

Abstract
The protein phosphatase 1-like gene (PPM1l) was identified as causal gene for obesity and metabolic abnormalities in mice. However, the underlying mechanisms were unknown. In this report, we find PPM1l encodes an endoplasmic reticulum (ER) membrane targeted protein phosphatase (PP2Ce) and has specific activity to basal and ER stress induced auto-phosphorylation of Inositol-REquiring protein-1 (IRE1). PP2Ce inactivation resulted in elevated IRE1 phosphorylation and higher expression of XBP-1, CHOP, and BiP at basal. However, ER stress stimulated XBP-1 and BiP induction was blunted while CHOP induction was further enhanced in PP2Ce null cells. PP2Ce protein levels are significantly induced during adipogenesis in vitro and are necessary for normal adipocyte maturation. Finally, we provide evidence that common genetic variation of PPM11 gene is significantly associated with human lipid profile. Therefore, PPM1l mediated IRE1 regulation and downstream ER stress signaling is a plausible molecular basis for its role in metabolic regulation and disorder.
Keywords: IRE1, PPM1l, Protein phosphatase, ER stress, Adipogenesis
Graphical abstract
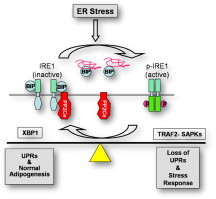
1. Introduction
Metabolic syndrome is a major risk factor for diabetes, cardiovascular diseases and cancer. Its manifestation involves complex cellular signaling pathways. Using a systems-based co-expression network analysis in mice, Chen et al. [1] has identified a number of sub-networks that are significantly associated with metabolic syndrome and obesity. Among the genes identified in this study, the Ppm1 gene was shown to exhibit a significant causal effect on obesity based on statistical modeling. This finding highlights the power of an unbiased systems genetics approach to uncover novel genes associated with complex phenotypic traits relevant to human diseases. However, the molecular mechanism of Ppml1 gene function in obesity, and the role of the Ppml1 gene in human metabolic disorders, remains unestablished. In this report, we employed a comprehensive set of experimental tools, including proteomics, cell biology and genetic analyses, to uncover the molecular basis and the functional relevance of the Ppml1 gene product. Our results indicate that the Ppml1 gene encodes an endoplasmic reticulum (ER) targeted protein phosphatase with high specificity for an ER membrane localized kinase/endoribonuclease inositol-requiring protein-1 (IRE1), a key regulator in the ER stress signaling pathway.
The endoplasmic reticulum (ER) is an essential organelle that carries out several biological processes, including protein and lipid synthesis, secretion, and post-translational protein modification [2–4]. Increases of unfolded or misfolded ER proteins leads to compensatory induction of ER chaperones and reduction of protein translation, collectively termed the unfolded protein response (UPR), through an integrated signaling network also known as ER stress signaling [5,6]. ER stress signaling has a major role in normal physiology of endocrine tissues, such as the pancreas and adipocytes, as well as other cell types having a major load of protein synthesis and secretion such as the lactating mammary gland, the central nervous system (CNS) and antibody-producing B cells. Abnormalities in the UPR, including dysregulated IRE1 activity, have been implicated in neurodegenerative diseases, diabetes, obesity, inflammation and heart disease [7–12]. However, it remains unclear which component(s) of ER stress signaling are critical in the pathogenesis of human metabolic diseases.
The ER membrane localized kinase/endoribonuclease (IRE1) is one of three key sensors which transmit ER stress from the ER lumen to downstream UPR elements [5,13,14]. Under ER stress conditions, IRE1 oligomerizes to activate its RNase activity, leading to specific splicing of X-box binding protein 1(XBP1) mRNA and 28S rRNA cleavage; these, in turn, result in the induction of UPR response genes and inhibition of protein synthesis, respectively [15–20]. In addition, activated IRE1 can also cause cell death by recruiting TRAF2 to the ER membrane and activating the pro-apoptotic ASK1-JNK pathway and subsequent cleavage of procaspase 12 [21–24]. Therefore, IRE1 mediated UPR can function as an adaptive response to ER stress to maintain ER homeostasis or serve as a signaling gateway to trigger ER-stress induced apoptosis. A recent study by Sha et al. [25] also showed that IRE1-XBP1 mediated signaling is critical to normal adipocyte differentiation, yet a more recent report by Han et la [26] showed adipogenesis regulation by eIF2α and CHOP, but not IRE1α. However, it is not clear if IRE1-XBP1 signaling contributes to genetic basis of metabolic disorders in human population.
IRE1 oligomerization and subsequent downstream RNase activation is initiated and maintained by trans-autophosphorylation of IRE1. Although IRE1 autophosphorylation has been characterized in great detail, the mechanisms involved in its dephosphorylation remain unclear as does the functional significance of this event [13]. BAX Inhibitor-1 (BI-1) is a specific negative regulator of IRE1α, and appears to modulate the signaling amplitude of IRE1α by suppressing its ribonuclease activity and downstream XBP-1 dependent cell survival [27,28]. However, no players have been identified to regulate the qualitative outcome of IRE1 signaling between adaptive UPR vs. stress kinase activation and cell death remain unclear.
Using a proteomic approach, we show that PP2Ce is an intrinsic component of the IRE1 signaling complex in mammalian cells. PP2Ce specifically interacts with IRE1 and efficiently dephosphorylates phosphor-Ser-724 of IRE1 without affecting other ER stress signaling branches. PP2Ce expression selectively attenuates IRE1 mediated XBP-1 splicing and UPR response following ER stress while inactivation of PP2Ce significantly increases IRE1α Ser724 phosphorylation, suggesting that PP2Ce is an endogenous IRE1-specific phosphatase. Unexpectedly, enhanced IRE1 phosphorylation resulting from PP2Ce inactivation reduced ER stress-stimulated XBP-1 induction and downstream UPR. Instead, PP2Ce inactivation triggered stress kinase activation and CHOP induction. More relevantly, we observed that PP2Ce protein is significantly induced during adipocyte differentiation and that PP2Ce deficiency impaired normal adipogenesis. Furthermore, a cross-species analysis of human GWAS data for associations between UPR genes and metabolic traits revealed that SNPs near Ppml1 are enriched for suggestive p-values with both plasma low density lipoprotein and triglyceride levels. Our study establishes a mechanistic basis for PPM1l mediated signaling in metabolic regulation, and reveals a previously uncharacterized important role for its encoded protein phosphatase PP2Ce in ER stress regulation through direct targeting to IRE1.
2. Results
2.1. Proteomic analysis identified Ppm1l as a molecular component of the IRE1 signaling complex
The Inositol Requring Protein-1 (IRE-1) is a highly conserved ER stress signal transducer. In order to investigate the molecular mechanisms of IRE1 regulation and function, the IRE1 signaling complex was isolated from a pancreatic insulinoma INS-1 cell line expressing an IRE1 dominant-negative mutant (IRE1α-K599A) fused with both FLAG and HA epitopes at its carboxyl terminus by binding to anti-FLAG and anti-HA immunoaffinity columns. The complexes were separated by SDS-PAGE (Figure 1A) and the molecular identities of the components were determined by LC/MS/MS tandem mass spectrometry as described [29]. A number of these proteins are known IRE1-interacting proteins, including BIP and ERdj3 (Figure 1A) [13,30]. Among the uncharacterized IRE1 interacting proteins, we identified the product of the Ppml1 gene based on extensive peptide coverage (Supplemental Figure 1A).
Figure 1.
