field gel electrophoresis; CAST, computer assisted stereology toolbox.
Keywords: Developmental programming, Ubiquinone, Oxidative-stress, DNA damage, Telomere length, Cellular senescence
Graphical abstract

1. Introduction
It has been known for several years that low birth weight babies have significantly increased risk of developing the metabolic syndrome and cardiovascular disease (CVD) in later life [1–4]. These findings led to the proposal of the ‘Thrifty Phenotype Hypothesis’ [5], which postulates that under conditions of sub-optimal in-utero nutrition, the fetus permanently alters its organ structure and function to ensure immediate survival of the organism. Although beneficial in continued conditions of poor postnatal nutrition, such programming is proposed to be detrimental in postnatal conditions of adequate or over nutrition. This hypothesis is supported by both human [6,7] and animal studies [8,9], which have demonstrated that accelerated postnatal growth exaggerates the detrimental effect of low birth weight.
The concept of developmental programming has been demonstrated in a diverse range of animal models including rodents [8,9], sheep [10] and non-human primates [11,12]. Such models have been particularly important for dissecting the underlying mechanisms linking suboptimal early nutrition and long-term health. Emerging mechanisms mediating the effects of a suboptimal early environment on cardiovascular disease include a reduction in cardiomyocyte number at birth [13], structural changes in the aorta [14,15], increased cardiac oxidative-stress [16] and accelerated telomere shortening [17]; a robust measure of cellular aging [18]. A growing body of evidence suggests that telomere shortening plays a pivotal role in vascular senescence and CVD risk [19,20].
Reactive oxidative species (ROS) and reactive nitrogen species (RNS) are collectively known as reactive inflammatory species (RIS) [21]. These can accumulate during cardiovascular aging [22,23] and can cause damage to DNA, protein and lipids. Additionally, impairment in the antioxidant defense network can contribute to the age-associated increase in RIS. Telomeric DNA is particularly susceptible to RIS damage [24,25] and this damage can lead to accelerated telomere shortening. Critically short telomeres are known to activate a series of DNA damage checkpoint proteins including p53, p21 and p16INK, which can induce cellular senescence and apoptosis [26]. In the heart, one pathological consequence of increased RIS is congestive heart failure [27].
Oxidative-stress has been suggested to play a pivotal role in developmental programming [28–31]. We recently reported that animals that are born small as a result of maternal protein restriction, but grow rapidly during lactation, demonstrate increased nitrosative and oxidative-stress, up-regulation of the base excision repair (BER) machinery and increased DNA damage in heart tissue by weaning [16]. These observations are all consistent with an accelerated cardiac cellular aging phenotype. Furthermore, they suggest that increased oxidative stress leading to DNA damage is a very early consequence of suboptimal nutrition in-utero and accelerated postnatal growth and therefore may be an important therapeutic target.
Maternal antioxidant supplementation can reduce some of the effects of adverse conditions in-utero. Sen and Simmons [32] reported a reduction in adiposity and improved glucose tolerance in offspring from high-fat fed pregnancies, supplemented with a mixture of vitamins A, C, E and Selenium. Giussani et al. [33] demonstrated that maternal treatment with vitamin C protects against programmed cardiac and peripheral vascular endothelial dysfunction in the adult offspring of hypoxic pregnancy. Additionally, hypertension, vascular dysfunction and microvascular rarefaction, in a rat protein restriction model can be prevented by antenatal treatment with the antioxidant Lazaroid [34].
Although these studies show proof of principle that maternal antioxidants can prevent detrimental programming effects, the doses used to achieve these effects are not recommended for use in humans and therefore unlikely to ever be feasible to administer to pregnant women. Importantly, these studies focus on interventions to the mother. In many cases, evidence is not present for suboptimal in-utero exposure until at the time of, or just after delivery. Therefore it is also important to address the potential beneficial effects of targeted postnatal antioxidant supplementation.
Ubiquinone or Coenzyme Q (CoQ) encompasses a group of homologous molecules that are present in almost all tissues. They consist of a benzoquinone ring linked to an isoprenoid side chain, the length of which varies between species. In rodents, the most common form is CoQ9, containing 9 isoprenoid units, whereas CoQ10 (containing 10 isoprenoid units) is the most common isoform in humans. It is known that rodents can convert dietary CoQ10 into CoQ9. CoQ has been shown in humans to be a safe supplement with minimal side effects [35–37]. In its quinone form, CoQ acts as an electron carrier, transferring electrons in the mitochondrial electron transport chain (ETC) between complexes I and III and complexes II and III. In its quinol form (the reduced form of quinone), it acts as the most abundant and efficient antioxidant in the body [38]. CoQ has a pivotal role as a redox link between flavoproteins and cytochromes in the mitochondrial respiratory chain. CoQ10 at high doses has also been shown to act as a regulator of gene transcription in vivo, including those genes important in PPARα signaling and lipid metabolism and it has been hypothesized that CoQ10 can trigger ROS-sensitive intracellular pathways that regulate the induction of specific genes [39].
In-vitro, mitoQ a modified form of CoQ, prevented telomere shortening and increased the lifespan of fibroblasts under conditions of mild oxidative-stress [40]. In-vivo supplementation of CoQ10 to rats fed a high-fat diet reduced DNA double stranded breaks in peripheral blood mononuclear cells and increased lifespan [41], protected against age-related oxidative-stress and improved mitochondrial function in cardiac tissue [42], liver and skeletal muscle [43]. Furthermore, MitoQ supplementation has recently been shown to decrease features of the metabolic syndrome in ATM+/− and ApoE−/− mice [44]. In humans, several meta-analyses have demonstrated that CoQ10 supplementation improves clinical outcome in patients with heart failure [45–47]. Therefore there is good evidence from studies in humans, animal models and cell culture systems, that CoQ is an effective antioxidant.
The current study therefore tested the hypothesis that supplementation with a clinically relevant dose of CoQ10 would prevent the detrimental effects of nutritional programming on cardiac aging. We used model of nutritionally induced low birth-weight and accelerated postnatal growth (recuperated animals) that demonstrates cardiac mitochondrial dysfunction, increased oxidative-stress and accelerated cellular aging. We assessed the potential of postnatal CoQ10 supplementation to prevent the molecular and structural cardiac defects in both young and old offspring.
2. Materials and methods
2.1. Animal experimental groups
All procedures involving animals were conducted under the British Animals (Scientific Procedures) Act (1986). Pregnant Wistar rats were maintained on a 20% protein diet (control) or, an isocaloric low protein (LP) (8%) diet fed ad libitum, as previously described [48]. Both diets were purchased from Arie Blok, The Netherlands. The day of birth was recorded as day 1 of postnatal life. Pups, born to LP diet-fed dams were cross fostered to control-fed mothers on postnatal day 3, in order to create a recuperated litter. Each recuperated litter (R) was culled to 4 male pups at random to maximize their plane of nutrition. The control (C) group was the offspring of mothers fed the 20% protein diet and suckled by 20% protein fed dams. Each control litter was culled to 8 pups as a standard. To prevent any stress or rejection to the animals when cross fostered, pups were transferred with some of their own bedding. Body weights were recorded at postnatal days 3, 7, 14, 21, 3 months and 12 months. At 21 days, 2 males per litter were weaned onto standard laboratory chow (SDS) and the other 2 littermates were weaned onto the same diet supplemented with CoQ10 to give a dose of 1 mg/kg/body weight/day (group abbreviations; CQ and RQ). Animals were maintained on these diets until 3 or 12 months of age. All animals were killed by CO2 asphyxiation. At post mortem, hearts were either removed, weighed, and snap frozen in liquid nitrogen and then stored at −80°C or fixed in formaldehyde until analysis. For all measurements, one pup per litter was used, thus N indicated throughout represents number of litters.
