

Abstract
Medicines that decrease body weight and restore nutrient tolerance could improve human diabetes and obesity treatment outcomes. We developed lipid–acylated glucagon analogs that are co-agonists for the glucagon and glucagon-like peptide 1 receptors, and stimulate weight loss and plasma glucose lowering in pre-diabetic obese mice. Our studies identified lipid acylation (lipidation) can increase and balance in vitro potencies of select glucagon analogs for the two aforementioned receptors in a lipidation site-dependent manner. A general capacity for lipidation to enhance the secondary structure of glucagon analogs was recognized, and the energetics of this effect quantified. The molecular structure of a lipid–acylated glucagon analog in water was also characterized. These results support that lipidation can modify biological activity through thermodynamically-favorable intramolecular interactions which stabilize structure. This establishes use of lipidation to achieve specific pharmacology and implicates similar endogenous post-translational modifications as physiological tools capable of refining biological action in means previously underappreciated.
Abbreviations: Glucagon Aib2,16 amide, Aib2,16a
Keywords: Glucagon, Lipid, Diabetes, Obesity, Peptide, Structure
1. Introduction
Acylation of peptides and proteins with long-chain, saturated lipids (i.e. lipidation) has been demonstrated to extend biological action [1,2] and induce membrane association, respectively [3,4]. The former is accomplished by facilitating binding with carrier proteins particularly serum albumin and through self-aggregation, both of which delay the appearance of the lipidated peptide or protein in the plasma following subcutaneous administration. Previous work from this group and others established that glucagon-based peptides with balanced potency for the glucagon and glucagon-like peptide 1 (GLP-1) receptors (GCGR and GLP-1R, respectively) lowered body weight in rodents with greater efficacy than peptides predominantly activating the GLP-1R [5,6]. Importantly this was achieved without adverse glycemic effects traditionally observed with unopposed glucagon agonism. More recently the importance of relative balance in activity at each receptor for maximizing weight loss and preventing hyperglycemia in mice was reported [7].
In the pursuit of long-acting lipidated GCGR/GLP-1R co-agonists to be used as therapeutics for obesity and associated diseases such as type 2 diabetes, we observed that site-specific lipidation alone could generate balanced, high potency co-agonism in glucagon-based peptides. Through combined studies directed at chemical refinement, pharmacology and biophysical assessment the ability of site-specific lipidation to simultaneously increase potency and equalize selectivity for the glucagon and GLP-1 receptors was investigated. Direct physical interactions between the lipid unit and peptide functional groups were identified and appear responsible for directing the enhanced co-agonism in these lipidated peptides. Lipidation is a common endogenous post-translational modification [3,4,8] and our observations demonstrate how similar chemistry can elicit autonomous structural changes which improve pharmacodynamics for medicinal purposes. These results suggest that nature may use analogous lipidation approaches to refine the biological action of peptides and proteins.
2. Materials and methods
2.1. Peptide synthesis
Peptides were synthesized on a CSBio 336 peptide synthesizer using tert-butyloxycarbonyl (BOC) chemistry [9] for the linear sequence assembly and 9-fluorenylmethoxycarbonyl (FMOC) chemistry [10] for side chain acylation (amino acids and coupling reagents were purchased from Midwest Biotech, Chemimpex or Aaptec). Deprotection of Lys(Fmoc) was accomplished with 20% piperidine/DMF in a 30 min reaction. For spacing group additions the free lysine ϵ-amino group was coupled to a 10-fold molar excess of the appropriate FMOC-derivatized amino acids (e.g., FMOC-Glu-Otbu for γGlu) over 24 h with the PyBOP coupling reagent (1:1 M ratio in DIEA (1 mL)/DMF).
Acylation reactions with C8 (myristic acid, Sigma Aldrich), C16 (palmitic acid) or C18 (stearic acid) lipids used a 10-fold molar excess of lipid and DEPBT as the coupling reagent for a 24 h reaction. Deprotected peptide resins were dried with nitrogen and the peptide cleaved from the resin with HF/p-cresol 95:5 at 0 °C for 1 h. The reaction solvent was evaporated and the remaining resin–peptide mixture was precipitated with ice cold diethyl ether in an ice bath, filtered and further washed with cold ether. Samples were solubilized with a 5% AcOH/10% ACN/H2O solution prior to electrospray ionization mass spectrometry (ESI-MS) analysis (APIII). Correct molecular weight peptides were purified on a Waters reverse phase high performance liquid chromatography (HPLC) system running a Phoenomenex Luna C5 column and A buffer of 10% ACN/0.1% TFA/H2O and B buffer 0.1% TFA/ACN. Chromatography fractions were checked for purity with a Waters analytical HPLC and by ESI-MS.
2.2. In vitro receptor profiling
HEK293 cells were co-transfected with either the glucagon or GLP-1 receptor, and a luciferase gene linked to a cAMP responsive element for the bioassay. Cells were incubated with serum for 16 h in Dulbecco's modified Eagle medium (DMEM)/0.25% Bovine Growth Serum (HyClone) and then for 5 h with serial dilutions of the selected peptides at 37 °C under 90% humidity and 5% carbon dioxide. After the 5 h incubation, LucLite (Perkin-Elmer) luminescence substrate reagent was added to the plate and the light output was measured in a Wallac Trilux luminescence counter (Perkin-Elmer). 50% effective concentration (EC50) values were calculated using the logistic function in Origin 8.6 (Origin Lab).
Peptide concentrations were quantified using Beer's Law and UV absorbance measurements at 280 nm on a Thermo Electron Genesys 6 Spectrophotometer. Stock peptide samples were dissolved initially in 0.01 or 0.005 N NaOH (pH 11) at 50–150 µM and diluted into DMEM/0.5% Bovine Serum Albumin (BSA) to achieve desired concentrations for the assay.
2.3. In vivo pharmacology
C57B1/6 mice (n=8 per group) were obtained from Jackson Laboratories and single or group-housed on a 12:12 h light–dark cycle at 22 °C with free access to food and water. The mouse studies were performed under the guidelines sanctioned by the Institutional Animal Care and Use Committee of the University of Cincinnati. Mice were fed a high-lipid diet (Research Diets, high sucrose with 58% kcal from lipids) and treated with 100 nmol/kg (for the experiment in Figure 2A–E) or 10 nmol/kg (for the experiment in Figure 2F) of peptide or vehicle (saline) by daily (the first experiment) or bi-daily (the second experiment) injections for 7 or 6 days, respectively. Peptides were dissolved in saline (pH 7.4) or for peptides with very poor solubility (Lys12-γGlu-γGlu-C16 11 and Lys20-γGlu-γGlu-C16 17) in 0.001 N NaOH and saline (1:3). Body weight was monitored using nuclear magnetic resonance (NMR) technology (EchoMRI) [11] throughout the treatments and tail blood glucose levels measured using a hand-held glucometer (TheraSense Freestyle) on starting and end days. Graphs of the in vivo data were produced using GraphPad Prizm 5 software. The combined average starting weight for the mice was 49±1 g in the first study, and 46±2 g for the second.
Figure 2.