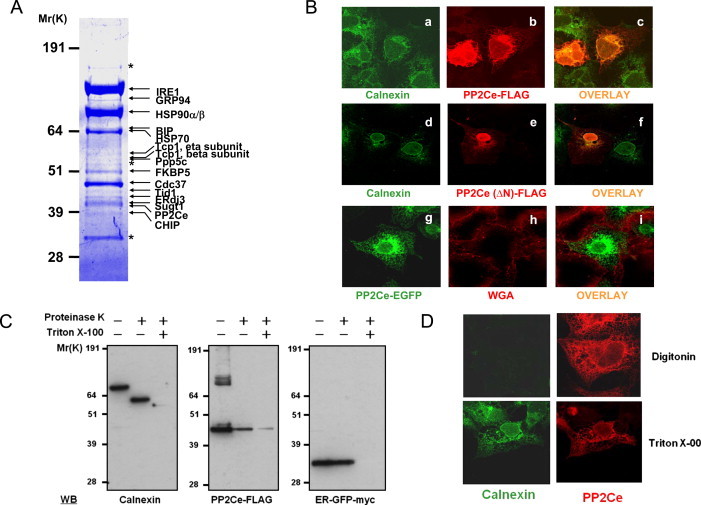
Identification of ER targeted PP2Ce in IRE1 signaling complex. (A) INS-1 pancreatic β-Cell lines expressing IRE1α–K599A C-terminally tagged with both FLAG and HA tags were subjected to sequential purification using anti-FLAG and anti-HA affinity columns. The IRE1α interacting proteins were detected by Coomassiee blue staining and further identified by LS/MS/MS mass spectrometry. The identities of IRE1 interacting proteins were indicated on the right. Asterisks marked the common nonspecific binding proteins observed in control immunoprecipitation assay using the parental INS-1 Flp-In T-REX cell line. (B) Confocal imaging of COS7 cells expressing PP2Ce-FLAG (panels a–c), or PP2Ce (∆N)- FLAG ( panels d–f), or PP2Ce-EGFP (panels g–i) and visualized by immunofluorescent staining as described in Experimental Procedures. ER and plasma membrane was visualized using Calnexin and wheat germ agglutinin (WGA) respectively. (C) ER fraction isolated from HEK293 cells expressing PP2Ce was treated with 50 μg/mL proteins K at 4 °C for 30 min in the absence or presence of 0.1% Triton X-100 followed by western blotting with the antibodies as indicated. (D) Confocal immunofluorescence images of COS7 cells expressing PP2Ce-FLAG after permeabilized with 25 μg/mL digitonin or 0.1% Triton X-100 and stained for calnexin (green) or FLAG (red) as indicated.
The Ppm1l gene is associated with metabolic syndrome based on an unbiased systems genetics analysis in mice [1]; however, the underlying molecular mechanism is unclear. The Ppm1l encoded protein is a member of the protein phosphatase 2C family and is highly conserved from fly to human (Supplemental Figure 1B). It contains a trans-membrane signal peptide at its N-terminus and a putative ER retention signal at its C-terminus (Supplemental Figure 1C and D). Therefore, we named the protein PP2Ce, as protein phosphatase 2C on endoplasmic reticulum. This protein was first reported by Li et al. and Saito et al. as a cytosolic protein phosphatase for negative regulation of TAK1 and ASK1 based on studies of a truncated form of the protein named as PP2Cε [31,32], but subsequently was reported by the same group to also act as an ER membrane protein phosphatase for the ceramide transport protein, CERT [33]. We found that the human PP2Ce full-length cDNA with its intact 5′ untranslated region encoded only a single protein product when ectopically expressed in HEK293 cells (Supplemental Figure 1). By immunofluorescence staining, we found that the full length FLAG-tagged PP2Ce protein was exclusively co-localized with Calnexin, an ER resident marker, whereas the N-terminal truncation mutant without the signal peptide region (PP2Ce-ΔN) displayed a nuclei and cytosolic distribution pattern (Figure 1B). Subcelluar fractionation also confirmed PP2Ce as an ER resident trans-membrane protein (Supplemental Figure 2). The Flag-tagged C-terminus of PP2Ce was found to be localized to the cytosol instead of the ER lumen based on a Proteinase K protection assay (Figure 1C) and immunostaining in partially permeabilized cells (Figure 1D). Thus, our data suggest that PP2Ce is an ER-membrane targeted protein with its phosphatase domain facing to the cytosol, and it is a component of the IRE-1 signaling complex in mammalian cells.
2.2. PP2Ce is a highly specific IRE1 phosphatase in vitro and in vivo
PP2Ce recombinant protein showed Ser/Thr phosphatase activity in a Mn2+ dependent manner comparable to that of PP2Cα against a generic phosphor-substrate, whereas mutation of the conserved aspartate 302 (D302A) abolished its phosphatase activity (Supplemental Figure 2), suggesting that PP2Ce has a characteristic metal dependent ser/thr phosphatase activity. Recombinant wildtype PP2Ce, but not a phosphatase-dead mutant (PP2Ce-D302A), effectively dephosphorylated the phosphor-Ser724 site of IRE1α protein (p-IRE1α) in vitro (Figure 2A). In contrast, recombinant PP2Cα did not dephosphorylate p-IRE1α in the same assay, suggesting that PP2Ce has intrinsic specific phosphatase activity towards IRE1. Over-expression of PP2Ce in INS-1 cells significantly attenuated the level of p-IRE1α-ser724 under both basal conditions and after ER stress induction (Figure 2B). Furthermore, when co-expressed with both IRE1α and PERK, another UPR-associated ER transmembrane kinase in the UPR signaling network, PP2Ce effectively reduced p-IRE1α but had no effect on the level of p-PERK (Figure 2C). These experiments establish that PP2Ce possesses intrinsic substrate specificity towards IRE1 Ser724 in vitro and in vivo.
Figure 2.
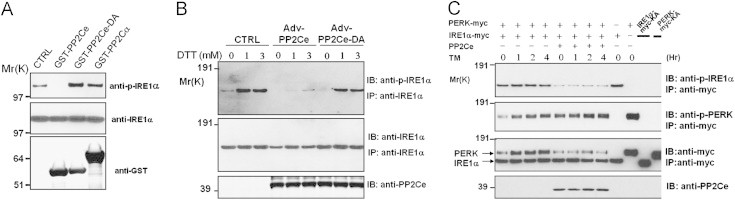
PP2Ce is an IRE1 specific phosphatase. (A) Phosphorylation status of purified hyperphosphorylated IRE1α after incubating with GST-PP2Ce, GST-PP2Ce-DA mutant and PP2Cα recombinant proteins as indicated. (B) INS-1 cells were infected with adenoviruses expressing PP2Ce (MOI 100) or PP2Ce-D302A (MOI 100) and then treated for 15 min with the indicated concentrations of DTT. Total cell lysates were analyzed with immuneprecipitation (IP) and immunoblot (IB) using the antibodies as indicated. (C) HEK293 cells were transiently transfected with the PERK-myc, IRE1α-myc and PP2Ce as indicated. 48 h later, the cells were treated with tunicamycin (10 μg/mL) for the 1, 2 and 4 h as indicated. Immunoprecipitates with anti-myc antibody or total cell extracts were subjected to IP and IB analysis with anti-p-PERK, anti-p-IRE1α, anti-myc and anti-PP2Ce as indicated.