2.2. CoQ10 dosage methodology
In order to determine the effectiveness of supplementation, and to test palatability of the supplemented diet, a 4-week pilot study was performed, using 1 mg/kg of body weight per day CoQ10 a dose that is tolerated by humans without any side effects [49–51]. This dose was achieved by appropriate CoQ10 supplementation of lab chow based on calculations of weekly food intake in relation to body weight. No significant difference in body weight gain (%) (13.9; 13.4; 14.3) or food intake (224.9 g±13.7; 214.9 g±12.9; 212.0 g±11.2) was observed between the CoQ10 treated, acetone only or control groups, respectively. Aortic CoQ9 was increased in the CoQ10 treated group compared to the control and acetone treated groups (approximately 50%), and serum CoQ10 was increased (approximately 200%) in the CoQ10 treated group compared to the control and acetone treated groups. CoQ10 was impregnated into the diet pellets by dissolving CoQ10 in acetone and mixing this with the diet pellets [52]. This mix was left in a fume hood overnight to allow evaporation of acetone. Diet was prepared twice a week throughout the study.
2.3. Telomere length analysis
High molecular weight DNA was extracted using The Wizard Genomic DNA Isolation kit (Promega) according to the manufacturer's instructions. DNA quantity and purity were determined using a Nanodrop spectrophotometer (Nanodrop Technologies) [17]. DNA (1.2 µg) was digested by Hinf1 and RSA1 restriction enzymes at 37 °C for 2 h and separated by pulsed field gel electrophoresis (PFGE) and transferred to nylon membranes by Southern Blotting [17]. Standard undigested and digested genomic DNA from a 3-month control animal was also included on each gel to verify digestion efficiency [17]. Telomere length was measured using Telo TAGGG telomere length assays (Roche Diagnostics). Telomere signals were analyzed using Adobe Photoshop (Adobe Systems Inc) and MacBas software (Fujifilm UK) [17]. Telomere length was quantified where the percentage intensity (% telomere length) of the telomeric signal was determined in 4 molecular size regions, as defined by molecular weight markers [17].
2.4. Nitrosative and oxidative-stress analysis
Protein oxidation was assayed using a Nitrotyrosine ELISA kit (MitoSciences), according to the manufacturer's instructions. Lipid peroxidation was analyzed using an OxiSelect HNE Adduct ELISA kit (Cambridge Biosciences), according to the manufacturer's instructions.
2.5. Total CoQ9 and CoQ10 measurements
Total tissue CoQ9 and CoQ10 status was quantified by reverse phase HPLC with UV detection at 275 nm according to the method of Duncan et al. [53]. CoQ10 was separated on a HPLC column (Techsphere ODS 5 µ, 150×4.6 mm2). The mobile phase consisted of ethanol:methanol:60% (v/v) perchloric acid; 700:300:1.2 (v:v) to which is added 7 g of sodium perchlorate [54]. The flow rate was maintained at 0.7 ml/min.
2.6. Gene expression analysis
RNA was extracted using the TRIzol kit (Sigma) following manufacturers' instructions. An additional DNaseI digestion step was carried out during the clean-up stage to ensure no genomic DNA contamination of the samples. RNA quantification was performed using a NanoDrop spectrophotometer (Nanodrop Technologies) and RNA integrity was assessed using an Agilent Bioanalyser (Agilent Technologies). One microgram RNA was used to synthesize cDNA using oligo-dT primers and M-MLV reverse transcriptase (Promega). Gene expression was determined using custom designed primers (Sigma) and SYBR Green reagents (Applied Biosystems). Primer sequences are presented in Supplemental Table 1A. Quantification of gene expression was performed using the Step One Plus RT-PCR machine (Applied Biosystems). Equal efficiency of the reverse transcription of RNA from all groups was confirmed through quantification of expression of the house-keeping gene ppia. Expression did not differ between groups at either 3 or 12 months, or with aging, (Supplemental Table 1B).
2.7. Protein analysis
Western blotting analysis was performed to determine protein expression of OGG-1, NTHL-1, NEIL-1, p47phox, p67phox, XO, MnSOD, CuZnSOD, catalase, HO-1, PRDX-3, GPx1, GR, p53, p21, p16INK, Bax, Bcl2 and caspase 3. Protein was extracted and assayed as described previously [16]. Protein (20 μg) was loaded onto polyacrylamide gels, and was electrophoresed and transferred to polyvinylidene fluoride membranes. OGG-1, NTHL-1, PRDX-3, HO-1, catalase, GPx1, GR, Bax, Bcl2, p21 (Abcam), XO (Santa Cruz), p67phox (Cell Signaling), MnSOD (Merck Millipore) and caspase-3 (Stressgen Biotechnologies) were all detected using anti-rabbit IgGs. p47phox, NEIL-1, p16INK (Santa Cruz) and CuZnSOD (R & D Systems) were measured using anti-goat IgGs. p53 (Abcam) was analyzed using anti-mouse IgGs. All IgGs were purchased from Jackson ImmunoResearch. Equal protein loading was confirmed by staining electrophoresed gels with Coomassie Blue to visualize total protein.
2.8. Stereological analysis
Twelve-month hearts were fixed in 10% neutral buffered formalin, processed, embedded in paraffin wax and cut exhaustively with all sections collected. Slides were stained with haematoxylin and eosin. Analysis was performed at 1.25× magnification using Computer Assisted Stereology Toolbox (CAST) software to quantify wall and lumen width and area. The observer was blinded to the treatment groups during analysis. Wall and lumen width was measured where the superimposed diagonal lines crossed the wall or lumen under analysis. Area was calculated using the Cavalieri principle.
2.9. Statistical analysis
All data were analyzed using a 3-way ANOVA with maternal diet, CoQ10 supplementation and age as the independent variables. Data are represented as mean±S.E.M. A value of p<0.05 was considered statistically significant. All statistical analyses were performed using Statistica 7 software (Statsoft Inc). In all cases, N refers to the number of litters.
3. Results
3.1. Recuperated animals are born small and undergo rapid postnatal catch-up growth
Recuperated animals were significantly (p<0.001) lighter compared to controls on postnatal day 3 [6.1±0.1 g vs 7.3±0.1 g] and day 7 [13.4±0.6 g vs 16.2±0.8 g]. However, by day 14, this group had undergone accelerated postnatal growth hence the body weights were similar between groups [32.5±1.3 g vs 32.0±1.3 g]. Body weight (Supplementary Table 2A), cardiac weight and cardiac wall width; measured using stereology (Supplementary Table 2B), were similar between groups at both 3 and 12 months of age. However, there was a significant (p<0.001) effect of age upon body weight (Supplementary Table 2A) and absolute cardiac weight (Supplementary Table 2B), with an increase in weight observed between 3 and 12 months of age.