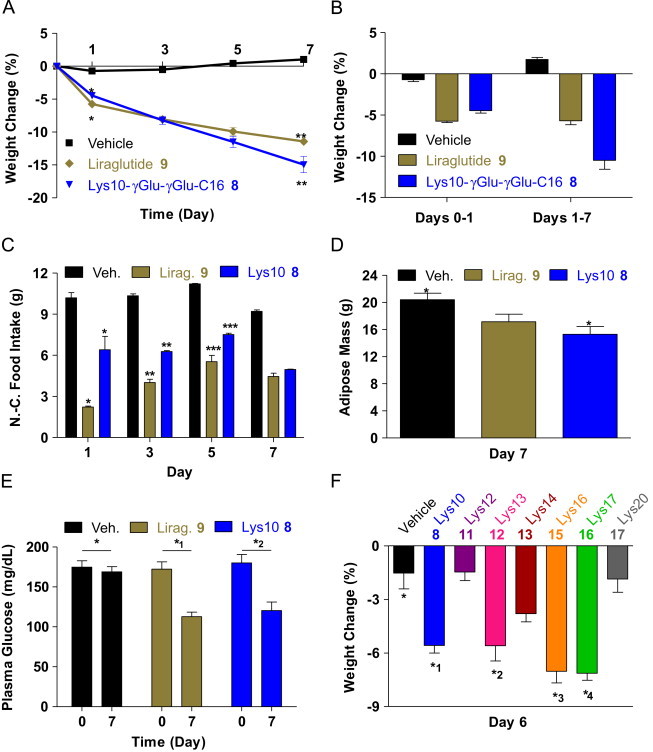
In vivo performance of lipidated Aib2,16a analogs. (A) Percent change in weight for DIO mice (N=8) given daily subcutaneous injections of vehicle (saline), liraglutide (9) (100 nmol/kg) or Lys10-γGlu-γGlu-C16 (8) (100 nmol/kg) for 7 days beginning at day zero (SEM shown). SEM, standard error of measurement. *P<0.01. **P<0.05. (B) Weight changes for the three groups at day one and from days one to seven (SEM shown). (C) Non-cumulative food intake by the three groups measured on days one, three, five and seven (SEM shown). N.-C., non-cumulative. *P<0.01. **P<0.001. ***P<0.001. (D) Adipose mass for the three groups at day seven. *P<0.01. (E) Plasma glucose levels for the three groups on days one and seven (SEM shown). *P<0.01. (F) Percent change in weight measured at day six for DIO mice (N=8) given subcutaneous injections on days zero, two and four of vehicle or a lipidated Aib2,16a analog (10 nmol/kg).
2.4. Circular dichroism (CD) spectroscopy and thermal denaturation
Spectra were collected on a JASCO J-715 Spectrometer in a 1 mm quartz cuvette (Hellma), or 2 mm with stir bar for thermal denaturation. For regular spectra, 3 scans were collected per sample. Data were smoothed followed by subtraction of the baseline buffer spectrum and unit conversion from millidegrees to molar ellipticity per residue [12]. Peptides were prepared as ~100 µM stock solutions in 0.001 N NaOH that were centrifuged to allow separation and removal of any aggregated material. After quantitation of the stock solution concentration by ultraviolet absorption spectroscopy, a sample was diluted into 20 mM sodium phosphate buffer (pH 7.4) or trifluoroethanol (TFE) to a final concentration of 10 µM. Thermal denaturation conditions were in 20 mM sodium phosphate and a 5 °C/min melting transition. Three consecutive heating and cooling cycles were performed on each peptide to ensure reversibility of the transition. The data was smoothed and baseline subtracted from each individual spectrum. Averaged data were fit to a two-state equilibrium folding model [13] using Origin 8.6 (OriginLab Corporation). For the lipidated peptides, the unfolding transition was split into two separate plots at 323 K and each fit individually to the two-state model [13].
2.5. Free energy of receptor activation
The free energy of receptor activation values were calculated using the equation ΔG=−RT ln Ka where R=1.98 kcal/mol, T=310 K and Ka=1/Kd, or 1 divided by the EC50 value from in vitro assays in Figure 1A and B [14].
Figure 1.
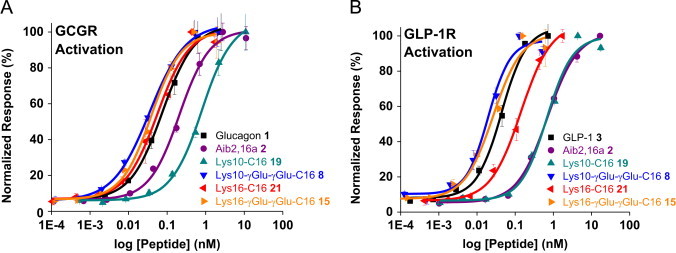
In vitro characterization of lipidated Aib2,16a analogs. (A) GCGR activation by lipidated Aib2,16a analogs in engineered HEK293 cells (N=2, SD shown). Data are fit to a dose–response function to provide the EC50 values. N, number of independent data points. SD, standard deviation. (B) The GLP-1R response measured using the same assay format and lipidated peptides as panel (A).
2.6. Dynamic light scattering
1 mg of peptide was dissolved in 1.0 mL 0.01 N NaOH in a 1.5 mL low protein retention tube by vortex, given 1 h of sitting time to allow particles in solution to settle to the bottom, and the top 700 µL carefully removed for placement into a plastic cuvette for the analysis using a Malvern Zetasizer Nano-S. These samples were not centrifuged to remove aggregate material because these (and the water solubility) results are viewed in the context of the in vivo studies, for which the peptide samples are not centrifuged prior to administration. Peptide samples for the 48 h, frozen at −20 °C for 1 day and frozen at −20 °C for 1 month data points were prepared using the same protocol. For the 1 month frozen samples, these were removed from the freezer once per 5 days, thawed and refrozen to instigate self-aggregation.
2.7. Water solubility
1 mg of peptide was weighed in a 1.5 mL low protein retention tube and to it 1.0 mL of PBS added, vortexed and then transferred to a 50 mL tube. This process was repeated with 0.5–1.0 mL of PBS until the peptide in the 50 mL tube was declared soluble by producing a clear solution equal to a PBS control tube and had no more than a few remaining visible fibrils in solution. The peptide sample was not moved for 30 min then a sample removed from the top for concentration determination by UV analysis to produce the final reported peptide solubility values.
2.8. NMR spectroscopy
For the water sample, 2 mg Lys13-γGlu-γGlu-C16 (12) was dissolved in 0.25 mL 0.001 NaOH (pH 11) and centrifuged at 15,000 RPM for 10 min. 167 µL of the centrifuged sample was then diluted 1:2 with 10 mM sodium phosphate (pH 4.7) in 5% D2O/H2O to provide the sample for NMR analysis at concentration of ~678 µM (2.67 mg/mL). For the deuterated trifluoroethanol (d-TFE) sample, 2 mg Lys13-γGlu-γGlu-C16 (12) was dissolved in 728 µL all d-TFE (1 g, sciencelab.com) after centrifugation at 15,000 RPM for 10 min to provide the NMR sample at ~694 µM (2.74 mg/mL). NMR experiments were performed at 298 K on a VNMRS 600 MHz (d-TFE sample) or 800 MHz (water sample) NMR spectrometer operating at magnetic field strengths of 14.1 T and 18.8 T, respectively, and using HCN cold probes (Agilent Technologies). For d-TFE, homonuclear 2D total correlation spectroscopy (TOCSY) used a relaxation delay of 1.5 s with 50 ms spin lock (mix) time, and 4 scans per free induction decay (FID) were used. Whereas for 2D nuclear Overhauser effect spectroscopy (NOESY), a relaxation delay of 1.5 s with 200 ms mixing time and 16 scans per FID was used. 2D TOCSY of the water sample used a relaxation delay of 1.5 s with 50 ms spin lock (mix) time and 64 scans per FID. For 2D NOESY, a relaxation delay of 1.5 s with 200 ms mixing time and 64 scans per FID was used.
NMR data was processed using the NMRPipe and NMRDraw software package [15]. The TOCSY and NOESY spectra (Figure S1 contains both NOESY spectra) were analyzed using Sparky 3 (T.D. Goddard and D.G. Kneller, University of California, San Francisco). Chemical shift statistics available from the BioMagResBank (www.bmrb.wisc.edu) were used to assist the assignment process [16]. The ChemBioDraw 1H NMR applet was used to predict the ppm values for C16 protons. Chemical shift resonance assignments for each data set are included in Tables S1 and S2.