2.3. Molecular basis for the specificity of PP2Ce activity towards IRE1
Using N-terminal deletion (ΔN) and C-terminal deletion (ΔC) mutants (Figure 3A), we showed that the N-terminal signal anchor (SA) was essential to dephosphorylate co-expressed IRE1α in cells (Figure 3B), while the putative ER retention signal in the PP2Ce C-terminus was dispensable. Moreover, wildtype PP2Cα protein (WT) and chimeric proteins containing the PP2Cα coding sequence fused with the PP2Ce N-terminal signal anchor sequence plus (PP2Cα−ER) or minus the PP2Cα C-terminus (PP2Cα−ERΔC) all failed to dephosphorylate IRE1α (Figure 3B). Therefore, ER membrane targeting alone is not sufficient to confer IRE1 specificity to other PP2C family members. To reveal the molecular motifs of IRE1 necessary for PP2Ce activity, we generated an IRE1/PERK chimeric protein containing an N-terminal ER-luminal domain from IRE1β and a cytosolic kinase domain from PERK (Figure 3C) which could undergo trans-autophosphorylation and subsequently phosphorylation of eIF2α when overexpressed in 293 cells (Figure 3D). However, co-expressed PP2Ce was not able to dephosphorylate the IRE1β-PERK chimera (Figure 3D), suggesting that the IRE1 cytosolic domain contains motifs necessary for PP2Ce mediated dephosphorylation. This hypothesis was supported by results obtained from co-immunoprecipitation assays. By co-expressing C-terminal tagged IRE1-myc and C-terminal tagged PP2Ce-Flag, we demonstrated that PP2Ce interacted with either IRE1α or IRE1β equally well but not PERK (Figure 4A and B). Using a series of PP2Ce truncation mutants, we identified an IRE1β binding domain in PP2Ce between amino acid residues 92 and 236 (Supplemental Figure 4). Conversely, we used a series of IRE1β deletion mutants in the same co-immunoprecipitation assay and determined the PP2Ce interaction domain in IRE1β to occur between amino acid residues 453 and 634 (Supplemental Figure 5). Therefore, IRE1 and PP2Ce interact via specific motifs residing in the cytosolic sides of both molecules.
Figure 3.
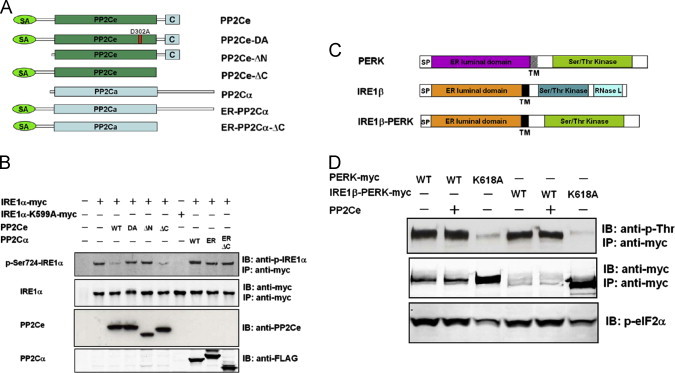
Molecular basis of PP2Ce specificity to IRE1. (A) Illustration of PP2Ce wildtype, phosphatase dead (D302A), N-terminal deletion (ΔN) and C-terminal deletion (ΔC) mutants, as well as PP2Cα widltype (WT), ER targeted (ER) and ER targeted combined with C-terminal deletion mutant (ER–ΔC). B. HEK293 cells were transiently transfected with plasmids expressing IRE1α-myc or kinase dead IRE1α-K599A-myc along with PP2Ce and PP2Cα expressing plasmid as illustrated in A. 48 h later, immunoprecipitates of cell lysates with anti-myc antibody or total cell extracts were subjected to immunoblotting analysis with anti-p-IREα, anti-myc, anti-PP2Ce and anti-Flag as indicated. (C) Illustration of the structural domains of PERK, IRE1β as well as IRE1β–PERK chimera. SP, signal peptide; TM, transmembrane domain. (D) HEK293 cells were transiently transfected with the plasmids expressing PP2Ce, PERK, IRE1β and chimera as illustrated in (C). Total cell extracts or immunoprecipitates using anti-myc antibody were subjected to immunoblotting analysis with anti-p-Thr, anti-myc, anti-p-eIF2α, anti-eIF2α and anti-PP2Ce as indicated.
Figure 4.
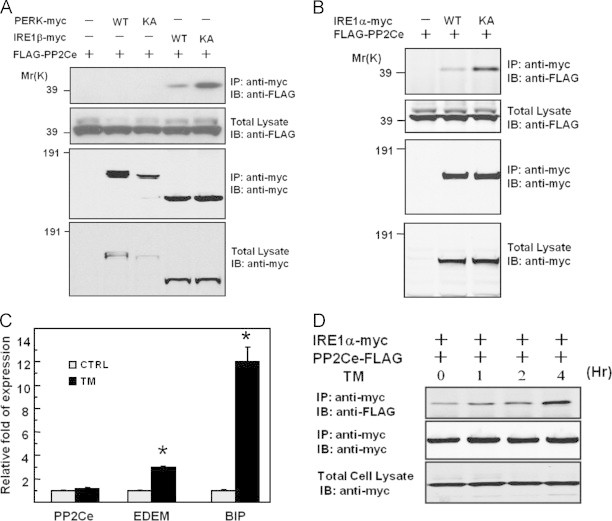
Specific interactions between PP2Ce and IRE1. (A–B) HEK293 cells were transiently transfected with the plasmids expressing Flag-PP2Ce with PERK-myc (widltype or kinase dead KA mutant), IRE1β-myc (wildtype or kinase dead KA mutant) in (A), or with IRE1α-myc (wildtype or kinase dead KA mutant) in (B). Total cell extracts or immunoprecipitates using anti-myc antibody were subjected to immunoblotting analysis with anti-Flag or anti-myc antibodies as indicated. (D) HEK293 cells were transiently transfected with the plasmids as indicated for 48 h, followed by the treatment of tunicamycin (10 μg/mL) for 1–4 h as indicated. Relative amount of IRE1α co-immunoprecipitated with PP2Ce was determined.
While mRNA levels for UPR genes such as BIP or EDEM were significantly induced upon ER stress, PP2Ce mRNA levels remained unchanged in response to ER stress (Figure 4C, Supplemental Figure 6). However, the interaction between IRE1 and PP2Ce was significantly enhanced upon ER-stress stimulation (Figure 4D). These data suggest that PP2Ce may function as a negative feedback regulator of IRE1 by counter-acting IRE1 autophosphorylation in response to ER stress.
2.4. PP2Ce is a negative regulator of IRE1 phosphorylation that modulates the functional outcome of UPR
Co-expression of IRE1β with wildtype PP2Ce or PP2Ce C-terminal truncation mutant (PP2Ce-ΔC), but not a phosphatase-dead PP2Ce mutant (PP2Ce-D302A) or a N-terminus truncation PP2Ce mutant (PP2Ce-ΔN),was sufficient to decrease the levels of phosphorylated IRE1β and significantly attenuated XBP-1 splicing as measured at both mRNA and protein levels (Figure 5). Primary embryonic fibroblasts (MEFs) prepared from P12 PP2Ce-/- mouse embryos showed significantly elevated p-IRE1 and JNK activity under basal conditions or in response to DTT treatment compared to the control wildtype MEF cells (Figure 6). A similar observation was made in primary spleenocytes isolated from PP2Ce-/- mice (Supplemental Figure 7). These data establish that PP2Ce is an endogenous IRE1 phosphatase necessary for maintaining basal IRE1 phosphorylation. As expected from increased IRE1 phosphorylation, expression levels of UPR genes XBP-1, BiP and CHOP were significantly elevated in PP2Ce-/- MEFs compared to wildtype controls in the basal state (Figure 6). However, under DTT stimulation, normal UPR response was impaired in PP2Ce-/- MEFs. While CHOP expression and stress-kinase JNK were induced, induction of XBP-1 and BiP was completely blunted in PP2Ce-/- MEFs compared to wild-type controls (Figure 6). These data establish that PP2Ce not only regulates the amplitude of basal IRE1 activity, but more importantly has a critical role in the functional outcome of UPR in response to ER stress.
Figure 5.