3.2. Dietary CoQ10 supplementation prevents CoQ10 deficits in the recuperated offspring
Dietary supplementation with CoQ10 [1 mg/kg of body weight/day] led to an approximate doubling of plasma CoQ10 at both 3 (p<0.05) and 12 months (p<0.01) (Figure 1A). No effect of maternal diet was observed on plasma CoQ10 levels (Figure 1A); however plasma CoQ9 (p<0.001) and CoQ10 (p<0.001) levels increased with age in both groups (Supplementary Figure 1 and Figure 1A). No significant effect of maternal diet was observed on plasma CoQ9 at 3 months (Supplementary Figure 1). At 12 months, cardiac CoQ9 and CoQ10 were reduced (p<0.05) in the recuperated group compared to controls, however no significant effect of maternal diet was observed upon cardiac CoQ9 or CoQ10 at 3 months (Figure 1B). Dietary CoQ10 supplementation increased cardiac CoQ10 (p<0.05) and CoQ9 (p=0.06) in recuperated animals at 12 months (Figure 1B), but supplementation did not alter cardiac CoQ10 levels in control animals (Figure 1B). Consistent with the plasma data, cardiac CoQ10 and CoQ9 levels increased with age (Figure 1B).
Figure 1.

The effect of in-utero protein restriction and accelerated postnatal growth upon cardiac and plasma CoQ9 and CoQ10 levels in 3 and 12 month rat hearts. CoQ9 and CoQ10 levels were measured using HPLC. Results are expressed as mean±S.E.M. * p<0.05 and ** p<0.01 compared to control/recuperated CoQ. (N=10 per group). C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (See also Supplementary Figure 1). (A) Plasma CoQ10, (B) Cardiac CoQ9 and CoQ10.
3.3. CoQ10 supplementation prevents the effects of maternal diet on telomere shortening
Telomere length was assessed as a marker of cardiovascular aging. At 3 months, shorter cardiac telomeres were observed in the recuperated group compared to controls, as reflected by fewer long telomeres [145–48.5 kb and 48.5–8.6 kb] (p<0.05 and p<0.01 respectively) and more short [4.2–1.3 kb] telomeres (p<0.01). CoQ10 supplementation prevented these changes by increasing the percentage of long telomeres [145–48.5 kb and 48.5–8.6 kb] (p<0.01 and p<0.05 respectively) and reducing (p<0.01) the number of short telomeres [4.2–1.3 kb], (Figure 2A). At 12 months, again fewer long [145–48.5 kb] (p<0.05) and more short (p<0.05) [4.2–1.3 kb] telomeres were observed in the recuperated group compared to controls (Figure 2B). CoQ10 supplementation ameliorated this shortening. Age-dependent telomere shortening was apparent in both groups with fewer long [145–48.5 kb] (p<0.001) and more short [4.2–1.3 kb] (p<0.01) telomeres at 12 months compared to 3 months (Figure 2A and B).
Figure 2.

The effect of in-utero protein restriction and accelerated postnatal growth upon telomere length in 3 and 12 month rat hearts. Telomere length was measured using Pulsed Field Gel Electrophoresis (PGFE) and Southern Blotting. Results are expressed as mean±S.E.M. * p<0.05 and ** p<0.01 compared to control/recuperated CoQ. N=6 per group; C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (A) 3 months and (B) 12 months.
3.4. CoQ10 supplementation prevents the effects of maternal diet on 3-NT levels
ROS and RNS accumulation is a major mechanism that leads to accelerated telomere shortening [23,24]. We therefore investigated the effects of early nutrition upon indices of cellular stress. Levels of 4-HNE (a marker of lipid peroxidation) did not differ between any of the groups (Supplementary Figure 2A). In contrast, 3-NT levels (a marker of peroxynitrite-mediated nitration) were significantly (p<0.05) increased in recuperated animals at both 3 and 12 months, with the effect becoming more prominent with age. This damage was prevented by CoQ10 supplementation at 3 months of age (Figure 3A).
Figure 3.

The effect of in-utero protein restriction and accelerated postnatal growth upon 3-NT, XO and p47phox levels in 3 and 12 month rat hearts. 3-NT and 4-HNE levels were measured using ELISA. XO and p47phox protein expressions were analyzed using Western blotting. Results are expressed as mean±S.E.M. * p<0.05 and *** p<0.001 compared to control/recuperated CoQ. N=10 per group (3-NT and 4-HNE levels). N=6 per group (XO protein expression). C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (See also Supplementary Figure 2). (A) 3-NT, (B) XO and (C) p47phox.
3.5. CoQ10 supplementation prevents the effects of maternal diet on xanthine oxidase
Xanthine oxidase (XO) and NADPH oxidase-2 (NOX-2) are well-established sources of cardiac ROS generation [55]. To investigate the potential sources of the increased cellular stress in the recuperated group, we measured protein levels of XO and NOX-2. XO expression was increased at 3 months (p<0.01) and 12 months (p<0.001) in the recuperated group compared to controls (Figure 3B) and therefore may contribute to the increased 3-NT levels. This increase was prevented by CoQ10 supplementation (Figure 3B). p67phox and p47phox were the only components of NOX-2 that could be detected by Western blotting. p67phox protein expression was not different between any of the groups (Supplementary Figure 2B). p47phox protein levels were increased (p<0.05) in recuperated animals, however, this effect was not prevented by CoQ10 supplementation (Figure 3C).
3.6. CoQ10 supplementation prevents the effects of maternal diet on base excision repair enzymes
As a response to oxidative damage to DNA, enzymes in the base excision repair (BER) DNA damage pathway can be activated [56]. Therefore, we investigated the potential consequences of increased cellular stress on DNA damage repair. Protein levels of the DNA glycosylases OGG-1, NEIL-1 and NTHL-1 were measured. Protein levels of OGG-1 were increased (p<0.001) in the recuperated group at 3 months (Figure 4A). CoQ10 intervention reduced (p<0.001) OGG-1 expression in the recuperated group at this age, restoring it back to the levels observed in the control group (Figure 4A). Protein levels of NTHL-1 were increased (p<0.01) in recuperated animals, however CoQ10 supplementation did not alter NTHL-1 levels (Figure 4B). No effect of maternal diet or CoQ10 intervention upon NEIL-1 protein expression was observed (Supplementary Figure 3).
Figure 4.

The effect of in-utero protein restriction and accelerated postnatal growth upon expression of DNA repair enzymes in 3 and 12 month rat hearts. Protein expression was analyzed using Western blotting. Results are expressed as mean±S.E.M. * p<0.05 and *** p<0.001 compared to control/recuperated CoQ. N=6 per group. C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (See also Supplementary Figure 3). (A) OGG1 and (B) NTHL1.
3.7. CoQ10 supplementation prevents the effects of maternal diet on antioxidant defense capacity
DNA can be damaged if there is an imbalance between cellular RIS levels and antioxidant defenses. We investigated whether increased cellular stress in recuperated animals was accompanied by differences in antioxidant defense capacity, by measuring protein expression of antioxidant enzymes. These included the superoxide dismutase (SOD) enzymes [manganese superoxide-dismutase (MnSOD), and copper-zinc superoxide dismutase (CuZnSOD)] and the peroxidases [catalase, glutathione peroxidases (GPxs), glutathione reductase (GR) and the peroxiredoxins (PRDXs)]. MnSOD (p<0.001), CuZnSOD (p<0.01) and catalase (p<0.001) were all increased in recuperated animals at 12 months (Figure 5A–C). CoQ10 supplementation prevented the increase in MnSOD at 3 months and CuZnSOD and catalase levels at 12 months (Figure 5A–C). Heme oxygenase-1 (HO-1), an inducer of MnSOD, was also increased in recuperated animals (p<0.001) at 12 months, however CoQ10 supplementation did not alter HO-1 levels (Figure 5D). Peroxiredoxin-3 (PRDX-3) levels were lower in the recuperated group compared to controls at 12 months, an effect that was prevented by CoQ10 supplementation (Figure 5E). GPx1 levels were increased (p<0.05) in the recuperated group compared to controls at 12 months and CoQ10 intervention increased (p<0.05) GPx1 levels in the control group (Figure 5F). No statistically significant effect of maternal diet or CoQ10 intervention upon HO-1 [100±17; 96±6; 93±9; 89±6], CuZnSOD [100±6; 107±8; 77±5; 152±10], PRDX-3 [100±11; 90±10; 99±8; 126±13], or GPx1 [100±8; 133±19; 76±12; 166±10] protein levels was observed at 3 months [C vs R vs CQ vs RQ]. No difference in GR protein expression was observed at either 3 months; [100±5; 88±5; 89±6; 88±4] or 12 months [113±6; 131±10; 121±6; 125±7]; [C vs R vs CQ vs RQ].