Solution structures were calculating using xplor-NIH [17]. To produce the structural models, the NOESY cross peaks were divided into groups based on their signal intensity in SPARKY, where intensity values less than 50,000 were classified as 4.0 Å with possible deviation of –2.2 Å or +1.0 Å, intensity from 50,000 to 100,000 was set as 3.0–1.2 Å or+1.0 Å and 100,000 or greater is 2.5–0.7 Å or+0.5 Å. Restraints that were violated based on these classifications had their maximum deviation values increased conservatively as needed up to 6.0 Å. Hydrogen bond restraints were added to the water model corresponding to the i, i+3 or 4 helical NOEs assigned (Figure S2) [18]. For the final calculations, none of the structures shown in Figures 4 and 5 had any distance restraint violations. Structural images were produced using PyMOL (Schrödinger, Inc.).
Figure 4.
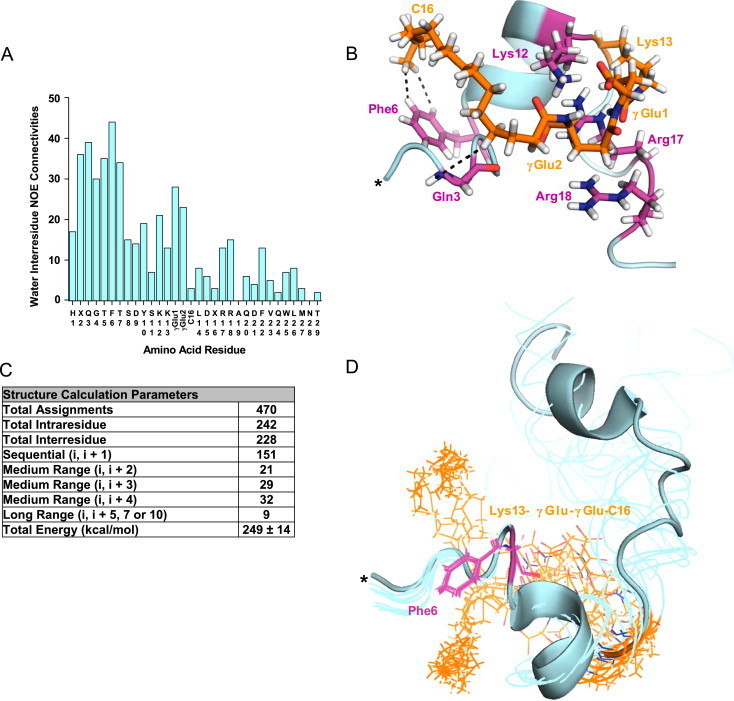
1H NMR-based structural model of Aib2,16a Lys13-γGlu-γGlu-C16 (12) in water. (A) Bar graph displaying the number of interresidue NOE connectivities per amino acid residue identified through the water NMR experiments. (B) One of the 10 lowest energy conformers from a 100 structure calculation in xplor-NIH [17] using the NOESY chemical shift resonance assignments as structural distance restraints is shown with the NOE interactions between C16 (HC4 and HC16, respectively) and residues Gln3 (HN) or Phe6 (Hδ and Hε) indicated by dotted lines. *, N-terminus. (C) Structure calculation parameters. Twelve additional hydrogen bond restraints were used for the calculation corresponding to the six helical NOEs assigned (Figure S3A) [17,18]. (D) Overlay of the 10 lowest energy conformers from the calculation. A single conformer is shown in cartoon form and the other nine as ribbons.
Figure 5.
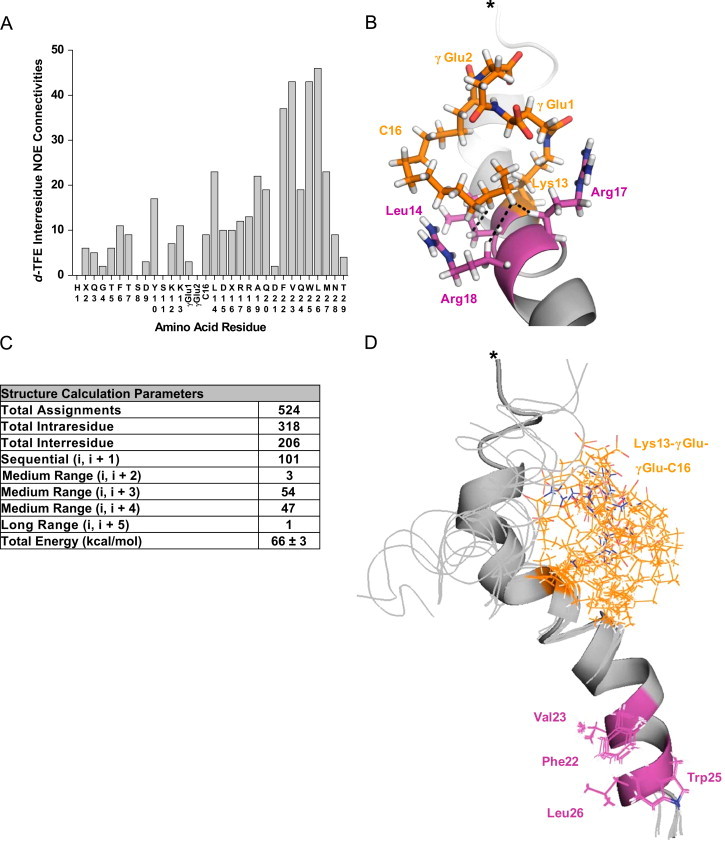
1H NMR-based structural model of Aib2,16a Lys13-γGlu-γGlu-C16 (12) in d-TFE. (A) Bar graph displaying the number of interresidue NOE connectivities per amino acid residue identified through the d-TFE NMR experiments. (B) One of the 10 lowest energy conformers from a 100 structure calculation in xplor-NIH [17] using the NOESY chemical shift resonance assignments as structural distance restraints is shown with the NOE interactions between C16 and residues Leu14 (HC14–Hβ), Arg17 (HC15–Hβ) or Arg18 (HC15–Hβ) indicated by dotted lines. *N-terminus. (C) Structure calculation parameters. (D) Overlay of the 10 lowest energy conformers from the calculation. A single conformer is shown in cartoon form and the other nine as ribbons.
3. Results
3.1. Lipidation enhances glucagon pharmacology
Tyr10 of glucagon (Table 1A) was substituted with lysine to provide a functional side chain for site-specific lipid acylation. The site selection was based upon the belief that a lipid would be least disruptive of physical and biological character when placed where the hormone is naturally hydrophobic. Tyr10 initially emerged from the study of glucagon and GLP-1 analogs substituted with leucine or tryptophan (Figure S2A) at the helical, hydrophobic face constituted by positions 6, 10 and 13. The purpose was to determine which, if any, of these residues could be modified with nonnative hydrophobic residues without losing receptor potency. This would identify positions that might be amenable with larger lipid-functionalized side chains. Bioactivity measurement in a HEK293 cell-based luciferase reporter assay, stably-transfected with either the GCGR or GLP-1 R was used to assess the receptor activation potency of various peptide analogs. Position 10 was observed to be the most accepting of mutation, without any appreciable change in potency at either the GCGR or GLP-1 R (Figure S2A).
Table 1.
Summary of lipidated glucagon-based peptides. (A) Important peptide sequences for this study. The differences of Aib2,16a and GLP-1 with the glucagon sequence are underlined. The N- and C-terminal hydrophobic helical faces in all three peptides consist of residues 6, 10 and 13, and residues 22, 23, 25 and 26, respectively [23]. X, aminoisobutyric acid (Aib). (B) Master table for peptides used in this study. EC50 values were obtained from experiments in Figure 1A and B for peptides 1, 2, 3, 8, 15, 20 and 21. For peptides 4, 5, 6, 10, 11, 12, 13, 14, 16, 17 and 19, the EC50 values were obtained from Figures S5 and S6, and normalized against standards from Figure 1A and B. As reference, liraglutide (9) has in vitro potency equal to GLP-1 (3) at the GLP-1R [2]. ΔGGLP-1R was calculated using Gibbs equation [14]. ΔGHelix was calculated using thermodynamic parameters (Table S3) obtained from the thermal denaturation experiments in Figure 3C. The aqueous and 90% TFE helical content relative to 2 was determined by dividing the 222 nm CD signal for each indicated peptide by the equivalent signal for Aib2,16a (2) (Figures 4A and S7A–C). PBS solubility was measured by ultraviolet absorption spectroscopy. Self-aggregation tendency was determined by dynamic light scattering experiments in 0.01 N sodium hydroxide (Figure S7D). Peptides were classified as strong if they aggregated immediately after sample preparation, moderate if aggregated after a single 24-h freeze cycle, weak if aggregated after 1 month of freezing or no apparent if they had no detected aggregation throughout these trials. No. peptide number. [Θ], mean residue ellipticity. Calc. pI, calculated isoelectric point. (C) Chemical structure of a Lys-γGlu-γGlu-C16 side chain at neutral pH.
