

Glucocorticoids lack the capacity to further augment neutrophil survival, in severe hypoxia.
Keywords: inflammation, apoptosis, steroids, hypoxia
Abstract
GCs are highly effective in treating a wide range of inflammatory diseases but are limited in their ability to control neutrophilic lung inflammation in conditions such as COPD. Neutrophil apoptosis, a central feature of inflammation resolution, is delayed in response to microenvironmental cues, such as hypoxia and inflammatory cytokines, present at inflamed sites. GCs delay neutrophil apoptosis in vitro, and this may therefore limit the ability of GCs to control neutrophilic inflammation. This study assesses the effect GCs have on hypoxia- and inflammatory cytokine-induced neutrophil survival. Human neutrophils were treated with GCs in the presence or absence of GM-CSF or inflammatory macrophage-CM at a range of oxygen concentrations (21–1% oxygen). Neutrophil apoptosis and survival were assessed by flow cytometry and morphological analysis and neutrophil function, by stimulus-induced shape change and respiratory burst. Dexamethasone promoted neutrophil survival at 21%, 10%, and 5% oxygen but not at 1% oxygen. Interestingly, GM-CSF and inflammatory CM increased neutrophil survival significantly, even at 1% oxygen, with cells remaining functionally active at 96 h. Dexamethasone was able to reduce the prosurvival effect of GM-CSF and inflammatory CM in a hypoxic environment. In conclusion, we found that GCs do not augment neutrophil survival in the presence of severe hypoxia or proinflammatory mediators. This suggests that GCs would not promote neutrophil survival at sites of inflammation under these conditions.
Introduction
GCs are used extensively and are highly effective in a wide range of inflammatory conditions, including polymyalgia rheumatica, ulcerative colitis, and asthma. However, neutrophilic inflammation, such as that seen in COPD and emphysema, is relatively GC-resistant, although the reasons for this remain unclear.
As key effector cells of the innate inflammatory response, neutrophils migrate rapidly to the site of tissue injury to contain and clear the initiating noxious stimulus. Timely resolution of this proinflammatory response is crucial for the restoration of tissue homeostasis, with neutrophil apoptosis and subsequent nonphlogistic removal by surrounding phagocytes central to this resolution process. In the circulation, neutrophils appear to have a relatively short lifespan of only a few hours [1, 2]. However, at an inflammatory site, neutrophil survival increases to several days, as a result of delayed apoptosis induced by environmental factors, such as hypoxia, pyrexia, and low pH, as well as proinflammatory mediators, including bacterial products, such as LPS and cytokines, including GM-CSF and TNF-α [3–6]. These serve to increase the expression of several survival proteins, such as the Bcl-2 family member Mcl-1, resulting in reduced downstream caspase activation [2, 7–12]. This increase in longevity provides neutrophils with an appropriate time-frame in which to perform their inflammatory functions (including phagocytosis, degranulation, and ROS production) and for the recruitment of macrophages for subsequent phagocytosis of effete and apoptotic neutrophils [13]. However, an inappropriate delay in neutrophil apoptosis can propagate the proinflammatory response with increased release of histotoxic mediators, resulting in local tissue damage [13, 14].
GCs modulate the inflammatory response through a variety of mechanisms, including alteration of cytokine production, increased macrophage phagocytic capacity, transactivation of anti-inflammatory genes, and transrepression of key proinflammatory transcription factors [15]. GCs are, however, powerful inducers of neutrophil survival in standard laboratory conditions in vitro [16] through a poorly described mechanism involving PI3K signaling and subsequent up-regulation of Mcl-1 [12]. Along with increasing recruitment from bone marrow and marginated neutrophil pools, this delay in apoptosis has been suggested as an important factor in GC-induced peripheral blood neutrophilia and their relative ineffectiveness in controlling lung neutrophilic inflammation [17–19].
Physiological oxygen levels vary between 5–11% in the circulation and 3–5% in normal tissue [20]. However, in inflamed tissues, oxygen levels fall below 2%, as a result of a disrupted blood supply and increased cellular metabolic activity and with regards to the normal tissue oxygenation cited above, are regarded as hypoxic [20–22]. The change in oxygen tension that occurs as neutrophils migrate into an area of inflammation is a major factor in their survival [6]. Primarily, this is through HIF-1α-NF-κB-dependent mechanisms involving prolyl hydroxylase 3, resulting in a variety of effects, including the induction of Mcl-1 expression [8, 9, 23].