Figure 5.

The effect of in-utero protein restriction and accelerated postnatal growth upon antioxidant protein expression in 3 and 12 month rat hearts. Protein expression was analyzed using Western blotting. Results are expressed as mean±S.E.M. * p<0.05, ** p<0.01 and *** p<0.001 compared to control/recuperated CoQ. N=6 per group. C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (A) MnSOD, (B) CuZnSOD (12 months), (C) Catalase, (D) HO-1 (12 months), (E) PRDX3 (12 months) and (F) GPx1 (12 months).
3.8. CoQ10 supplementation prevents the effects of maternal diet on markers of cellular senescence and apoptosis
Telomeric DNA which has been exposed to high levels of cellular stress and/or is critically short, can activate a series of DNA damage checkpoint proteins including p53, p21 and p16INK which can induce cellular senescence and/or apoptosis [26]. Therefore, cellular senescence and apoptosis markers were measured in order to investigate consequences of accelerated cardiac telomere shortening in the recuperated group. p53 and p21 protein levels were substantially increased (p<0.001) in recuperated animals compared to controls at 12 months. This difference was prevented by CoQ10 supplementation [Figure 6A (i) and (ii)]. No significant effect of maternal diet or CoQ10 supplementation upon p53 [100±12.8; 101.7±13.0; 93.6±5.2; 98.9±6.6] or p21 [100±22.7; 149.1±22.8; 89.3±8.8; 148.14.0] protein levels was observed at 3 months of age. p16INK levels increased modestly in the recuperated group compared to controls at 3 months (Supplementary Figure 4A). p16INK levels were similar between groups at 12 months of age. Bcl2-associated X protein (Bax) is a transcriptional target of p53 and a major pro-apoptotic molecule, conversely Bcl2 is a major anti-apoptotic protein; hence the ratio of Bax/Bcl2 is a well used measure of apoptosis [57]. This ratio was increased (p<0.01) in recuperated animals compared to controls and CoQ10 intervention abolished the increase at both ages (Figure 6B). This was primarily driven by an up-regulation of Bax in recuperated offspring (Supplementary Figure 4B). Under cellular stress, Bax promotes a major apoptotic molecule; caspase-3. This was increased (p<0.001) in the recuperated group compared to controls and was attenuated by CoQ10 supplementation [Figure 6A (iii)]. No effect of maternal diet or CoQ10 supplementation upon caspase-3 was observed at 3 months [100±2.5; 109.9±5.4; 89.3±3.6; 97.6±1.9]; [C vs R vs CQ vs RQ].
Figure 6.
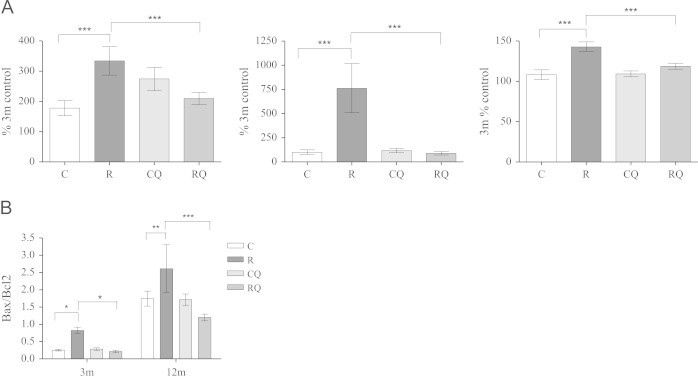
The effect of in-utero protein restriction and accelerated postnatal growth upon apoptosis and senescence marker protein expression in 3 and 12 month rat hearts. Protein expression was analyzed using Western blotting. Results are expressed as mean±S.E.M. * p<0.05, ** p<0.01 and *** p<0.001 compared to control/recuperated CoQ. N=6 per group. C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (See also Supplementary Figure 4). (A) (i) p53 (12 months), (ii) p21 (12 months), Caspase 3 (12 months) and (B) Bax /Bcl2.
3.9. CoQ10 supplementation prevents the effects of maternal diet upon cardiac fetal gene expression
The cardiac fetal genes such as natriuretic peptide A (nppa), natriuretic peptide B (nppb), actin, alpha-1 (acta-1) and sarco endoplasmic reticulum Ca(2+) ATPase (atp2a2) are re-expressed when the heart is under pathological stress. The sarcomere proteins (myosin heavy chains), undergo major isoform switches at birth. In postnatal healthy hearts of rodents, the ‘fast twitch’ myosin heavy chain 6 (myh6) isoform is predominant over the ‘slow twitch’ myosin heavy chain 7 (myh7). However, in failing hearts, a switch occurs, so that the myh7 isoform is predominant. In recuperated animals, the ratio of (myh7/myh6) expression was increased (p<0.05) compared to controls. Nppb was also increased (p<0.05) in the recuperated group. CoQ10 supplementation restored both levels to that of controls (Figure 7A and B). These data may suggest increased cardiac stress in the recuperated group, which is normalized by CoQ10 supplementation. GATA-4, a transcription factor which can induce multiple fetal cardiac genes, was reduced (p<0.05) in the recuperated group compared to controls (Figure 7C). No significant effect of maternal diet or CoQ10 intervention upon nppa [438.3±101.7; 433.6±94.9; 339.4±73.7; 240.1±34.4], acta-1 [10588.4±1379.2; 9063.1±1313.6; 8069.2±1313.6; 11059.6±2127.2] or atp2a2 [42256.6±4034.7; 39197.4±6698; 53012.9±6595.6; 43390.1±3778.2] expression was observed; [C vs R vs CQ vs RQ].
Figure 7.

The effect of in-utero protein restriction and accelerated postnatal growth upon fetal cardiac genes in 3 and 12 month rat hearts. Fetal cardiac genes were measured by RT-PCR. Results are expressed as mean±S.E.M. * p<0.05 and ** p<0.01 compared to control/recuperated CoQ. N=10 per group. C=control; CQ=control CoQ; R=recuperated and RQ=recuperated CoQ. (A) Myh7/Myh6, (B) Nppb and (C) GATA4.
All results are summarized in Supplementary Table 3.
4. Discussion
Oxidative-stress is emerging as a common mechanism underlying the effects of a suboptimal environment in early life on long-term metabolic health. It is therefore becoming increasingly apparent that antioxidants may represent potential therapeutic agents to prevent the detrimental effects of suboptimal early life exposures. Although in experimental animals, maternal antioxidant supplementation in adverse pregnancy has helped to define the causal effects of oxidative-stress on programmed outcomes in the offspring, the challenge has been to identify suitable antioxidants translatable to human treatment, in terms of appropriate dosing regimens and treatment periods.