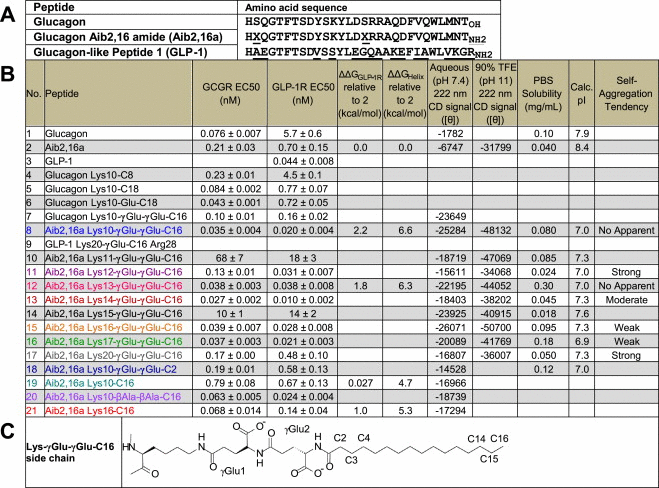 |
Lys10 acylated peptides were synthesized as either native glucagon (1) or the glucagon Aib2,16 amide analog (2) (hereafter referred to as Aib2,16a) (Table 1A). To aid with peptide identification, each peptide is assigned a boldface number upon introduction in Section 3. These numbers are listed next to their corresponding peptide in Table 1B and the peptide names in all of the figures, and also after introduction in the text where useful. The change at position 16 and the C-terminal amide are incorporated to increase the backbone helical structure (Figure S2B), which enhances the weak inherent agonist potency of glucagon by nearly tenfold for the GLP-1R (EC50=0.044±0.008 nM, 0.70±0.15 nM and 5.7±0.6 nM for GLP-1 3, Aib2,16a 2 and glucagon 1, respectively) (Table 1B) [5]. Resistance to N-terminal degradation by plasma dipeptidyl peptidase-IV (DPP-IV) is provided by the Aib2 substitution, although it also decreases GCGR potency by approximately threefold (EC50=0.21±0.03 nM versus 0.076±0.007 nM for Aib2,16a 2 and glucagon 1, respectively) [19]. The receptor activity profile of Aib2,16a is intermediate between native glucagon and a fully potent, equally balanced co-agonist being more than tenfold deficient in GLP-1R potency which remains the preferred molecular profile for the treatment of diabetes and obesity [5–7].
The first three Lys10 acylated glucagon analogs studied had addition of C8 (4) (C# designates the number of carbon atoms in the lipid; all lipids studied were saturated), C18 (5) or Glu-C18 (6) (i.e. lipidation with alpha-glutamic acid as a spacer) [20]. The Glu-C18 acylation alone improved native glucagon potency at the GCGR by twofold, while both C18-acylated analogs were increased approximately sixfold in potency at the GLP-1R (Table 1B). In contrast, C8 acylation had no apparent benefit at either receptor and selectively diminished potency at the GCGR by threefold (Table 1B). Further extension of the lipid from the backbone peptide through insertion of a structurally flexible two-amino acid spacer was explored in C16-acylated peptides. Glucagon acylated at Lys10 with γGlu-γGlu-C16 (7) (Table 1C) demonstrated potency slightly weaker at the GCGR (EC50=0.10±0.01 nM) but increased by thirty-fivefold at the GLP-1R (EC50=0.16±0.02 nM) when compared to the native hormones (Table 1B). Remarkably this single lipidation of native glucagon yields a balanced, high potency co-agonist at the GCGR and GLP-1R.
An identical γGlu-γGlu-C16 acylation at position 10 was explored in the Aib2,16a (2) backbone to generate Aib2,16a Lys10-γGlu-γGlu-C16 (8). For the remainder of the paper, this peptide and similar Aib2,16a analogs are referred to by the site and structure of the acylated side chain. Lys10-γGlu-γGlu-C16 (8) is a balanced co-agonist (EC50=0.035±0.004 nM and 0.020±0.004 nM at the GCGR and GLP-1R, respectively) of greater potency than either of the native ligands at their cognate receptors (Figure 1A and B). Use of the Aib2,16a sequence therefore permits the discovery of a second γGlu-γGlu-C16-acylated co-agonist that has superior potency, an equally-balanced receptor selectivity ratio (0.97:1) GCGR to GLP-1R selectivity with respect to the native hormones for Lys10-γGlu-γGlu-C16 (8) versus 2.91:1 GCGR to GLP-1R selectivity provided by the equivalent acylation in the native glucagon backbone (7) and is designed for sustained in vivo duration of action.
The comparative efficacy of this novel co-agonist (Lys10-γGlu-γGlu-C16 8) at promoting weight loss and plasma glucose control relative to a lipidated selective GLP-1R agonist (liraglutide 9 i.e. GLP-1 Lys20-γGlu-C16 Arg28) [2,21] was studied in diet-induced obese (DIO) mice. After 1-week of daily peptide administration the co-agonist (8) demonstrated enhanced loss of body weight (Figure 2A). The immediate weight loss on day one (Figure 2B) has consistency with the appetite suppressive effect characteristic of GLP-1R agonism [22]. This is reflected by the greater decrease in food intake observed for the liraglutide group on day one (Figure 2C), given that a selective GLP-1R agonist should cause roughly twice the amount of GLP-1R stimulation relative to a balanced GCGR/GLP-1R co-agonist. At day one and subsequently the weight change mediated by the peptide treatments is different (Figure 2B). For liraglutide (9) treatment the weight loss from days zero to one (−5.7±0.2%) is 108% greater than days one to seven (−5.3±0.2%). Lys10-γGlu-γGlu-C16 (8) however stimulated 233% more weight loss from days one to seven (−11±1%) compared to days zero to one (−4.5±0.3%) (Figure 2B). The differences in days one to seven can be attributed to the GCGR activity component of Lys10-γGlu-γGlu-C16 (8) enhancing energy expenditure to stimulate body weight loss more effectively than what can be accomplished through selective GLP-1R activation [5].
Additional support for this concept is contained in the non-cumulative food intake values. The liraglutide group ate less than the Lys10-γGlu-γGlu-C16 group over each measurement interval (Figure 2C) despite Lys10-γGlu-γGlu-C16 (8) causing 208% more weight loss from days one to seven than liraglutide (9) (Figure 2B). Furthermore, even though the Lys10-γGlu-γGlu-C16 group consumed more food, these mice had less adipose mass than the liraglutide group at the end of treatment (Figure 2D). For this to occur, the Lys10-γGlu-γGlu-C16 (8) treatment should be providing a second pharmacology (i.e. GCGR activation) besides GLP-1R stimulation. Plasma glucose as measured on day seven was normalized by both peptides (Figure 2E). Hence the relative GLP-1R activity in Lys10-γGlu-γGlu-C16 (8) was sufficient to counteract the inherent diabetogenic liability of unopposed GCGR agonism [5–7]. This in vivo study supports that lipidation represents a chemical modification which can be used to not only improve pharmacokinetics [1,2], but also enhance biological function. One potential counterpoint is that the treatments may have caused the mice to dislike the food through some type of illness brought on by the administered peptides. In this case, the resulting changes in food intake followed by body weight and plasma glucose might be occurring through peptide-induced conditions that are not related to the expected outcomes of treatment, e.g., increased nausea, satiety or energy expenditure. A conditioned taste aversion study could help rule out this possibility, but was not performed. Even if unrecognized conditions of malaise were causing observed responses, the unequal changes in food intake (Figure 2C) and adipose mass (Figure 2D) after liraglutide (9) or Lys10-γGlu-γGlu-C16 (8) treatment are still indicative of these two peptides having different mechanisms of action to produce the effects.