Within an inflammatory and hypoxic environment, there is little evidence regarding the effect of GCs on neutrophil survival, particularly, as hypoxia may alter GC function in other cells, including macrophages and lung epithelial cells [24]. Our study therefore aimed to assess the impact of GCs on neutrophil survival in a variety of oxygen tensions, along with concomitant exposure to inflammatory mediators. We investigated whether GCs could augment neutrophil survival during hypoxia in the presence or absence of other proinflammatory factors.
MATERIALS AND METHODS
Isolation of primary human neutrophils and monocytes
Neutrophils and PBMCs were isolated from the peripheral blood of healthy human volunteers using a discontinuous Percoll gradient as described [3, 25, 26]. Monocytes were subsequently purified from PBMCs by negative selection with magnetic beads (Miltenyi Biotec, Surrey, UK), according to the manufacturer's instructions. Neutrophil and monocyte purity was >96%, as assessed by analysis of cytocentrifuge preparations and flow cytometry. Ethical approval was obtained from the Lothian Research Ethics Committee (08/S1103/38).
Monocyte differentiation and generation of CM
Purified monocytes were differentiated into MDMs by culture in 96-well plates at 1.5 × 105 cells/well in 200 μl phenol red-free IMDM (Invitrogen, Paisley, UK) with 10% autologous serum [27]. The monocytes were cultured for 5 days with the media changed on Day 3. CM was generated by stimulating MDMs with 1 ng/ml LPS (Sigma-Aldrich, Dorset, UK) for 20 h. The resulting supernatant was removed and frozen at −20°C until use.
Neutrophil treatments
Purified neutrophils were cultured in 96-well plates at 7.5 × 105 cells/well in 150 μl phenol red-free IMDM (Invitrogen) with 10% autologus serum at various oxygen concentrations (1%, 5%, 10%, and 21%) using a BioSpherix OxyCycler C42 dual chamber (Wolf Laboratories, York, UK). Other standard culture parameters (5% CO2, 37°C with humidity) were maintained at all oxygen levels. Neutrophils were treated with or without GM-CSF (R&D Systems, Abingdon, UK), dexamethasone (Sigma-Aldrich), LY294002 (Merck Serono, Middlesex, UK), Akt inhibitor IV (Merck Serono), and CM at the concentrations and time-scales detailed in the individual figure legends.
Assessment of neutrophil survival, apoptosis, and necrosis
Neutrophil viability, apoptosis, and necrosis were measured by flow cytometry with Alexa Fluor 647-labeled AnnV (Cambridge Biosciences, Cambridge, UK) and PI (Sigma-Aldrich) or by hypodiploid DNA peak assay, as described [28], with results confirmed by morphological assessment of Diff-Quick-stained cytocentrifuge preparations (IBG Immucor, West Sussex, UK). Viable cells were defined as AnnV/PI-negative, apoptotic cells AnnV-positive/PI-negative, and necrotic cells PI-positive. Flow cytometry was performed on a FACSCalibur (BD Biosciences, Oxford, UK) with data analyzed using on FlowJo (TreeStar, Ashland, OR, USA).
Protein extraction and immunoblotting
Cell lysis was performed using a whole-cell lysis buffer, as described previously [5]. Protein concentrations were then determined by BCA assay using the manufacturer's instructions (Pierce, Thermo Fisher Scientific, Rockford, IL, USA). Immunoblotting was performed using equal amounts of protein denatured by SDS loading buffer (Invitrogen) and loaded onto 4–12% Bis-Tris gels (Invitrogen), which were transferred to Protran nitrocellulose (Whatman, GE Life Sciences, UK) and detection performed using ECL Prime (GE Life Sciences). Antibodies included anti-NF-κB, anti-Mcl-1, and anti-GR (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-actin (Sigma-Aldrich).