CoQ is an important inhibitor of oxidative damage [58] and has been shown to be a potential therapy for heart failure [45–47]. In addition to the cardioprotective effects, CoQ has also been widely reported have beneficial effects on aging [59,60]. Therefore, the current study aimed to test the hypothesis that poor maternal nutrition followed by accelerated postnatal growth increases RIS resulting in accelerated cardiac cellular aging during postnatal development and that dietary CoQ10 supplementation of the offspring postnatal diet would prevent this effect, thus ameliorating programmed premature cardiac aging, including accelerated cardiac telomere shortening, cellular senescence and apoptosis.
Deficits in cardiac CoQ9 and CoQ10 levels were observed in the recuperated animals compared to controls, reflecting our previous findings in the kidney [9]. Administration of a clinically relevant dose of CoQ10 restored cardiac CoQ9 levels in recuperated animals, but had no effect on these levels in the control group. This suggests that when no deficit in CoQ exists, the heart does not accumulate further CoQ10. Consistent with this observation CoQ10 supplementation had little effect on most of the parameters measured in control animals. These data support previous findings that cardiac CoQ did not increase in control rats after 13 weeks of dietary supplementation [59]. Plasma levels of CoQ9 and CoQ10 were, as expected, highly elevated in the CoQ10 treated groups, and it is known that administration of CoQ10 to patients with chronic heart failure increases plasma CoQ10 levels and improved the contractility of dysfunctional myocardium [61]. This may suggest that some of the current observed beneficial effects of CoQ10 treatment could partially stem from the increased plasma CoQ10 levels.
We have previously reported significantly shortened aortic telomere length at 12 months in recuperated offspring [17]. Here we demonstrate that recuperated offspring have shortened cardiac telomeres both at 3 and 12 months of age compared to controls, indicative of accelerated cardiac aging. Interestingly, this attrition occurs much earlier than in the aorta, despite their close proximity. Cardiac telomere length is related to increased CVD risk in humans [19,20]; therefore accelerated cardiac telomere shortening may link low birth-weight and catch-up growth to increased risk of CVD. DNA damage, such as single strand breaks (SSBs) is a major mechanism of telomere shortening [62]. We have previously demonstrated significantly increased DNA SSBs in the recuperated animals at 22 days of age [16], which may contribute to accelerated telomere shortening later in life. CoQ10 supplementation prevented accelerated telomere shortening in the recuperated group, suggesting that this intervention prevents accelerated cardiac cellular aging in recuperated animals.
The recuperated group had significantly increased 3-nitrotyrosine (3-NT) levels, which are generated by the accumulation of peroxynitrite, a highly reactive oxidant, formed by the combination of nitric oxide (NO) and superoxide (•O2−). This suggests that hearts of recuperated animals have permanently increased RIS. Dietary CoQ10 supplementation normalized 3-NT levels in recuperated animals, thus providing a mechanism by which it may prevent the accelerated telomere shortening. XO, the enzymatic form of xanthine oxidioreductase (XOR) activity is associated with the synthesis of large amounts of ROS and RNS. XO was increased in the recuperated animals and again CoQ10 supplementation prevented this. Additionally, p47phox protein expression was significantly higher in the recuperated group compared to controls, but this was not prevented by CoQ10 supplementation. These data suggest that XO generation, and to a lesser extent, NOX2; may be sources of cellular stress in recuperated cardiac tissue and this, in the case of XO, is corrected by CoQ10 administration. It has been shown that XO, but not NOX enzymes largely mediates O2− production after myocardial infarction in rats [63].
At 3 months of age, OGG-1, which repairs 8-hydroxy-deoxyguanine (8-OH-dG) DNA lesions, was markedly increased in the recuperated group compared to controls and CoQ10 supplementation restored these levels to that of the controls. Interestingly, incubation of isolated mitochondrion in the presence of succinate and antimycin (which maximize the reduced CoQ pool), has been shown to eliminate 8-OH-dG oxidative damage [58]. Additionally, as site-specific DNA damage at guanine rich sequences by oxidative-stress can accelerate telomere shortening, the increase in OGG-1 in the recuperated group may provide a partial mechanism underlying their accelerated telomere shortening. No difference in OGG-1 levels was observed between groups at 12 months that may suggest that the aging process has blunted up-regulation of some of the BER machinery. NTHL-1, which excises oxidized pyrimidines, was significantly up-regulated in recuperated animals compared to controls at both ages. CoQ10 supplementation did not alter NTHL-1 levels. These data suggest that both purine and pyrimidine-based lesions are increased in the recuperated group. However, CoQ10 supplementation is more effective in decreasing purine-based DNA lesions.
MnSOD and catalase protein levels were significantly increased in recuperated animals. These data are consistent with our previous findings at 22 days of age [16] and may suggest a compensatory up-regulation in response to the observed increased RIS. Others have shown that insults such as abdominal aortic coarctation in rats [64] and high-fat feeding in mice [65] causes a compensatory up-regulation of MnSOD and catalase to deal with excess ROS in the cardiovascular system. Additionally, peroxynitrite-mediated tyrosine nitration can cause post-translational modifications to human MnSOD whereby total protein increased after nitration [66]. CoQ10 supplementation reduced MnSOD and catalase protein levels at 3 months, and catalase at 12 months of age. There were also significant increases in CuZnSOD and GPx1 levels in the recuperated group compared to controls at 12 months. Again, these increases may be compensatory responses to increased RIS. CoQ10 supplementation significantly reduced CuZnSOD levels in the recuperated group, again indicating that CoQ10 may be ameliorating cellular stress in recuperated offspring. HO-1 was significantly increased in recuperated animals at 12 months; however CoQ10 intervention did not alter HO-1 levels. HO-1 is up-regulated under conditions of nitrosative stress [67] and can be induced by, and acts against oxidant-induced tissue injury [68]. There was a significant decrease in PRDX-3 levels in the recuperated group compared to controls at 12 months of age. PRDX-3 deficiency can increase susceptibility to oxidative-stress [69,70]; therefore this reduction may make recuperated animals even more vulnerable to oxidative-stress. CoQ10 supplementation restored these levels to those of the control group. Taken together these data suggest that CoQ10, through its own potent antioxidant properties, ameliorates RIS by altering antioxidant defense capacity in recuperated animals. The antioxidant properties of CoQ10 occur as a result of its redox active quinoid moiety that can relatively easily accept and donate electrons. Its hydrophobic tail means that it is soluble and mobile in lipid membranes, including the mitochondrial inner membrane, where it effectively protects the cell from phospholipid peroxidation and free-radical induced oxidative damage.
At 12 months of age, p53 and p21 protein levels were significantly increased in the recuperated group and CoQ10 supplementation normalized these levels. These data suggest that recuperated animals have increased cardiac cellular senescence that can be prevented by CoQ10 supplementation. There was only a modest effect of maternal diet upon p16INK protein levels; which may suggest that the mechanism of cellular senescence/apoptosis is mediated via a p53/p21 dependent pathway. Bax and caspase-3 were also significantly increased in cardiac tissue of the recuperated group and CoQ10 supplementation reduced both caspase-3 and Bax levels to that of controls. These data suggest that hearts of recuperated animals have an increased pro-apoptotic phenotype and CoQ10 supplementation reduces apoptosis levels to that of controls. In addition, GATA-4 was significantly down-regulated in the recuperated group. It is known that apoptosis, induced by cellular stress, decreases the expression of GATA-4 and restoration of GATA-4 via ectopic expression attenuates apoptosis [71,72]. These data suggest that the observed decrease in GATA-4 mRNA in recuperated animals may be associated with the observed cellular stress-induced apoptosis.