Another point of consideration is the possibility that the content of the food provided to the DIO mice may have participated in the ability of the peptides to produce the antidiabetic effects (Figure 2A, D and E). Specifically, after treatment has started the high lipid diet could be less preferred (Figure 2C) due to a synergistic effect of the peptide treatment to elicit nausea or satiety through GLP-1R stimulation (perhaps also the GCGR for the co-agonist) combined with a general uneasiness associated with having food largely comprised of lipids in the stomach. As a result, the mice would have less desire to eat because the provided food reinforced an unpleasant effect of the treatment. It may have been useful to determine the effect of Lys10-γGlu-γGlu-C16 (8) or a similar GCGR/GLP-1R co-agonist in lean mice that consume a protein- or carbohydrate-based diet to see if their food intake was equally affected. This analysis would help clarify if the food provided to the mice could have influenced the current experiment. Study of lean mice should also provide information regarding how their body will respond to a co-agonist if there is little or no adipose mass to lose. Although adipose mass data on day 0 was not collected, it appears that at least some of the weight lost by the Lys10-γGlu-γGlu-C16 group is attributable to changes in adipose tissue (Figure 2D). The remaining weight loss is then probably mostly protein and water content. One primary goal for GCGR/GLP-1R co-agonists is to provide a therapeutic route for removing excess body weight in humans. To understand how the lean mouse body would react to treatment with a co-agonist will help begin to identify whether a co-agonist could also be used to prevent human obesity from recurring.
3.2. Site-specific effects of lipidation on pharmacodynamics, pharmacokinetics and physical properties
Generality for the enhanced receptor potency observed with γGlu-γGlu-C16 acylation at position 10 was examined through the same modification of other residues (Ser11 10, Lys12 11, Tyr13 12, Leu14 13, Asp15 14, Aib16 15, Arg17 16 and Gln20 17) in Aib2,16a. Sites near the N- and C-terminus were avoided because these regions make contacts with receptor transmembrane and extracellular domains [23], respectively, and due to the closeness bulky side chains may not be tolerated very well. Direct receptor interactions with mid-region residues of glucagon and GLP-1 are less evident and as such represent preferred sites for lipidation. γGlu-γGlu-C16 acylation at positions 10 (8), 13 (12), 14 (13), 16 (15), and 17 (16) in Aib2,16a provided nearly equivalent increases in potency at the GCGR and GLP-1R, to a point in excess of the native hormones (Table 1B). The lesser activities through γGlu-γGlu-C16 acylation at position 11 (10), 12 (11), 15 (14), and 20 (17) is likely a function of the greater relative importance for receptor interactions involving these native amino acid side chains [24–27].
An in vivo study comparing the seven γGlu-γGlu-C16-acylated Aib2,16a analogs (8, 11, 12, 13, 15, 16 and 17) having in vitro potency equal to or surpassing the non-acylated form (2) was conducted in DIO mice. The total change in body weight measured at day seven after peptide administration on days zero, two and four is shown in Figure 2F. Four of the seven peptides (8, 12, 15 and 16) generated statistically significant (P<0.001) reductions in body weight relative to vehicle control. The Lys14-γGlu-γGlu-C16 (13) performance was intriguing in causing less weight loss than these four peptides, despite being of greatest in vitro potency at both receptors (Table 1B).
Inspection of the physical properties for each peptide tested in vivo revealed that solely the lipidation site imposes differing solubility in phosphate-buffered saline (PBS, pH 7.4) and propensity to self-aggregate (Table 1B). The Lys14-γGlu-γGlu-C16 analog (13) is less soluble and more prone to self-aggregate than the peptides equivalently acylated at positions 10 (8), 13 (12), 16 (15) or 17 (16). From this analysis, it seems plausible that the lower in vivo potency of Lys14-γGlu-γGlu-C16 results from unfavorable relative solubility and aggregation properties. This study establishes that the specific site of lipidation is of utmost importance to biophysical and in vitro biochemical properties that subsequently determine animal pharmacology, and presumably in vivo bioavailability.
3.3. Lipidation stabilizes helical structure
We hypothesized that the in vitro receptor potency enhancements (Table 1B) predominantly result from the lipidated side chain serving one of three possible functions; stabilization of peptide helical secondary structure, a constructive interaction with the cellular membrane, or the cellular receptor. All of the peptides with any form of C16 acylation exhibited considerable elevation in their aqueous helical content relative to Aib2,16a as measured by CD spectroscopy. Other groups have observed that lipidation using linear hydrocarbon chains ranging from C8 to C16 in peptides or proteins can enhance helical structure [28–32]. However, to our knowledge, the observations presented in this report are the first definitive examples of lipidation stabilizing structure and subsequently modifying biological function.
The magnitude of the increase in helicity was dependent on the specific site and chemical structure of the modification (Table 1B). Peptides with γGlu-γGlu-C16 acylation typically exhibited the highest aqueous helicity, with the Lys16 analog (15) being most helical. These results indicate that helix stabilization is the probable mediator of enhanced in vitro receptor potency, which matches with similar observations of helix stabilization through covalent bonding in glucagon analogs [5]. The diminished receptor potency of the γGlu-γGlu-C16 Lys11 (10) and Lys15 (14) analogs (Table 1B) despite their comparably enhanced helicity reflects the increased relative importance of these native amino acids side chains (Ser11 and Asp15) in receptor agonism [24,25]. A similar argument can be made for the Lys12 (11) and Lys20 (17) analogs (Table 1B), although the dependence is not as severe [26,27].
Aib2,16a is dramatically enhanced in potency at both receptors, most notably GLP-1R, following helix stabilization through γGlu-γGlu-C16 acylation at six of the nine lipidation sites studied (Table 1B). To explore the uniqueness of the structural enhancement derived from γGlu-γGlu-C16 acylation, a short-length acylation and a non-charged spacer were respectively studied in the Aib2,16a peptide. A position 10 γGlu-γGlu-C2 acylation (18) has an effect on helical structure that is similar but distinguishable from C16 acylation (19), where the CD signal at 190–210 nm has stronger negative absorbance (Figure 3A). The Lys10-βAla-βAla-C16 analog (20) has considerably reduced relative absorbance from 210 nm to 220 nm compared to the γGlu-γGlu-C16-counterpart (8), and the minimum is red-shifted by 10 nm (Figure 3A). Clearly both spacers impart unique secondary structure within this Lys10 set of analogs. The results illustrate the conformational sensitivity to relatively small changes in the overall primary chemical sequence of the peptide. C16 acylation of Aib2,16a at positions 10 (19) and 16 (21) resulted in almost equal helical structure enhancement, and insertion of γGlu-γGlu at either site (8 and 15, respectively) further augmented this effect (Figure 3A).
Figure 3.