Assessment of neutrophil activation
Neutrophils cultured in 96-well plates at 7.5 × 105/well with 20 ng/ml GM-CSF for 96 h at 1% oxygen were stained with AnnV and PI. Viable cells (AnnV−ve/PI−ve) were purified by cell sorting (FACSAria II cell sorter; BD Biosciences). More than 96% of sorted neutrophils were viable, as determined by AnnV/PI staining, hypodiploid DNA peak, and morphological analysis of cytocentrifuge preparations. Neutrophil activation was assessed by fMLF-induced respiratory burst and shape change. Superoxide anion generation was estimated as an increase in reactive oxygen intermediates, indicated by oxidation of the nonfluorescent DHR (Invitrogen) to the fluorescent rhodamine123. Briefly, neutrophils (2×106/ml) incubated with DHR (1μM) for 5 min at 37°C. Cells were then stimulated with fMLF (100 nM) for 15 min and placed on ice. The change in fluorescence and forward-scatter was measured by flow cytometry to determine superoxide anion generation and shape change, respectively.
Statistical analysis
Data are presented as mean ± sem. Statistical analysis was performed using ANOVA with a Tukey's post hoc test, and P < 0.05 was considered significant. Data analyses were performed using GraphPad Prism software (La Jolla, CA, USA).
RESULTS
GCs promote neutrophil survival at 21% oxygen
A number of GCs used in clinical practice, including the archetypal GC dexamethasone (Fig. 1A and B) as well as budesonide (Fig. 1C), beclamethasone (Fig. 1D), and 6α-methylprednisolone (Fig. 1E) all promote neutrophil survival in a concentration-dependent manner by delaying neutrophil apoptosis under standard culture conditions (21% oxygen). The promotion of neutrophil survival (AnnV and PI-negative cells) by dexamethasone is time-dependent, as it diminishes over time from ∼60% survival at 24 h to ∼30% survival at 48 h, although this is still significantly greater than untreated cells at 48 h (Fig. 1F). By 72 h, however, the survival effect of dexamethasone is lost, with <10% survival in dexamethasone-treated and in untreated control cells (Fig. 1F). During the first 24 h, cell death occurs predominantly through apoptosis (Fig. 1G) with subsequent secondary necrosis evident at later time-points (Fig. 1H).
Figure 1. GCs increase neutrophil survival by delaying apoptosis under standard culture conditions (21% oxygen).

(A) Representative cytocentrifuge images showing neutrophil morphology and AnnV/PI binding of neutrophils that have been freshly isolated or cultured for 24 h, with or without 100 nM dexamethasone (Dex). (B–E) Concentration responses (1 nM–10 μM) of (B) dexamethasone, (C) budesonide, (D) beclomethasone, and (E) 6α-methylprednisolone on neutrophil apoptosis. (F–H) Percentage of neutrophil (F) survival, (G) apoptosis, and (H) necrosis over a time-course of 72 h in standard culture conditions (21% oxygen), as assessed by flow cytometry of AnnV/PI binding. Open bars represent cells cultured without dexamethasone; closed bars represent cells cultured with 100 nM dexamethasone. Data are shown as mean ± sem; n = 6–9; *P < 0.05; ***P < 0.001.
GC-mediated neutrophil survival is P13K-dependent and is lost under hypoxia
In keeping with previous data [7], we demonstrate that increasing levels of hypoxia promotes neutrophil survival at 24 h in a concentration-dependent manner (Fig. 2A). Furthermore, hypoxia-mediated neutrophil survival (at 1% oxygen) is independent of the prosurvival PI3K/Akt signaling pathway, whereas GC-mediated survival (at 21% oxygen) is PI3K/Akt-dependent (Fig. 2B and C). Dexamethasone promotes neutrophil survival at 24 h at 21%, 10%, and 5% oxygen (Fig. 2A). At 1% oxygen, however, dexamethasone causes no further increase in neutrophil survival (Fig. 2A).
Figure 2. Neutrophil survival induced by dexamethasone and low oxygen levels is regulated by NF-κB and Mcl-1.

(A) Percentage of neutrophil survival at 24 h while cultured at various oxygen levels, with (closed bars) and without (open bars) 100 nM dexamethasone. (B) The impact of a selective PI3K inhibitor (10 μM LY294002) and a selective Akt inhibitor (10 μM Akt inhibitor V) on neutrophil survival after 24 h of culture under standard culture conditions (21% oxygen) with 100 nM dexamethasone (closed bars) or at 1% oxygen (open bars). (C) Representative immunoblots of the expression of NF-κB (p65; 65kDa) and Mcl-1 (40 kDa) induced by dexamethasone (Dex) and hypoxia. (D) Representative immunoblot of the expression of GR (GRα; 94 kDa) during hypoxia. β-Actin (42 kDa) expression shows equal protein loading. Data represent n = 3–8 and are shown as mean ± sem; *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05; ###P < 0.001 (compared with 21% oxygen); §P < 0.05 (compared with 21% oxygen). PI3Ki, PI3K inhibitor; Akti, Akt inhibitor; Cont, Control.