CoQ10 supplementation also prevented the observed increases in the ratio of myh7/myh6 and nppb in recuperated offspring. Reactivation of these cardiac fetal genes is initially an adaptive process to increase the contractility, excitability and plasticity of cardiomyocytes in response to pathological stress, which if sustained, will lead to heart failure. Therefore, the reactivation of cardiac fetal genes observed in the current study may suggest an increased risk of heart failure in later life.
It remains to be established if supplementation with CoQ10 prevents any of the other detrimental effects of low birth weight and catch up growth, such as a reduction in lifespan. The potential role of ROS in determination of lifespan is debated [73]. Initial evidence suggested that that increased ROS was important in the pathogenesis of aging [74–76]. However other studies have shown that antioxidant supplementation did not alter [77] or actually shortened lifespan [78]. The reasons for this are unclear but it could relate to the poor mitochondrial penetrance (a major site of ROS production) or lack of effectiveness of certain anti-oxidants in vivo.
5. Conclusions
Nutritionally induced in-utero growth restriction followed by accelerated postnatal growth results in increased oxidative and nitrosative stress, accelerated telomere shortening, altered DNA repair and antioxidant defense capacity, in addition to increased cellular senescence and apoptosis in hearts of adult offspring. These are all indicative of a premature cardiac aging phenotype. CoQ10 supplementation of the offspring postnatal diet corrected the cardiac cellular stress, telomere shortening, antioxidant defense alterations, and cellular senescence and apoptosis, thereby protecting against premature cardiovascular aging (please see Graphical Abstract). An important next step will be to establish if other suboptimal in utero exposures, such as fetal hypoxia lead to a similar phenotypic outcome in relation to accelerated cardiac aging. Such information will provide insight as to whether CoQ10 supplementation is likely to represent a widely applicable intervention strategy that is likely to prevent the increased risk of cardiovascular disease resulting from an array of different in utero insults. These findings would have important implications for translatable therapies targeted towards at-risk human populations.
Conflict of interest
The authors wish to declare that there are no conflicts of interest associated with this manuscript.
Acknowledgments
We would like to thank K. Phillips and A. Wayman for their technical expertise. This work was supported by The British Heart Foundation and the Medical Research Council. SEO is a British Heart Foundation Senior Fellow and a member of the MRC Metabolic Diseases Unit. DAG is a Lister Institute Fellow and a Royal Society Wolfson Research Merit Award Holder. IPH is supported by the Department of Health's NIHR Biomedical Research Centres funding scheme at UCLH/UCL.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.09.004.
Appendix A. Supplementary materials
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
References
- 1.Barker D.J.P., Winters P.D., Osmond C., Margetts B., Simmonds S.J. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;334:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 2.Barker D.J.P., Gluckman P.D., Godfrey K.M., Harding J.E., Owens J.A., Robinson J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 3.Leeson C.P.M., Kattenhorn M., Morley R., Lucas A., Deanfield J.E. Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation. 2001;103:1264–1268. doi: 10.1161/01.cir.103.9.1264. [DOI] [PubMed] [Google Scholar]
- 4.Barker D.J., Hales C.N., Fall C.H., Osmond C., Phipps K., Clark P.M. Type-2 (non-insulin-dependent) diabetes mellitus, hypertension, and hyperlipidemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;63:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 5.Hales C.N., Barker D.J.P. The thrifty phenotype hypothesis. British Medical Bulletin. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 6.Eriksson J.G., Forsen T., Tuomilehto J., Osmond C., Barker D. Fetal and childhood growth and hypertension in later life. Hypertension. 2000;36:790–794. doi: 10.1161/01.hyp.36.5.790. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson J.G., Forsen T., Tuomilehto J., Winter P.D., Osmond C., Barker D. Catch up growth in childhood and death from coronary heart disease: longitudinal study. British Medical Journal. 1999;318:427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarry-Adkins J.L., Chen J.H., Smith N.S., Jones R.H., Cherif H., Ozanne S.E. Poor maternal nutrition followed by accelerated postnatal catch up growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB Journal. 2009;23:1521–1528. doi: 10.1096/fj.08-122796. [DOI] [PubMed] [Google Scholar]
- 9.Shelley P., Tarry-Adkins J., Martin-Gronert M., Poston L., Heales S., Clark J. Rapid neonatal weight gain in rats results in a renal ubiquinone (CoQ) deficiency associated with premature death. Mechanisms of Ageing and Development. 2007;28:681–687. doi: 10.1016/j.mad.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Tarry-Adkins J.L., Ozanne S.E. Mechanisms of early life programming: current knowledge and future directions. American Journal of Clinical Nutrition. 2011;94:1765S–1771S. doi: 10.3945/ajcn.110.000620. [DOI] [PubMed] [Google Scholar]
- 11.Fan L., Lindsley S.R., Comstock S.M., Takahashi D.L., Evans A.E., He G.W. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. International Journal of Obesity (London) 2013;37:254–262. doi: 10.1038/ijo.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grayson B.E., Levasseur P.R., Williams S.M., Smith M.S., Marks D.L., Grove K.L. Changes in melanocortin expression and inflammatory pathways in fetal offspring in nonhuman primates fed a high fat diet. Endocrinology. 2010;151:1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corstius H.B., Zimanyi M.A., Maka N., Herath T., Thomas W., Van Der Laarse A. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatric Research. 2005;57:796–800. doi: 10.1203/01.PDR.0000157726.65492.CD. [DOI] [PubMed] [Google Scholar]
- 14.Skilton M.R., Gosby A.K., Wu B.J., Ho L.M.L., Stocker R., Caterson I.D. Maternal undernutrition reduces aortic wall thickness and elastin content in offspring of rats without altering endothelial function. Clinical Science (London) 2006;111:281–287. doi: 10.1042/CS20060036. [DOI] [PubMed] [Google Scholar]
- 15.Lim K., Zimanyi M.A., Black M.J. Effect of maternal protein restriction on rats on cardiac fibrosis and capilliarization in adulthood. Pediatric Research. 2006;60:83–87. doi: 10.1203/01.pdr.0000220361.08181.c3. [DOI] [PubMed] [Google Scholar]
- 16.Tarry-Adkins J.L., Martin-Gronert M.S., Fernandez-Twinn D.S., Hargreaves I., Alfaradhi M.Z., Land J.M. Poor maternal nutrition followed by accelerated postnatal catch up growth leads to alterations in DNA damage and repair, oxidative and nitrosative stress, and oxidative defense capacity in rat heart. FASEB Journal. 2013;27:379–390. doi: 10.1096/fj.12-218685. [DOI] [PubMed] [Google Scholar]
- 17.Tarry-Adkins J.L., Martin-Gronert M.S., Chen J.H., Cripps R.L., Ozanne S.E. Maternal diet influences DNA damage, aortic telomere length, oxidative stress and antioxidant defense capacity in rats. FASEB Journal. 2008;22:2037–2044. doi: 10.1096/fj.07-099523. [DOI] [PubMed] [Google Scholar]