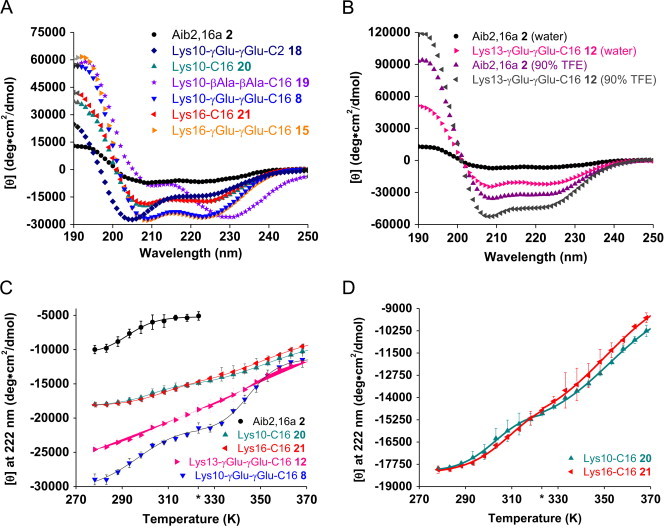
CD spectra and thermal denaturation of lipidated Aib2,16a analogs. (A) CD spectra (N=3) of lipidated Aib2,16a analogs (10 µM) in water (pH 7.4) at 298 K. [θ], mean residue ellipticity. (B) CD spectra of Lys13-γGlu-γGlu-C16 (12) (10 µM) compared to Aib2,16a (2) (10 µM) in water (pH 7.4) or 90% TFE/10% 0.01 N NaOH (pH 11) at 298 K. (C) Reversible thermal denaturation of Aib2,16a and lipidated Aib2,16a analogs in water (pH 7.4) (N=2 from consecutive experiments using the same sample, SD shown). Data are fit to a two-state folding equilibrium model where for the lipidated peptides the fit was performed as two individual transitions separated at 323 K (denoted by an * on the temperature axis) in each instance [13]. Thermodynamic parameters (Table S3) were obtained from the fit and used to calculate the helical free energy at 310 K (ΔG(310 K)), which was determined to be −2.8 kcal/mol, 1.9 kcal/mol, 2.5 kcal/mol, 3.5 kcal/mol and 3.8 kcal/mol for peptides 2, 20, 21, 12 and 8, respectively. These values were used to calculate ΔΔGHelix relative to 2 in Table 1B. (D) A second view of the data in panel (C) showing only peptides 20 and 21.
To determine whether the helix-stabilizing effect of lipidation persisted in a solvent capable of independently promoting secondary structure through stabilization of hydrophobic side chain interactions [33], the Lys-γGlu-γGlu-C16 Aib2,16a analogs were studied in dilute aqueous alkaline solutions containing 90% TFE. The non-acylated Aib2,16a peptide (2) exhibits strongly increased helicity in 90% TFE compared to in water (Figure 3B). All of the Aib2,16a analogs site-specifically acylated with γGlu-γGlu-C16 demonstrated higher helical content in 90% TFE than the non-acylated peptide backbone based on the strength of the 222 nm CD signal. The magnitude of the increase varies from 107% to 159% for the Lys12 (11) and Lys16 (15) analogs, respectively (Table 1B). Thus, the ability of the γGlu-γGlu-C16 acylation to enhance the core Aib2,16a helicity is site-dependent in both solvent conditions. However the effect is two to threefold greater in water at pH 7.4, where the increases vary from 231% (11) to 386% (15) in considering the same set of γGlu-γGlu-C16 analogs (Table 1B).
3.4. Thermodynamic analysis of lipidated peptides
CD thermal denaturation was performed to quantify the energetic contribution of the position 10 γGlu-γGlu-C16 acylation to Aib2,16a aqueous helical structure. Lys10-γGlu-γGlu-C16 (8) possesses 6.6 kcal/mol more helical free energy at 310 K than Aib2,16a (2) (Figure 3C). The relative difference in free energy of GLP-1R activation at 310 K for these two peptides was calculated to be 2.2 kcal/mol (Table 1B). Glucagon-related peptides bind target receptors in a primarily helical conformation despite having no preferred aqueous secondary structure [23]. Comparison of the calculated free energy differences (6.6 kcal/mol versus 2.2 kcal/mol) verifies that the conformational energy provided to the helical fold of Aib2,16a by the position 10 γGlu-γGlu-C16 acylation is sufficient to produce the increased in vitro potency recorded (Figure 1A and B).
Additional CD thermal denaturation experiments with three other lipidated Aib2,16a analogs permit further characterization of the energetics for lipidation to stabilize helical structure (Figure 3C and D). Lys10-C16 (19) and Lys16-C16 (21) gauge the free energy provided to the helical structure of Aib2,16a by lipidation without a spacer, which resembles cysteine palmitoylation commonly found in nature [4]. The increases in helical free energy at 310 K following C16 acylation at Lys10 and Lys16 were calculated to be 4.8 kcal/mol and 5.1 kcal/mol, respectively (Table 1B). Lys13-γGlu-γGlu-C16 (12) was studied because this peptide had been used for NMR spectroscopy experiments.
3.5. Molecular characterization of a lipidated peptide in water
The structural features of Lys13-γGlu-γGlu-C16 (12) were determined in water and in d-TFE using NOESY and TOCSY 1H NMR spectroscopy. This specific peptide was chosen for structure determination because it is the most soluble and resists self-aggregation to the greatest extent within the family of γGlu-γGlu-C16 analogs studied (Table 1B). Direct interactions between the γGlu-γGlu-C16 acylation and peptide backbone that could function to stabilize helical structure were identified in each solvent.
Qualitative analysis of the NOESY spectrum for Lys13-γGlu-γGlu-C16 (12) in water yielded 26 interresidue nuclear Overhauser effect (NOE) interactions that are indicative of secondary structure from N-terminal residues His1 to Lys13 (Figure S3A), including 4 of distance i, i+3 or 4 typical of helical structure beginning at Gly4. Gln3 and Phe6 accounted for the most total interresidue NOE connectivities per single residue (Figure 4A). Long range NOE interactions were identified between C16 HC4 and Gln3 HN, also C16 HC16 with Phe6 Hδ and Hε (Figures 4B, S3B and C). These observations strongly support the presence of a structure-stabilizing effect at the peptide N-terminus by C16, including for His1 to Gln which are evidently non-helical. It should be noted that the non-acylated version of this peptide (2) at approximately seventy-fold lower concentration and different pH has low-level helicity that was increased by 329% following γGlu-γGlu-C16 acylation (Table 1B and Figure 3B). Consequently the exact degree of enhancement at the N-terminus is unknown but expected to be substantial.
The HN, Hα, Hβ and Hγ protons in γGlu1 and γGlu2 were identified primarily through their extensive (27 total interactions) NOE connectivity with the Hδ, Hε, Hη and Hζ protons of the Lys12, Arg17 and Arg18 side chains (Figure S3C–F). These apparent electrostatically-driven connectivities seem to have less of an impact on helical structure than C16 due to the fewer number of NOEs indicative of secondary structure that were obtained from Leu14 to Arg18 (2 total interactions) (Figure S3A). Some synergism and/or structural overlap could exist between γGlu-γGlu and C16 to stabilize helical structure for residues located near the middle of His1 to Arg18, in particular Lys12 and Lys13, but it is difficult to precisely determine.
Overall, there was far less structural information for C-terminal residues than N-terminal (Figure 4A). The five NOE interactions representative of secondary structure, including 2 helical NOEs, identified beyond Asp21 (Figure S3A) are probably due to the C-terminal amide substitution aiding partial formation of the helical C-terminal hydrophobic face (five NOEs were identified between Phe22 & Trp25 aromatic, Val23 Hγ and Leu26 Hδ protons). However, it cannot be excluded that the lipidated side chain does not have some (expectedly minor) ability to stabilize C-terminal helical structure despite the absence of direct contacts with this region. An ensemble of 10 Lys13-γGlu-γGlu-C16 (12) conformers that were calculated using the water NOESY chemical shift resonance assignments (Figure 4C) as structural distance restraints is displayed in Figure 4D. The structural models depict how lipidation can function similar to lipid micelles or TFE [34–36] to permit NMR characterization of a peptide-based class B GPCR by promoting stabilization of intramolecular interactions. Yet, lipidation is distinct in that it creates a nucleation site within which nearby backbone residues can make long range physical contacts, thereby supporting the formation of local secondary structure (Figures 4 and S3).