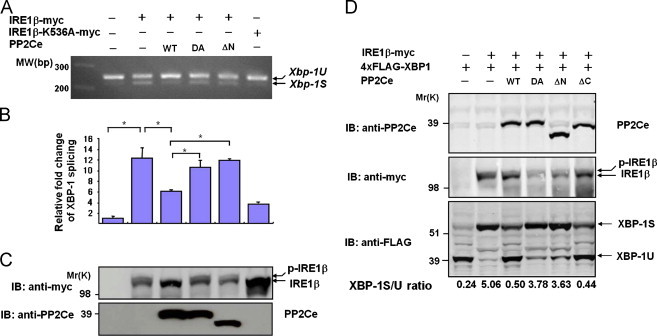
PP2Ce regulates IRE1 mediated ER stress signaling in vitro. A. Xbp-1S and Xbp-1U was detected by RT-PCR from HEK293 cells expressing IRE1β-myc, kinase dead IRE1β-K536A-myc mutant with or without PP2Ce as indicated. B. Xbp-1S/Xbp-1U ratio was quantified by density scan using Quantity One software (Bio-rad) from three experiments presented as mean±SD compared to the vector control. ⁎, p<0.01. (C) IRE-1β and PP2Ce immunoblots from the same HEK293 cells as in A.
Figure 6.
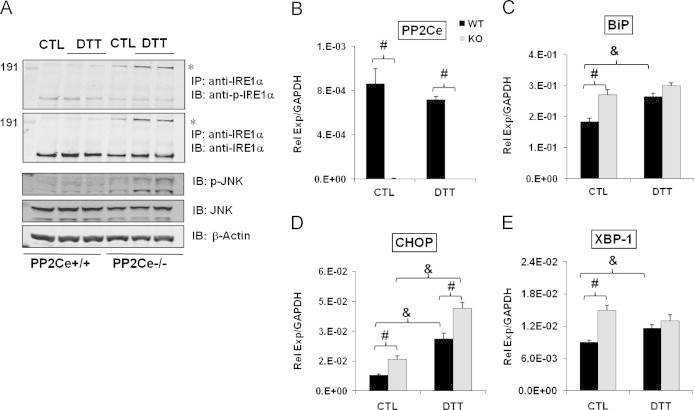
PP2Ce is essential to normal UPR in response to ER stress in vitro. (A) Immunoblots of phosphor—IRE1α and ΙΡΕ1α following immunoprecipitation with anti-IRE1α, or immunoblot for p-JNK, total JNK and β-actin from total protein extracts prepared from primary MEFs of P12 wildtype and PP2Ce-/- mouse embryo under control (CTL) or DTT treatment as described in Methods. ⁎ Indicates phosphorylated oligomerized IRE1 species. (B–E) Quantitative RT-PCR for mRNA levels of PP2Ce, BIP, CHOP and XBP-1 normalized against GAPDH as indicated in wildtype (WT) and PP2Ce-/- (KO) MEFs following control (CTL) and DTT treatment.
2.5. PP2Ce regulates UPR during adipogenesis
The IRE-1α-XBP1 pathway has been implicated in metabolic regulation and in particular normal adipogenesis [25,34]. Using hormonal induction of 3T3L1 cells as a well established in vitro model system for adipogenesis (Supplemental Figure 8), we found that XBP1, p-eIF2α, and BiP levels were all transiently induced with a peak at around day 3 post-induction and subsequently reduced in fully differentiated adipocytes (Figure 7A and B). In contrast, PP2Ce expression was induced at the mRNA and protein levels in later stages of differentiation and its expression was sustained in mature adipocytes (Figure 7A and C). Using an adenoviral vector expressing siRNA against PP2Ce, we significantly attenuated the PP2Ce induction in 3T3L1 cells during adipocyte differentiation (Figure 7C). Loss of PP2Ce expression led to defective adipogenesis as demonstrated by diminished PPARγ expression and a lack of lipid droplet staining (Figure 7C and D). As expected, loss of PP2Ce expression significantly and specifically enhanced p-IRE1α level without affecting PERK-mediated eIF2α phosphorylation (Figure 7E). Consistent with what was observed in MEFs, CHOP expression and p-JNK level were elevated while the expression of BiP was reduced in the PP2Ce deficient cells. These data suggest that PP2Ce induction is a necessary step for normal adipogenesis and its activity affects the dynamic regulation of UPR during adiopocyte differentiation.
Figure 7.
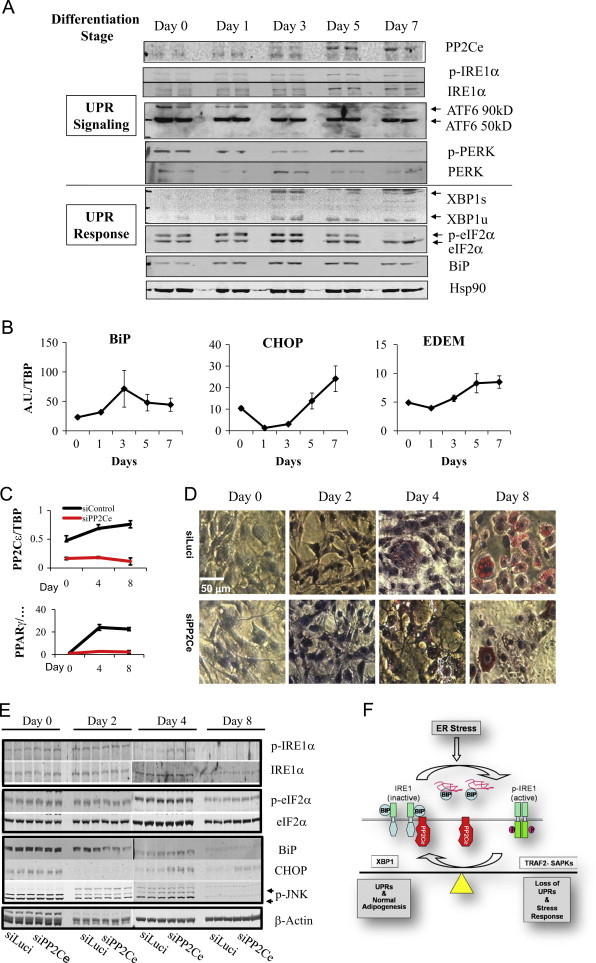
PP2Ce and UPR signaling during adipogenesis. (A) Immunoblot of IRE1α phosphorylation, ATF cleavage, and PERK phosphorylation at specified time points during adipogenesis induction in 3T3L1 cells. (B) Quantitative RT-PCR for UPR genes, BiP, CHOP and EDEM as indicated. (C) Quantitative RT-PCR for PP2Ce mRNA in 3T3L1 cells treated with siRNA for luciference as control and PP2Ce at different tie points after adipogenic induction. (D) Oil red staining in 3T3L1 cells as described in C. (E) Immunoblot of BiP, CHOP, phospho-and total IRE1α, phospho- and total eIF2α, phospho-JNK, β-actin as indicated from 3T3L1 cells treated with siLuci vs. siPP2Ce at different time points of adipocyte differentiation as indicated. (F) Schematic illustration of the function of PP2Ce in IRE1 mediated ER stress regulation and adipogenesis.
2.6. Genome wide association of Ppml1 with metabolic traits in human and mice
The mouse PP2Ce coding gene, Ppml1 was found to be associated with metabolic traits as reported previously [1]. Our current study implies that PP2Ce is an important regulator of ER stress signaling essential for normal UPR and adipogenesis. Although ER stress signaling has been implicated in metabolic regulation, the specific players involved in human metabolic disorders remain controversial and unclear [25,26]. To further explore the role of the ER stress pathway in human metabolic regulation, we examined a human GWAS dataset [35] for associations between plasma lipid levels and Ppml1 and 11 other genes involved in all three major branches of ER stress responses. For each gene, we identified each SNP within 1 Mb of the transcript, and used a permutation test (details in Methods) to determine whether nominally significant GWAS p-values (P<0.01) were enriched compared to a randomized distribution of p-values (Table 1). Among the 12 genes examined, Ppml1 was the only gene that showed highly significant association with LDL levels (p=0.0061) and triglyceride levels (p<0.000001). Interestingly, the functional target of PP2Ce IRE1β encoding gene Ern2 also showed a significant association with triglyceride levels (p=0.0028). This targeted analysis of SNP for UPR related genes in a prior human association analysis supports the significant contribution of the PP2Ce-IRE1β signaling axis to lipid level traits, and suggests a mechanism by which variations in activity of this pathway could influence metabolic disorders in humans.