- 18.Haussman M.F., Winkler D.W., O′Reilly K.M., Huntingdon C.E., Nisbet I.C., Vleck C.M. Telomeres shorten more slowly in long-lived bird and mammals than in short-lived ones. Proceedings of the Biological Society. 2003;270:1387–1392. doi: 10.1098/rspb.2003.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minamino T., Komuru I. Role of telomeres in vascular senescence. Frontiers in Bioscience. 2008;13:2971–2979. doi: 10.2741/2902. [DOI] [PubMed] [Google Scholar]
- 20.Wong L.S., van der Harst P., de Boer R.A., Huzen J., van Gilst W.H., van Veldhuisen D.J. Aging, telomeres and heart failure. Heart Failure Reviews. 2010;15:479–486. doi: 10.1007/s10741-010-9173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Tocchetti C.G., Krieg T., Moens A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radical Biology and Medicine. 2012;53:1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Csiszar A., Ungvari Z., Edwards J.G., Kaminski P.M., Wolin M.S., Koller A. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circulation Research. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 23.van der Loo B., Labugger R., Skepper J.N., Bachschmid M., Kilo J., Powell J.M. Enhanced peroxynitrite formation is associated with vascular aging. Journal of Experimental Medicine. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oikawa S., Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Letters. 1999;453:365–368. doi: 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- 25.Richter T., von Zglinicki T. A continuous correlation between oxidative stress and telomere shortening. Experimental Gerontology. 2007;42:1039–1042. doi: 10.1016/j.exger.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Sharpless N.E., DePinho P.A. Telomeres, stem cells, senescence and cancer. Journal of Clinical Investigation. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ide T., Tsutsui H., Kinugawa S., Suematsu N., Hayashidani S., Ichikawa K. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circulation Research. 2000;86:152–157. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- 28.Battista M.C., Calvo E., Chorvatova A., Compt B., Corbeil J., Brochu M. Intra-uterine growth restriction and the programming of left ventricular remodelling in female rats. Journal of Physiology. 2005;565:197–205. doi: 10.1113/jphysiol.2004.078139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simmons R.A. Developmental origins of diabetes: the role of oxidative stress. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26:701–708. doi: 10.1016/j.beem.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yzydorczyk C., Comte B., Cambonie G., Lavoie J.C., Germain N., Ting Shun Y. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension. 2008;52:889–895. doi: 10.1161/HYPERTENSIONAHA.108.116251. [DOI] [PubMed] [Google Scholar]
- 31.Giussani D.A., Davidge S.T. Developmental programming of cardiovascular disease by prenatal hypoxia. Journal of Developmental Origins of Health and Disease. 2013 doi: 10.1017/S204017441300010X. [DOI] [PubMed] [Google Scholar]
- 32.Sen S., Simmons R.A. Maternal antioxidant supplementation prevents adiposity in Western diet fed rats. Diabetes. 2010;59:3058–3065. doi: 10.2337/db10-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giussani D.A., Camm E.J., Nui Y., Richter H.G., Blanco C.E., Gottschalk R. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One. 2012;7(2):e31017. doi: 10.1371/journal.pone.0031017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cambonie G., Comte B., Yzdorczyk C., Ntimbane T., Germaine N., Le N.L.O. Antenatal oxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. American Journal of Physiology – Regulatory Integrative and Comparative Physiology. 2006;292:R1236–R1245. doi: 10.1152/ajpregu.00227.2006. [DOI] [PubMed] [Google Scholar]
- 35.Hidaka T., Fuji K., Funahashi I., Fukutomi N., Hoseo K. Safety assessment of coenzyme Q10 (CoQ10) Biofactors. 2008;32:199–208. doi: 10.1002/biof.5520320124. [DOI] [PubMed] [Google Scholar]
- 36.Ikematsu H., Nakamura K., Harashima S., Fujii K., Fukutomi N. Safety assessment of coenzyme Q10 (Kaneka Q10) in healthy subjects: a double-blind, randomized placebo-controlled trial. Regulatory Toxicology and Pharmacology. 2006;44:212–218. doi: 10.1016/j.yrtph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Hathcock J.N., Shao A. Risk assessment for coenzyme Q (ubiquinone) Regulatory Toxicology and Pharmacology. 2006;45:282–288. doi: 10.1016/j.yrtph.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Bentinger M., Brismar K., Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;S7:S41–S50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Schmelzer C., Kitano M., Hosoe K., Dorling F. Ubiquinol affects the expression of genes involved in PPARα signalling and lipid metabolism without changes in methylation of CpG promoter islands in the liver of mice. Journal of Clinical Biochemistry and Nutrition. 2012;50:119–126. doi: 10.3164/jcbn.11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saretzki G., Murphy M.P., von Zglinicki T. MitoQ counteracts telomere length and elongates lifespan of fibroblasts. Aging Cell. 2003;2:141–143. doi: 10.1046/j.1474-9728.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- 41.Quiles J.L., Ochoa J.J., Huertas J.R., Mataix J. Coenzyme Q supplementation protects from age-related DNA double-strand breaks and increases lifespan in rats fed on a PUFA-rich diet. Experimental Gerontology. 2004;39:189–194. doi: 10.1016/j.exger.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Ochoa J.J., Quiles J.L., Heurtas J.R., Mataix J. Coenzyme Q10 protects from age-related oxidative stress and improves mitochondrial function in hearts of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. Journal of Gerontology: Biological Sciences and Medical Sciences. 2005;60:970–975. doi: 10.1093/gerona/60.8.970. [DOI] [PubMed] [Google Scholar]
- 43.Ochoa J.J., Quiles J.L., Lopez-Frias M., Heurtas J.R., Mataix J. Effect of lifelong coenzyme Q10 supplementation on age-related oxidative stress and mitochondrial function in liver and skeletal muscle of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. Journal of Gerontology: Biological Sciences and Medical Sciences. 2007;62:1211–1218. doi: 10.1093/gerona/62.11.1211. [DOI] [PubMed] [Google Scholar]
- 44.Mercer J.R., Yu E., Cheng K.K., Prime T.A., Griffin J.L., Masoodi M. The mitochondrial-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/−/ApoE−/− mice. Free Radical Biology and Medicine. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 45.Molyneux S.L., Florkowski C.M., Richards A.M., Lever M., Young J.M., George P.M. Coenzyme Q10; an adjuctive therapy for congestive heart failure? New Zealand Medical Journal. 2009;122:74–79. [PubMed] [Google Scholar]
- 46.Fotino A.D., Thompson-Paul A.M., Bazzano L.A. Coenzyme Q supplementation on heart failure: a meta-analysis. American Journal of Clinical Sciences. 2013;97:268–275. doi: 10.3945/ajcn.112.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sander S., Coleman C.I., Patel A.A., Kluger J., White C.M. The impact of coenzyme Q10 on systolic function in patients with chronic heart failure. Journal of Cardiac Failure. 2006;12:464–472. doi: 10.1016/j.cardfail.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Snoeck A., Remacle C., Reusens B., Hoett J.J. Effect of low protein diet during pregnancy on the fetal rat endocrine pancreas. Biology of the Neonate. 1990;57:107–118. doi: 10.1159/000243170. [DOI] [PubMed] [Google Scholar]