3.6. Molecular characterization of a lipidated peptide in d-TFE
Analysis of the Lys13-γGlu-γGlu-C16 (12) 1H NMR TOCSY and NOESY spectra derived from d-TFE yielded 21 total NOEs of distance i, i+3 or 4 that are representative of helical secondary structure throughout nearly the entire linear backbone from Gln3 to Asn28 (Figure S4). The Phe22 & Trp25 aromatic, Val23 Hγ and Leu26 Hδ protons produced 21 total NOE interactions with each other (Figure 5A) indicating stabilization of the C-terminal hydrophobic face. Together these data show that d-TFE stabilized a conformation that is similar to the purported receptor-active state, which is predominantly a linear helix through a C-terminal hydrophobic face [23].
NOE connectivity was identified for C16 with Lys13 and γGlu1 (five total interactions), and perhaps more importantly for helix stabilization C16 HC14 with Leu14 Hβ, and C16 HC15 with Arg17 Hβ and Arg18 Hβ (Figures 5B and S4B–E). γGlu1 had two additional NOE interactions with Lys13 and Leu14, but no other interresidue connectivities for γGlu1 or γGlu2 could be identified. The d-TFE NOESY chemical shift resonance assignments (Figure 5C) were used to provide structural distance restraints for generating an ensemble of Lys13-γGlu-γGlu-C16 (12) models, 10 of which are shown in Figure 5D. Together, the Lys13, Leu14, Arg17 and Arg18 side chains appear to form a binding interface for γGlu1 and C16 (Figure 4B), that could serve to stabilize helical structure (Figure 3B) in the mid-region of the peptide.
3.7. Lipid–peptide structural relationships influence receptor potency
Glucagon-related peptides are believed to bind their class B G protein-coupled receptors through conserved C-terminal helical, hydrophobic faces constituted by residues 22, 23, 25 and 26 (Table 1A) [23]. Since d-TFE induces a similar conformation (Figure 5D), the paired water and d-TFE NMR data sets can exemplify the structural change that occurs as the Lys13-γGlu-γGlu-C16 analog (12) transitions between receptor unbound and bound forms. The vastly altered structural interactions observed in each solvent condition (Figures 4B and 5B) help to conceptualize how the γGlu-γGlu-C16 acylation will modify physical behavior with the peptide in response to altered environments to support different secondary structures.
Interestingly, the role of γGlu-γGlu to enhance in vitro potency at Lys10 where direct C16 acylation without spacer provides an Aib2,16a analog of approximately tenfold lower potency at both receptors (Figure 1A and B) (ΔΔG=2.2 kcal/mol between Lys10-γGlu-γGlu-C16 8 and Lys10-C16 19 at the GLP-1R) is inconsistent with the sufficiently elevated helical free energy calculated for both peptides (ΔΔG=6.6 kcal/mol and 4.7 kcal/mol, respectively) (Table 1B). The spacer may be enabling structural flexibility such that the entire peptide can effectively transition to the receptor-bound state, utilizing the helix stabilization energy provided by the lipidated side chain to enhance receptor potency. It seems that the helix stabilization in Lys10-C16 (19) might reflect a counterproductive interaction as viewed by the relative inability to efficiently transition to the receptor-active conformation.
Moving C16 closer to the middle of the Aib2,16a backbone devoid of spacer was investigated to determine whether receptor potency could be enhanced by direct acylation when attached at a point more distant from the N-terminus than Lys10. Acylation at Lys16 with C16 (21) provided equal potency as the γGlu-γGlu-C16 modification (15) at the GCGR, each analog being approximately fourfold greater than the non-acylated peptide (2) and more than tenfold improved from Lys10-C16 (19) (Figure 1A). Additionally, Lys16-C16 (21) exhibited fivefold greater potency relative to the Lys10 derivative (19) and the non-acylated peptide (2) at the GLP-1R (Figure 1B). These results highlight the sensitivity of the two receptors to the location and chemical structure in which the lipid is covalently fixed to the peptide. In each instance the helical content of the two C16-acylated Aib2,16a analogs (19 and 21) is enhanced to nearly equivalent amounts (Figure 3A). More subtle differences in peptide structure are evidently producing the sizable potency differences in receptor co-agonism, demonstrating intricacies of the inherent structural propensity for lipidation to modulate biological function.
4. Discussion
Single molecule peptides capable of balanced action at multiple receptors should provide enhanced efficacy in the treatment diabetes, obesity, their associated metabolic syndrome [5,6] and potentially other diseases. We observed that peptide acylation with a long-chain saturated lipid can enhance pharmacodynamics (Table 1B) to achieve this goal. The ability of lipidation to increase receptor potency and equalize receptor selectivity appears to be accomplished through non-covalent stabilization of secondary structure (Table 1B and Figures 3A, B, and 4), in a way similar to previous results obtained by covalent backbone coupling [5]. Lipidation also modified biophysical properties of studied peptides, including their water solubility, self-aggregation propensity and thermal stability (Table 1B and Figure 3C). All of the changes in peptide functionality were observed to be dependent on the lipidation chemical structure and site of attachment to the peptide backbone (Table 1B). The helix stabilization derived from lipidation provided the opportunity to visualize the solution structure of a single glucagon-based co-agonist in aqueous and non-aqueous solvents, through NMR spectroscopy. Two profoundly different helix-stabilized structures were characterized (Figures 4 and 5) that typify the dynamic ability of lipidation to support the transition from a preferred aqueous conformation in the absence of receptor, relative to a structure that is predictably more suitable to receptor binding. These observations expand the prior application of lipidation as a modification suitable for prolonging pharmacokinetics to a more potent and versatile tool in optimization of peptide and protein structure–function.
Acylation of cysteine and N-terminal amines with lipids such as myristate and palmitate are common post-translational modifications observed in various natural biological settings [3,4,8,28,30,32,37–39]. In analogy, protein phosphorylation is a reversible, hydrophilic chemical modification that is capable of modifying structure and subsequent biological activity [40,41]. The extension of these glucagon-based observations with lipidation (Table 1B and Figures 3C and 4) suggests analogous structural changes are possible through endogenous post-translational lipidation, that could lead to the enhancement or redirection of biological function [3,4,28,30,32,37–39]. Furthermore, phosphorylation and lipidation occur in single proteins, in some cases residing within five residues of each other [38,39]. The two modifications cooperate to provide control of function and cellular localization [37–39]. We close in having demonstrated that lipidation can stabilize helical structures to refine biological function in an energetically efficient manner and propose that lipidation may be nature's hydrophobic counterpart to phosphorylation as a means to reversibly change macromolecule structure and ultimately function.
Conflict of interest
The research reported within was partially funded by Marcadia Biotech. Richard DiMarchi serves as a scientific consultant to Roche Pharmaceuticals in areas associated with the reported subject matter.
Acknowledgments
We thank Dr. David Giedroc for useful insight to the NMR experiments and critical revision of this manuscript, Mr. Jay Levy, Dave Smiley and Dr. Bin Yang for synthesis of supplemental peptides, Mr. James Ford, Ms. Yin-Chin Chen and Dr. Brian Finan for support with cell culture and Dr. Todd Stone for instruction in CD and dynamic light scattering spectroscopy. A special thanks to Dr. Paul Pfluger, and Ms. Jazzmin Hembree, Christine Raver, Jenna Holland and Kristy Heppner for their expert contributions to in vivo studies. The NMR solution structures of Aib2,16a Lys13-γGlu-γGlu-C16 (12) were deposited to the Protein Data Bank under accession codes 2m5p (water) and 2m5q (d-TFE).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2013.08.008.