Table 1.
Genome-wide association of ER stress response network genes with plasma triglyceride (A) and LDL (B) levels. Mouse chromosome number of the gene, number of SNPs identified within +/− 1 MB of the locus, number of SNPs with significant association with each traits based on GWAS analysis, and significance based on permutation analysis. p<0.01 was highlighted.
| (A) Association of ER stress response network genes with plasma triglyceride level | ||||
|---|---|---|---|---|
| Gene symbol | Chromosome | SNPs within 1 MB | Number of SNPs withp<0.01 | Significancep-value after permutation |
| Atf6 | 1 | 140 | 12 | 0.8333 |
| Ppp1r15b | 1 | 135 | 11 | 0.867 |
| Hspa5 | 2 | 121 | 20 | 0.0407 |
| Nfe2l2 | 2 | 170 | 28 | 0.022 |
| Ppm1l | 3 | 84 | 23 | <0.000001 |
| Eif2a | 3 | 64 | 0 | 0.9997 |
| Eif2ak3 | 6 | 24 | 0 | 0.9525 |
| Bbc3 | 7 | 28 | 3 | 0.4349 |
| Ern2 | 7 | 71 | 16 | 0.0028 |
| Ddit3 | 10 | 49 | 0 | 0.9983 |
| Dnajc3 | 14 | 84 | 19 | 0.0016 |
| Atf4 | 15 | 56 | 5 | 0.6539 |
| (B) Association of ER stress response network genes with plasma LDL level | ||||
| Atf6 | 1 | 107 | 8 | 0.918 |
| Ppp1r15b | 1 | 78 | 3 | 0.9891 |
| Hspa5 | 2 | 117 | 5 | 0.9975 |
| Nfe2l2 | 2 | 166 | 14 | 0.9241 |
| Ppm1l | 3 | 24 | 7 | 0.0061 |
| Eif2a | 3 | 76 | 3 | 0.9865 |
| Eif2ak3 | 6 | 17 | 0 | 0.9005 |
| Bbc3 | 7 | 26 | 5 | 0.0985 |
| Ern2 | 7 | 79 | 9 | 0.5074 |
| Ddit3 | 10 | 57 | 5 | 0.7133 |
| Dnajc3 | 14 | 69 | 5 | 0.8646 |
| Atf4 | 15 | 111 | 18 | 0.0791 |
3. Discussion
3.1. Conclusion
Metabolic dysregulation and obesity are major risk factors for cancer, diabetes, and cardiovascular diseases. In an earlier study using an unbiased genetic approach in mice, a novel gene Ppm1l was implicated in metabolic traits [1], but the underlying mechanism was unclear. In this report, we used a proteomic approach to identify the Ppm1l gene product as a molecular component of the IRE1 signaling complex. Based on comprehensive biochemical and molecular characterization, we establish that Ppm1l encodes an ER membrane anchored protein phosphatase 2C family member (PP2Ce) that has highly specific activity towards IRE1. PP2Ce expression suppresses basal and ER-stress induced IRE1 phosphorylation and downstream XBP-1 splicing, while inactivation of endogenous PP2Ce increases basal IRE1 phosphorylation leading to downstream UPR gene expression in the absence of ER stress. Unexpectedly, however, PP2Ce deficiency leads to impaired ER stress-induced UPR, and induction of the CHOP-mediated stress pathway. PP2Ce expression is highly induced during adipogenesis and PP2Ce inactivation leads to defective adipocyte differentiation and maturation in vitro. Therefore, our data has established a critical role for PP2Ce in ER stress regulation as a negative regulator of IRE1 phosphorylation. More importantly, disruption of this negative feedback regulation for IRE1 leads to a qualitatively different outcome of the ER stress response, changing from physiological UPR and survival to the activation of stress signaling and loss of UPR (Figure 7F). Finally, using both in vitro studies and targeted genome-wide association analysis, we demonstrated the strong correlation between PP2Ce and its target, IRE1, in adipogenesis and metabolic regulation. Overall, our study establishes that PP2Ce mediated IRE1 regulation in ER stress signaling is a potentially important molecular basis for its genetic contribution to metabolic syndrome.
3.2. Adaptive and pathological outcome of ER stress signal
Lin et al. [36] have reported that different durations and composition of ER stress signaling pathways can dictate cell fate and the balance between survival and death. IRE1 mediated signaling plays an important role in the physiological UPR, in which protein folding capacity is increased by enhanced expression of ER targeted chaperones while protein synthesis is attenuated and ER targeted protein degradation is elevated. However, constitutive induction of IRE1 can also induce a pathological form of ER stress involving CHOP, stress kinases and caspase-12 dependent apoptosis. A number of negative regulators have been implicated in ER stress regulation including PERK inhibitors p58IPK, CreP and GADD35 [37–40], and Bax Inhibitor-1, a negative regulator of the amplitude of IRE1 activity. However, the molecular mechanisms involved in the qualitative regulation of IRE1 signaling between compensatory UPR and pathological stress remain unclear. Our study provides the first molecular evidence that an ER targeted and IRE1 specific protein phosphatase may serve as an essential negative modulator for both the amplitude of IRE1 phosphorylation and activity and the functional outcome as physiological or pathological UPR. Preliminary observation from PP2Ce deficient mice showed defective mammary gland maturation and induced apoptosis in lactating females (unpublished results), suggesting that the pathological outcome resulted from defects in PP2Ce/IRE1 signaling may be contributed by the level and cellular content of ER stress response. Clearly, further studies are needed to fully understand the molecular basis of such modulation.
3.3. Unique specificity of PP2Ce towards IRE1
Typically, Ser/Thr protein phosphatases exhibit limited specificity towards their target substrates, and are often considered ubiquitous and promiscuous. In contrast, PP2Ce possesses a highly restricted ER targeted intracellular localization and a remarkable specificity to IRE1. IRE1 exhibits a reciprocal specificity for PP2Ce, since PP2Ce cannot desphosphorylate another ER transmembrane stress signaling molecule, PERK. Furthermore, another member of the PP2C family, PP2Cα, cannot dephosphorylate IRE1 even when the protein is artificially targeted to the ER membrane. Binding assays suggest that PP2Ce requires both ER targeting as well as specific cytosolic motifs to achieve direct interaction with IRE1. Based on recently revealed structural features [41,42], cytoplasmic domains of IRE1 may be inaccessible for inter-molecular interactions in clustered IRE1 polymers, and therefore monomeric IRE1 is likely a preferred binding partner of PP2Ce. This is supported by our observation that PP2Ce binds more strongly to a monomeric kinase-dead mutant of IRE1 (KA) relative to the wildtype IRE1 (Figure 3A/B), while enhanced IRE1 phosphorylation and dimerization is detected in PP2Ce-/- MEFs. Although we have provided evidence that the PP2Ce/IRE1 interaction is enhanced under ER stress stimulation, more studies will be needed to establish the molecular mechanisms underlying PP2Ce recruitment to the IRE1 signaling complex upon induction of ER stress. Post-translational modifications of either partners or association with other proteins, such as Hsp90/Cdc37 [43], may modulate such interaction in response to ER stress
A truncated form of PP2Ce (named PP2Cε) has been reported to bind to and dephosphorylate transforming growth factor-beta-activated kinase 1 (TAK1) and apoptosis signal-regulating kinase 1 (ASK1) [32,44]. However, as our results indicated that this truncated form is not generated from the full-length PP2Ce cDNA and most likely resulted from a cloning artifact. Interestingly, it is well established that the oligomerized form of IRE1 can recruit TRAF2 to the ER membrane via direct association, thereby activating the ASK1-JNK MAPK kinase pathway [22]. Stress kinase activation was observed in PP2Ce deficient cells upon ER stress or adipocyte differentiation. However, ASK1 was not identified in the IRE1 signaling complex based on our proteomic analysis (Figure 1 and data not shown). In short, we cannot exclude the possibility of other substrates of PP2Ce that can also contribute to its metabolic regulation, however, our molecular, cellular and genome-wide association analyses indicate that PP2Ce/IRE1 mediated ER stress regulation is a very plausible one.