- 49.Santoz-Gonzalez M., Gomez-Diaz C., Nava P., Villalba J.M. Modifications of plasma proteome in long-lived rats fed on a coenzyme Q10-supplemented diet. Experimental Gerontology. 2007;42:798–806. doi: 10.1016/j.exger.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 50.Bello R.I., Gomez-Diaz C., Buron M.I., Alcain F.J., Navas P., Villalba J.M. Enhanced antioxidant protection of liver membranes in long lived rats fed on a coenzyme Q10-supplemented diet. Experimental Gerontology. 2005;40:694–706. doi: 10.1016/j.exger.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Lonnrot K., Holm P., Lagerstedt A., Huhtala H., Alho H. The effects of lifelong ubiquinone supplementation on the Q9 and Q10 tissue concentrations and life span of male rats and mice. Biochemistry and Molecular Biology International. 1998;44:727–737. doi: 10.1080/15216549800201772. [DOI] [PubMed] [Google Scholar]
- 52.Grunler J., Dallner G. Investigation of regulatory mechanisms in coenzyme Q metabolism. Methods in Enzymology. 2004;378:3–17. doi: 10.1016/S0076-6879(04)78001-5. [DOI] [PubMed] [Google Scholar]
- 53.Duncan A.J., Heales S.J.R., Mills K., Eaton S., Land J.M., Hargreaves I.P. Determination of coenzyme Q10 in blood mononuclear cells, skeletal muscle and plasma by HPLC using di-propoxy-Coenzyme Q10 as an internal standard. Clinical Chemistry. 2005;51:2380–2382. doi: 10.1373/clinchem.2005.054643. [DOI] [PubMed] [Google Scholar]
- 54.Boitier E., Degoul F., Desquerre I., Charpentier C., Francois D., Ponsot G. A case of mitochondrial encephalomyopathy associated with Coenzyme Q10 deficiency. Journal of the Neurological Sciences. 1998;156:41–46. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 55.Sugamura K., Keaney J.F. Reactive oxygen species in cardiovascular disease. Free Radical Biology and Medicine. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harzra T.K., Das A., Das A., Choudhury S., Kow Y.W., Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amsterdam) 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teixeira M., Rodrigues-Santos P., Garrido P., Costa E., Parada B., Sereno J. Cardiac antiapoptotic and proproliferative effect of recombinant human erythropoietin in a moderate stage of chronic renal failure in the rat. Journal of Pharmacy and Bioallied Sciences. 2012;1:76–83. doi: 10.4103/0975-7406.92743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ernster L., Dallner G. Biochemical, physical and medical aspects of ubiquinone functions. Biochimica et Biophysica Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 59.Kwong L.K., Kamzalov S., Rebrin I., Bayne A.V., Jana C.K., Morris P. Effects of coenzyme Q10 administration on its tissue concentrations, mitochondrial oxidant generation and oxidative stress in the rat. Free Radical Biology and Medicine. 2002;33:627–638. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 60.Huertas J.R., Martínez-Velasco E., Ibáñ̆ez S., López-Frías M., Ochoa J.J., Quiles J.L., Mataix J. Virgin olive oil protect heart mitochondria from peroxidative damage during aging. Biofactors. 1999;9:337–343. doi: 10.1002/biof.5520090233. [DOI] [PubMed] [Google Scholar]
- 61.Belardinelli R., Mucaj A., Lacalprice F., Solenghi M., Principi F., Tiano L. Coenzyme Q improves contractility of dysfunctional myocardium in chronic heart failure. Biofactors. 2005;25:137–145. doi: 10.1002/biof.5520250115. [DOI] [PubMed] [Google Scholar]
- 62.von Zglinicki T., Pilger R., Sitte N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radical Biology and Medicine. 2000;28:64–74. doi: 10.1016/s0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 63.Lee T.M., Chen C.C., Hsu Y.J. Differential effects of NADPH oxidase and xanthine oxidase inhibition on sympathetic reinnervation in postinfarct rat hearts. Free Radical Biology and Medicine. 2011;50:1461–1470. doi: 10.1016/j.freeradbiomed.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 64.Sindhu R.K., Roberts C.K., Ehdaie A., Zhan C., Vaziri N.D. Effects of aortic coarctation on aortic antioxidant enzymes and NADPH oxidase protein expression. Life Sciences. 2005;76:945–953. doi: 10.1016/j.lfs.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 65.Rindler P.M., Plafker S.M., Szweda L.I., Kinter M. High dietary fat selectively increases catalase expression within cardiac mitochondria. Journal of Biological Chemistry. 2012;28:1979–1990. doi: 10.1074/jbc.M112.412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacMillan-Crow L.A., Crow J.P., Thompson J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 67.Castilho A., Avelera C.A., Leal E.C., Simoes N.F., Fernandes C.R., Meirinhos R.I. Heme oxygenase-1 protects retinal epithelial cells against high glucose and oxidative/nitrosative stress-induced toxicity. PLoS One. 2012;7:1–10. doi: 10.1371/journal.pone.0042428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hangaishi M., Ishizaka N., Aizawa T., Kurihara Y., Taguchi J., Nagai R. Induction of heme oxygenase-1 can act protectively against cardiac ischemia/reperfusion in vivo. Biochemical and Biophysical Research Communications. 2000;279:582–588. doi: 10.1006/bbrc.2000.3973. [DOI] [PubMed] [Google Scholar]
- 69.Li L., Shoji W., Takano H., Nishimura N., Aoki Y., Takahashi R. Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS-induced oxidative stress. Biochemical and Biophysical Research Communications. 2007;13:715–721. doi: 10.1016/j.bbrc.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 70.Li L., Kaifu T., Obinata M., Takai T. Peroxiredoxin III-deficiency sensitizes macrophages to oxidative stress. Journal of Biochemistry. 2009;145:425–427. doi: 10.1093/jb/mvp011. [DOI] [PubMed] [Google Scholar]
- 71.Suzuki Y.J., Evans T. Regulation of cardiac myocyte apoptosis by the GATA-4 transcription factor. Life Science. 2004;74:1829–1838. doi: 10.1016/j.lfs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki Y.J. Stress-induced activation of GATA-4 in cardiac muscle cells. Free Radical Biology and Medicine. 2003;15:1589–1598. doi: 10.1016/s0891-5849(03)00208-9. [DOI] [PubMed] [Google Scholar]
- 73.Speakman J.R., Selman C. The free-radical damage theory: accumulating evidence against a simple link of oxidative stress and lifespan. Bioessays. 2011;33:255–259. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- 74.El-Assar M., Angulo J., Rodriguez-Manas L. Oxidative stress, and vascular inflammation in aging. Free Radical Biology and Medicine. 2013;65C:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 75.Hemiki S., Lapointe J., Wen Y. Taking a good look at free radicals in the aging process. Trends in Cell Biology. 2011;10:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vina J., Borras C., Abdelaziz K.M., Garcia-Valles R., Gomez-Cabrera M.C. The free radical theory of aging revisited: the cell-signaling disruption theory of aging. Antioxidants and Redox Signaling. 2013;19:779–787. doi: 10.1089/ars.2012.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Selman C., McLaren J.S., Meyer C., Duncan J.S., Redman P., Collins A.R. Life-long vitamin C supplementation in combination with cold exposure does not affect oxidative damage or lifespan in mice, but decreases expression of antioxidant protection genes. Mechanisms of Ageing and Development. 2006;127:897–904. doi: 10.1016/j.mad.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 78.Selman C., McLaren J.S., Collins A.R., Duthie G.G., Speakman J.R. Deleterious consequences of antioxidant supplementation on lifespan in a wild-derived mammal. Biology Letters. 2013;9:1–4. doi: 10.1098/rsbl.2013.0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data