Appendix A. Supplementary materials
Supplementary Data
References
- 1.Havelund S., Plum A., Ribel U., Jonassen I., Vølund A., Markussen J. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharmaceutical Research. 2004;21:1498–1504. doi: 10.1023/b:pham.0000036926.54824.37. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen L.B., Nielsen P.F., Huusfeldt P.O., Johansen N.L., Madsen K., Pedersen F.Z. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. Journal of Medicinal Chemistry. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 3.Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A.O. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;18:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aicart-Ramos C., Valero R.A., Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochimica et Biophysica Acta. 2011;12:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Day J.W., Ottaway N., Patterson J.T., Gelfanov V., Smiley D., Gidda J. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nature Chemical Biology. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 6.Pocai A., Carrington P.E., Adams J.R., Wright M., Eiermann G., Zhu L. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day J.W., Gelfanov V., Smiley D., Carrington P.E., Eiermann G., Chicchi G. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers. 2012;98:443–450. doi: 10.1002/bip.22072. [DOI] [PubMed] [Google Scholar]
- 8.Martin B.R., Cravatt B.F. Large-scale profiling of protein palmitoylation in mammalian cells. Nature Methods. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrifield B. Concept and early development of solid-phase peptide synthesis. Methods in Enzymology. 1997;289:3–13. doi: 10.1016/s0076-6879(97)89040-4. [DOI] [PubMed] [Google Scholar]
- 10.Fields G.B., Noble R.L. Solid phase synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. International Journal of Peptide and Protein Research. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 11.Tinsley F.C., Taicher G.Z., Heiman M.L. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obesity Research. 2004;12:150–160. doi: 10.1038/oby.2004.20. [DOI] [PubMed] [Google Scholar]
- 12.Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nature Protocols. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenfield N.J. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nature Protocols. 2006;1:2527–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Holde K.E., Johnson W.C., Ho P.S. 2nd ed. Prentice Hall; Upper Saddle River, New Jersey: 1998. Principles of physical biochemistry. [Google Scholar]
- 15.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 16.Ulrich E.L., Akutsu H., Doreleijers J.F., Harano Y., Ioannidis Y.E., Lin J. BioMagResBank. Nucleic Acids Research. 2008;36:D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwieters C.D., Kuszewski J.J., Tjandra N., Clore G.M. The Xplor-NIH NMR molecular structure determination package. Journal of Magnetic Resonance. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 18.Wütrich K. Wiley; New York: 1986. NMR of proteins and nucleic acids. [Google Scholar]
- 19.Patterson J.T., Day J.W., Gelfanov V.M., DiMarchi R.D. Functional association of the N-terminal residues with the central region in glucagon-related peptides. Journal of Peptide Science. 2011;17:659–666. doi: 10.1002/psc.1385. [DOI] [PubMed] [Google Scholar]
- 20.Madsen K., Knudsen L.B., Agersoe H., Nielsen P.F., Thøgersen H., Wilken M. Structure–activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. Journal of Medicinal Chemistry. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 21.Russell-Jones D. Molecular, pharmacological and clinical aspects of liraglutide, a once-daily human GLP-1 analogue. Molecular and Cellular Endocrinology. 2009;297:137–140. doi: 10.1016/j.mce.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Kanoski S.E., Rupprecht L.E., Fortin S.M., De Jonghe B.C., Hayes M.R. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology. 2012;62:1916–1927. doi: 10.1016/j.neuropharm.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parthier C., Reedtz-Runge S., Rudolph R., Stubbs M.T. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends in Biochemical Sciences. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Unson C.G., Wu C.R., Fitzpatrick K.J., Merrifield R.B. Multiple-site replacement analogs of glucagon. A molecular basis for antagonist design. Journal of Biological Chemistry. 1994;269:12548–12551. [PubMed] [Google Scholar]
- 25.Krstenansky J.L., Trivedi D., Hruby V.J. Importance of the 10–13 region of glucagon for its receptor interactions and activation of adenylate cyclase. Biochemistry. 1986;25:3833–3839. doi: 10.1021/bi00361a014. [DOI] [PubMed] [Google Scholar]
- 26.Unson C.G., Wu C.R., Cheung C.P., Merrifield R.B. Positively charged residues at positions 12, 17, and 18 of glucagon ensure maximum biological potency. Journal of Biological Chemistry. 1998;273:10308–10312. doi: 10.1074/jbc.273.17.10308. [DOI] [PubMed] [Google Scholar]
- 27.Adelhorst K., Hedegaard B.B., Knudsen L.B., Kirk O. Structure–activity studies of glucagon-like peptide-1. Journal of Biological Chemistry. 1994;269:6275–6278. [PubMed] [Google Scholar]
- 28.Johansson J., Nilsson G., Strömberg R., Robertson B., Jörnvall H., Curstedt T. Secondary structure and biophysical activity of synthetic analogues of the pulmonary surfactant polypeptide SP-C. Biochemical Journal. 1995;307:535–541. doi: 10.1042/bj3070535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qahwash I.M., Boire A., Lanning J., Krausz T., Pytel P., Meredith S.C. Site-specific effects of peptide lipidation on beta-amyloid aggregation and cytotoxicity. Journal of Biological Chemistry. 2007;282:36987–36997. doi: 10.1074/jbc.M702146200. [DOI] [PubMed] [Google Scholar]
- 30.Pylypenko O., Schönichen A., Ludwig D., Ungermann C., Goody R.S., Rak A. Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. Journal of Molecular Biology. 2008;377:1334–1345. doi: 10.1016/j.jmb.2008.01.099. [DOI] [PubMed] [Google Scholar]
- 31.Poschner B.C., Langosch D. Stabilization of conformationally dynamic helices by covalently attached acyl chains. Protein Science. 2009;18:1801–1805. doi: 10.1002/pro.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y., Kahn R.A., Prestegard J.H. Dynamic structure of membrane-anchored Arf⁎GTP. Nature Structural and Molecular Biology. 2010;17:876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiersen H., Rees A.R. Trifluoroethanol may form a solvent matrix for assisted hydrophobic interactions between peptide side chains. Protein Engineering. 2000;13:739–743. doi: 10.1093/protein/13.11.739. [DOI] [PubMed] [Google Scholar]
- 34.Braun W., Wider G., Lee K.H., Wüthrich K. Conformation of glucagon in a lipid–water interphase by 1H nuclear magnetic resonance. Journal of Molecular Biology. 1983;69:921–948. doi: 10.1016/s0022-2836(83)80143-0. [DOI] [PubMed] [Google Scholar]
- 35.Thornton K., Gorenstein D.G. Structure of glucagon-like peptide (7−36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 36.Alaña I., Malthouse J.P., O'Harte F.P., Hewage C.M. The bioactive conformation of glucose-dependent insulinotropic polypeptide by NMR and CD spectroscopy. Proteins. 2007;68:92–99. doi: 10.1002/prot.21372. [DOI] [PubMed] [Google Scholar]
- 37.Moffett S., Adam L., Bonin H., Loisel T.P., Bouvier M., Mouillac B. Palmitoylated cysteine 341 modulates phosphorylation of the beta2-adrenergic receptor by the cAMP-dependent protein kinase. Journal of Biological Chemistry. 1996;271:21490–21497. doi: 10.1074/jbc.271.35.21490. [DOI] [PubMed] [Google Scholar]
- 38.Charych E.I., Jiang L.X., Lo F., Sullivan K., Brandon N.J. Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: implications for the treatment of schizophrenia. Journal of Neuroscience. 2010;30:9027–9037. doi: 10.1523/JNEUROSCI.1635-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin D.T., Makino Y., Sharma K., Hayashi T., Neve R., Takamiya K. Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nature Neuroscience. 2009;12:879–887. doi: 10.1038/nn.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szilák L., Moitra J., Krylov D., Vinson C. Phosphorylation destabilizes alpha-helices. Nature Structural Biology. 1997;4:112–114. doi: 10.1038/nsb0297-112. [DOI] [PubMed] [Google Scholar]
- 41.Andrew C.D., Warwicker J., Jones G.R., Doig A.J. Effect of phosphorylation on alpha-helix stability as a function of position. Biochemistry. 2002;41:1897–1905. doi: 10.1021/bi0113216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data