ER stress regulation is an important cellular signaling event and is involved in numerous physiological processes including inflammation and metabolic regulation [45–47]. Dysfunction of ER stress signaling is implicated in human diseases such as cancer, obesity, diabetes, neurodegeneration and atherosclerosis [48]. Therefore, the characterization of PP2Ce as an important ER stress regulator provides new molecular insights for a spectrum of human diseases and suggests a potential molecular target for therapeutic intervention.
4. Materials and methods
4.1. Plasmid and adenoviral constructs
Full-length mouse PP2Ce was amplified from a mouse EST clone (BC096031) by PCR and then cloned into pcDNA3, pGEX5.3 and pShuttle-CMV vectors with or without 3XFLAG tag at the C-terminus. PP2Ce truncation mutant lacking the coding sequence for the first 57 amino acids was created by PCR and subcloned into pShuttle-CMV vector tagged with or without 3XFLAG at the C terminus. PP2Ce point mutant D302A was constructed by site directed PCR mutagenesis and cloned into pcDNA3 and pGEX5.3 vectors and pShuttle-CMV vectors with or without 3XFLAG tag at the C-terminus. Mouse PP2Cα was amplified from a mouse heart cDNA library by PCR and cloned into pcDNA3 and pGEX5.1 vector with or without N-terminal FLAG tag. Plasmids encoding PERK, IRE1β and XBP1 were kindly provided by Dr. David Ron (Skirball Institute, New York University School of Medicine). Human IRE1a cDNAs were a kind gift from Dr. Laurie H. Glimcher (Harvard Medical School). Mouse IRE1α full length coding sequence was derived from a mouse ETS clone (CF172635) which lacks the first 60 nucleotides from the translation initiation site at the 5′ end and also contains several mis-sense point mutations which were corrected by overlapping PCR according to the GENEBANK sequence (NM_023913). A C-terminal myc tag was linked to the mouse IRE1α coding sequence by overlapping PCR. mIRE1α–K599A mutants was generated by site directed PCR mutagenesis and cloned into pcDNA3 or pcDNA5/FRT/TO vectors with a myc tag or FLAG-HA tandom tag, respectively. Constructs of IRE1β-PERK chimera, ER-PP2Cα chimeras, IRE1β and PP2Ce truncation mutants were generated by overlapping PCR. All adenoviruses were concentrated with a cesium chloride gradient, and further purified on PD10 gel filtration column (Amersham), and preserved in PBS plus 10% glycerol.
4.2. RNAi mediated gene silencing
To achieve efficient know-down of endogenous PP2Ce, synthetic 64-nt oligonucleotide pairs targeting mouse and rat PP2Ce were annealed and ligated into pcDNA3-SUPER, which was constructed by subcloning the H1-RNA expression cassette into pcDNA3. As a control, shRNA targeting firefly luciferase was constructed into pcDNA3-SUPER as described above. The targeting sequences were listed as follows: PP2Ce shRNA1, 5′-caagacgcacccgtccatc-3′; PP2Ce shRNA2, 5′-gtcatggtggtgaagttca-3′; PP2Ce shRNA3, 5′-CCAAGACCTGGGAATTCAA-3′; Luciferase shRNA, 5′-CTGACGCGGAATACTTCGA-3′.
4.3. Antibodies
Polyclonal antibody (Abgent) against mouse PP2Ce were generated by immunizing rabbits with synthetic peptide corresponded to PP2Ce(97-113). Antibody was further affinity purified using Protein G column. Polyclonal antibody against β-Actin (SC-1616), Calnexin (SC-11397), XBP1(SC-7160), IRE1α (SC-20790), p-S724-IRE1α (NB100–2323, Novus Biologicals), BIP(#SPA826, Stressgen), p-eIF2a (#44-728G, Biosource), eIF2a(#9722, Cell signaling), p-JNK(#9251, Cell Signaling), p-T980-PERK(#3129, Cell Signaling), myc tag(#2276, Cell Signaling), myc tag (#2272, Cell Signaling) and anti-FLAG M2(Sigma) were purchased from the indicated suppliers. Polyclonal antibodies against IRE1α and p-S724-IRE1α, were a kind gift from Dr. Fumihiko Urano (UMASS Medical School).
4.4. Protein phosphatase assays
Protein phosphatase activity was determined by using synthetic phosphopeptide H–Arg–Arg–Ala–pThr–Val–Ala–OH (#p-152, BIOMOL) and BIOMOL GreenTM Rreagent(#AK111, BIOMOL). Briefly, 1 μg phosphopeptide was incubated with 50 ng purified GST recombinant proteins in 50 μL phosphatase buffer [50 mM Tris–HCl (pH 7.0), 0.1 mM EDTA, 2 mM DTT, and 0.01% Brij35, 10 mM MnCl2 (or 10 mM MgCl2)] at 30 °C for 30 min. The reaction was terminated by the addition of 100 μL BIOMOL green dye for 30 min and the released Pi was measured by fluorometer at 620 nm. For in vitro dephosphorylation assay of IRE1α, 0.2 μg purified hyperphosphorylated IRE1α was incubated with 20 ng GST fusion proteins in 25 μL phosphatase buffer at 30 °C for 10 min and the reaction was stopped by the addition of 4X SDS loading dye, followed by SDS-PAGE and immunoblotting analysis.
4.5. Cell cultures
HEK293, COS7 and NIH3T3 cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/ml streptomycin. NIH3T3 stable cell lines expressing. NIH3T3 stable cell lines expressing either PP2Ce specific shRNAs or luciferase shRNA were established by using G418(400 μg/mL) selection and then maintained in regular NIH3T3 culturing medium plus G418(400 μg/mL). Pancreatic β-cell lines INS-1 and INS-1Flp-In T-REX were a kind gift from Dr. Gerhart U Ryffel (Institut für Zellbiologie, Universitätsklinikum Essen). INS-1 cell were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, 2 mM l-glutamine and 50 µM β-mercaptoethanol, 100 U/ml penicillin and 100 µg/ml streptomycin. INS-1Flp-In T-REX cell was maintained in the same culture medium used for INS-1 except the additional antibiotics including 10 μg/ml blasticidine and 200 g/ml Zeocin. INS-1 stable cell lines expressing IRE1α-K599A was generated according to manufacture's instruction and maintained in the INS-1 culturing medium supplemented with 10 µg/ml blasticidine and 50 µg/ml hygromycin.
4.6. Subcelluar fractionation, alkaline extraction and protease protection assay
HEK293 cells were homogenized in buffer A (250 mM sucrose, 5 mM Tris, 1 mM EGTA, pH 7.4) using Teflon-glass Dounce homogenizer. Unbroken cells and cellular debris were removed by centrifugation at 1000×g for 3 min. Mitochondria enriched fraction was further obtained by centrifugation at 6000×g for 10 min. Finally, supernatants were subjected to centrifugation at 100,000×g for 1 h to separate the cytosolic soluble and microsome enriched membrane fractions. The fractions were analyzed using the standard immunoblotting method. Alkaline extraction was performed by incubating microsomes in 0.1 M Na2C03 (PH 11.0) or 0.1 M NaOH (PH 11.0) on ice for 20 min, followed by centrifugation at 100,000×g for 10 min, to separate the insoluable ER integral membrane protein form soluble ER lumenal and peripheral membrane proteins. Purified microsomes were incubated on ice in buffer A for 30 min in the presence or absence of 50 μg/mL Proteinase K, followed by 1 mM PMSF treatment for 10 min. The addition of 0.1% Triton-X-100 was used as a positive control. Then, microsomes were separated by centrifugation at 100,000×g for 10 min and subjected to SDS-PAGE analysis.
4.7. Immunoblotting
Cells were washed twice with ice cold PBS, and harvested in Buffer B (50 mM HEPES [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM glycerophosphate, 2.5 mM sodium pyrophosphate 1 mM Na3VO4, 20 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 μg/mL of aprotinin, leupeptin, and pepstatin). Total cell lysates were subjected to SDS-PAGE on 6%Tris-Glycine, or 4–12% Bis-Tris or 12% Bis-Tris gels and analyzed with indicated primary antibodies. Protein signals were detected using enhanced chemiluminescence (ECL) western blotting detection regents (Pierce).
4.8. Confocal microscopy
HEK293 cells were cultured on 12 mm coverslips in 24-well plate. Forty eight hours after transfection, cells were washed twice with PBS, fixed with 10% formalin for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked in PBS with 3%BSA and 5% donkey serum for 1 h. After incubation with anti-FLAG M2 antibody (1:2000) and anti-Calnexin(1:200) for 2 h, cells were washed four times with PBS and then incubated with Alexa568 conjugated Donkey anti-Mouse IgG (1:1000 Molecular Probes) and Alexa488 conjugated Donkey anti-Rabbit IgG (1:1000, Molecular Probes) for 2 h. The plasma membrane was detected using Alexa568 conjugated wheat germ agglutinin. Lastly, coverslips were extensively rinsed with PBS and mounted onto glass slides with Anti-Fade regents (Molecular Probes). Images were acquired by a laser scanning confocal microscope (Olympus Fluoview) and analyzed with MetaMorph (Universal Imaging Corp).
4.9. Realtime RT-PCR analysis
Total RNA was extracted from NIT3T3 cells using Trizol Reagent (Invitrogen) according to manufacturer's instructions. Five µg total RNA was reverse transcribed into the first-strand cDNA using Superscript First-strand synthesis Kit (Invitrogen) with oligo-dT primers. Then, cDNA transcripts were quantified by iCycler iQ Real-Time PCR Detection System (Bio-RAD) using iQ SYBR Green Supermix (Bio-RAD). Each reaction was performed in duplicate and values were averaged to calculate the relative expression level.
4.10. Glycerol-gradient sedimentation assay
INS-1 total cell extract (500 μL) was loaded onto a 4.5 mL 20−40% glycerol gradient in Buffer B. After centrifugation at 160,000 g for 20 h, 330 μL individual fractions were collected from top to bottom of the glycerol gradient and subjected to SDS−PAGE analysis.
4.11. Adipocyte differentiation
3T3L1 cells were cultured in DMEM with 10% CS, l-glutamine, sodium pyruvate, and pen/strep. Cells are grown for 2 days after they are fully confluent (Day 0), and cells are stimulated with MDI medium (DMEM, 10%FBS, insulin, pen/strep, sodium pyruvate, l-glutamine dexamethasone, and IBMX) for two days. On day 2, cells are treated with insulin medium (10% FBS/DMEM+insulin) for two days. Differentiation of MEF cells were done at P2 following similar protocol with some modifications including rosglitazone in MDI medium, and insulin in both MDI and insulin media. Cells were rinsed in ice-cold PBS and fixed in 10% formalin at room temperature for 45 min. Cells were rinsed with water three times, and rinsed once in 60% isopropanol. Cells were stained for lipid droplets with filtered Oil-Red-O for 3 min at room temperature, and rinsed in water. Nuclei were stained with hematoxylin (Sigma) for 2 min at room temperature.
4.12. Comparison to human genome wide association study
Syntenic regions for twelve genes involved in ER stress response (Atf4, Atf6, Bbc3, Ddit3, Dnajc3, Eif2a, Eif2ak3, Ern2, Hspa5, Nfe2l2, Ppm1l and Ppp1r15b) were identified using the NCBI Human-Mouse homology tool (http://www.ncbi.nlm.nih.gov/projects/homology/maps/). For each trait (HDL, LDL, TG), SNPs which were located between 1 Mb 3′ of the transcription start site and 1 Mb 5′ of the end of the transcript and whose p-value for the trait of interest was less than 0.01 were counted. Significance was determined by 10,000 genome-wide permutations of SNP p-values followed by counting as described above. Significance was equal to the number of trials which contained more SNPs with p-values less than 0.01 than our unpermuted data divided by the total number of permutations (10,000).
Conflict of interest
None.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.07.005.
Appendix A. Supplementary materials
Supplementary Material
References
- 1.Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452(7186):429–435. doi: 10.1038/nature06757. (PubMed PMID: 18344982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumann O., Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. International Review of Cytology. 2001;205:149–214. doi: 10.1016/s0074-7696(01)05004-5. (PubMed PMID: 11336391) [DOI] [PubMed] [Google Scholar]
- 3.Berridge M.J. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32(5–6):235–249. doi: 10.1016/s0143416002001823. (PubMed PMID: 12543086) [DOI] [PubMed] [Google Scholar]
- 4.Harding H.P., Calfon M., Urano F., Novoa I., Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annual Review of Cell and Developmental Biology. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. (PubMed PMID: 12142265) [DOI] [PubMed] [Google Scholar]
- 5.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. (PubMed PMID: 17565364) [DOI] [PubMed] [Google Scholar]
- 6.Schroder M., Kaufman R.J. The mammalian unfolded protein response. The Annual Review of Biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. (PubMed PMID: 15952902) [DOI] [PubMed] [Google Scholar]
- 7.Kapoor A., Sanyal A.J. Endoplasmic reticulum stress and the unfolded protein response. Clinical Liver Disease. 2009;13(4):581–590. doi: 10.1016/j.cld.2009.07.004. (PubMed PMID: 19818306. Epub 2009/10/13. eng) [DOI] [PubMed] [Google Scholar]
- 8.Minamino T., Kitakaze M. ER stress in cardiovascular disease. Journal of Molecular and Cellular Cardiology. 2010;48(6):1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. (PubMed PMID: 19913545. Epub 2009/11/17. eng) [DOI] [PubMed] [Google Scholar]
- 9.Minamino T., Komuro I., Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circulation Research. 2010;107(9):1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. (PubMed PMID: 21030724. Epub 2010/10/30. eng) [DOI] [PubMed] [Google Scholar]
- 10.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation Research. 2010;107(7):839–850. doi: 10.1161/CIRCRESAHA.110.224766. (PubMed PMID: 20884885. Pubmed Central PMCID: 2951143. Epub 2010/10/05. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas S.E., Dalton L.E., Daly M.L., Malzer E., Marciniak S.J. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metabolism Research and Reviews. 2010;26(8):611–621. doi: 10.1002/dmrr.1132. (PubMed PMID: 20922715. Epub 2010/10/06. eng) [DOI] [PubMed] [Google Scholar]
- 12.Vidal R., Caballero B., Couve A., Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington's disease. Current Molecular Medicine. 2011;11(1):1–12. doi: 10.2174/156652411794474419. (PubMed PMID: 21189122. Epub 2010/12/30. eng) [DOI] [PubMed] [Google Scholar]
- 13.Bertolotti A., Zhang Y., Hendershot L.M., Harding H.P., Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2(6):326–332. doi: 10.1038/35014014. (PubMed PMID: 10854322) [DOI] [PubMed] [Google Scholar]
- 14.Wu J., Kaufman R.J. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differentiation. 2006;13(3):374–384. doi: 10.1038/sj.cdd.4401840. (PubMed PMID: 16397578) [DOI] [PubMed] [Google Scholar]
- 15.Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. doi: 10.1038/415092a. (PubMed PMID: 11780124) [DOI] [PubMed] [Google Scholar]
- 16.Lee A.H., Iwakoshi N.N., Glimcher L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. (PubMed PMID: 14559994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes and Development. 2002;16(4):452–466. doi: 10.1101/gad.964702. (PubMed PMID: 11850408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oda Y., Okada T., Yoshida H., Kaufman R.J., Nagata K. Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. Journal of Cell Biology. 2006;172(3):383–393. doi: 10.1083/jcb.200507057. (PubMed PMID: 16449189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. (PubMed PMID: 11779464) [DOI] [PubMed] [Google Scholar]
- 20.Iwawaki T., Hosoda A., Okuda T., Kamigori Y., Nomura-Furuwatari C., Kimata Y. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nature Cell Biology. 2001;3(2):158–164. doi: 10.1038/35055065. (PubMed PMID: 11175748) [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. (PubMed PMID: 10638761) [DOI] [PubMed] [Google Scholar]
- 22.Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. (PubMed PMID: 12050113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H.P. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. (PubMed PMID: 10650002) [DOI] [PubMed] [Google Scholar]
- 24.Yoneda T., Imaizumi K., Oono K., Yui D., Gomi F., Katayama T. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. Journal of Biological Chemistry. 2001;276(17):13935–13940. doi: 10.1074/jbc.M010677200. (PubMed PMID: 11278723) [DOI] [PubMed] [Google Scholar]
- 25.Sha H., He Y., Chen H., Wang C., Zenno A., Shi H. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metabolism. 2009;9(6):556–564. doi: 10.1016/j.cmet.2009.04.009. (PubMed PMID: 19490910. Pubmed Central PMCID: 2963107. Epub 2009/06/06. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han J., Murthy R., Wood B., Song B., Wang S., Sun B. ER stress signalling through eIF2alpha and CHOP, but not IRE1alpha, attenuates adipogenesis in mice. Diabetologia. 2013;56(4):911–924. doi: 10.1007/s00125-012-2809-5. (PubMed PMID: 23314846. Pubmed Central PMCID: 3606029. Epub 2013/01/15. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castillo K., Rojas-Rivera D., Lisbona F., Caballero B., Nassif M., Court F.A. BAX inhibitor-1 regulates autophagy by controlling the IRE1alpha branch of the unfolded protein response. EMBO Journal. 2011;30(21):4465–4478. doi: 10.1038/emboj.2011.318. (PubMed PMID: 21926971. Epub 2011/09/20. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lisbona F., Rojas-Rivera D., Thielen P., Zamorano S., Todd D., Martinon F. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Molecular Cell. 2009;33(6):679–691. doi: 10.1016/j.molcel.2009.02.017. (PubMed PMID: 19328063. Pubmed Central PMCID: 2818874. Epub 2009/03/31. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu G., Sun H., She P., Youn J.Y., Warburton S., Ping P. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. Journal of Clinical Investigation. 2009 doi: 10.1172/JCI38151. (May 1. PubMed PMID: 19411760) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Y. Hendershot L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Molecular Biology of the Cell. 2005;16(1):40–50. doi: 10.1091/mbc.E04-05-0434. (PubMed PMID: 15525676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M.G., Katsura K., Nomiyama H., Komaki K., Ninomiya-Tsuji J., Matsumoto K. Regulation of the interleukin-1-induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cepsilon) Journal of Biological Chemistry. 4. 2003;2003;278(1):12013–12021. 12013–12021. doi: 10.1074/jbc.M211474200. (PubMed PMID: 12556533) (PubMed PMID: 12556533) [DOI] [PubMed] [Google Scholar]
- 32.Saito J.I., Toriumi S., Awano K., Ichijo H., Sasaki K., Kobayashi T. Regulation of apoptosis signal-regulating kinase 1 by protein phosphatase 2Cepsilon. Biochemical Journal. 2007 doi: 10.1042/BJ20070231. (Apr 25. PubMed PMID: 17456047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito S., Matsui H., Kawano M., Kumagai K., Tomishige N., Hanada K. Protein phosphatase 2Cepsilon is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. Journal of Biological Chemistry. 7. 2008;2008;283283(10)(10):6584–6593. 6584–6593. doi: 10.1074/jbc.M707691200. (PubMed PMID: 18165232) (PubMed PMID: 18165232) [DOI] [PubMed] [Google Scholar]
- 34.Cho Y.M., Kim D.H., Kwak S.N., Jeong S.W., Kwon O.J. X-box binding protein 1 enhances adipogenic differentiation of 3T3-L1 cells through the downregulation of Wnt10b expression. FEBS Letters. 2013 doi: 10.1016/j.febslet.2013.04.005. (Apr 19. PubMed PMID: 23603388. Epub 2013/04/23. Eng) [DOI] [PubMed] [Google Scholar]
- 35.Willer C.J., Sanna S., Jackson A.U., Scuteri A., Bonnycastle L.L., Clarke R. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature Genetics. 2008;40(2):161–169. doi: 10.1038/ng.76. (PubMed PMID: 18193043. Epub 2008/01/15. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin J.H., Li H., Yasumura D., Cohen H.R., Zhang C., Panning B. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318(5852):944–949. doi: 10.1126/science.1146361. (PubMed PMID: 17991856) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Huizen R., Martindale J.L., Gorospe M., Holbrook N.J. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. Journal of Biological Chemistry. 2003;278(18):15558–15564. doi: 10.1074/jbc.M212074200. (PubMed PMID: 12601012) [DOI] [PubMed] [Google Scholar]
- 38.Yan W., Frank C.L., Korth M.J., Sopher B.L., Novoa I., Ron D. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):15920–15925. doi: 10.1073/pnas.252341799. (PubMed PMID: 12446838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jousse C., Oyadomari S., Novoa I., Lu P., Zhang Y., Harding H.P. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. Journal of Cell Biology. 2003;163(4):767–775. doi: 10.1083/jcb.200308075. (PubMed PMID: 14638860) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novoa I., Zeng H., Harding H.P., Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. Journal of Cell Biology. 2001;153(5):1011–1022. doi: 10.1083/jcb.153.5.1011. (PubMed PMID: 11381086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K.P., Dey M., Neculai D., Cao C., Dever T.E., Sicheri F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132(1):89–100. doi: 10.1016/j.cell.2007.10.057. (PubMed PMID: 18191223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korennykh A.V., Egea P.F., Korostelev A.A., Finer-Moore J., Zhang C., Shokat K.M. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457(7230):687–693. doi: 10.1038/nature07661. (PubMed PMID: 19079236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ota A., Wang Y. Cdc37/Hsp90 protein-mediated regulation of IRE1alpha protein activity in endoplasmic reticulum stress response and insulin synthesis in INS-1 cells. Journal of Biological Chemistry. 2012;287(9):6266–6274. doi: 10.1074/jbc.M111.331264. (PubMed PMID: 22199355. Pubmed Central PMCID: 3307264. Epub 2011/12/27. eng) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamura S., Toriumi S., Saito J., Awano K., Kudo T.A., Kobayashi T. PP2C family members play key roles in regulation of cell survival and apoptosis. Cancer Science. 2006;97(7):563–567. doi: 10.1111/j.1349-7006.2006.00219.x. (PubMed PMID: 16827794) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ron D., Hubbard S.R. How IRE1 reacts to ER stress. Cell. 2008;132(1):24–26. doi: 10.1016/j.cell.2007.12.017. (PubMed PMID: 18191217) [DOI] [PubMed] [Google Scholar]
- 46.Schroder M. Endoplasmic reticulum stress responses. Cellular and Molecular Life Sciences. 2008;65(6):862–894. doi: 10.1007/s00018-007-7383-5. (PubMed PMID: 18038217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi: 10.1038/nature07203. (PubMed PMID: 18650916) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin J.H., Walter P., Yen T.S. Endoplasmic reticulum stress in disease pathogenesis. Annual Review of Pathology. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. (PubMed PMID: 18039139) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material