Both dexamethasone and hypoxia induce the expression of NF-κB, as well as the prosurvival factor Mcl-1 (Fig. 2C). Hypoxia also increased the expression of the GR (Fig. 2D). Indeed, there was no change in the expression of NF-κB or Mcl-1 with dexamethasone treatment under hypoxia (Fig. 2C). Consistent with this, dexamethasone does not alter the rate of neutrophil apoptosis over 96 h at 1% oxygen (Fig. 3A).
Figure 3. Neutrophil survival mediated by low oxygen levels, GCs, and GM-CSF.
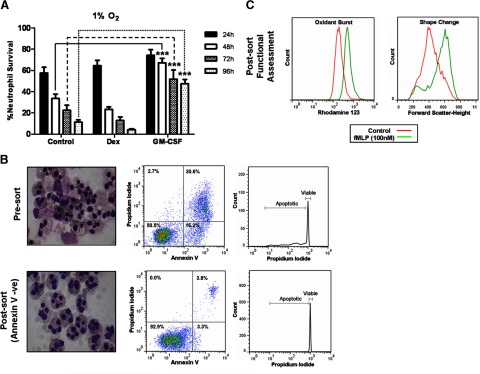
(A) Percentage of neutrophil survival when cultured at 1% oxygen at 24 h (closed bars), 48 h (open bars), 72 h (cross-hatched bars), and 96 h (dotted bars) and cultured with or without 100 nM dexamethasone or 20 ng/ml GM-CSF. (B) Representative cytocentrifuge images, flow cytometry plots of AnnV/PI binding, and DNA integrity of 96-h GM-CSF-treated neutrophils, pre- and postcell sorting for viable cells (AnnV/PI-negative). (C) Representative histograms of aged, GM-CSF-treated, viable neutrophil response to fMLF (100 nM) with oxidative burst measured by the oxidation of DHR to rhodamine 123 and shape change assessed by change in forward-scatter. Data represent n = 6 and are shown as mean ± sem. ***P < 0.001.
GM-CSF-mediated neutrophil survival is reduced by GCs only in hypoxic conditions
GM-CSF is a powerful neutrophil prosurvival factor. In contrast to dexamethasone, GM-CSF increases neutrophil survival significantly throughout 96 h incubation at 1% oxygen (Fig. 3A). By 96 h, there is ∼50% neutrophil survival compared with <10% in control or dexamethasone-treated cells (Fig. 3A). Importantly, these cells remain functionally active, with fMLF inducing shape change and oxidative burst (Fig. 3C). Dexamethasone has little effect on the prosurvival effects of GM-CSF at 21% or 5% oxygen (Fig. 4A and B), although it transiently increases survival at 21% oxygen (Fig. 4A). Interestingly, dexamethasone reduces significantly the GM-CSF-mediated increase in neutrophil survival from 48 h onward when cultured at 1% oxygen (Fig. 4C and D).
Figure 4. GCs attenuate GM-CSF-mediated neutrophil survival in hypoxic conditions.
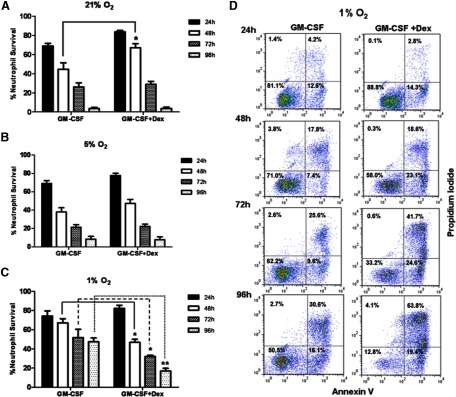
(A–C) The percentage of neutrophil survival when cultured with GM-CSF (20 ng/ml), with or without dexamethasone (100 nM) at (A) 21% oxygen, (B) 5% oxygen, or (C) 1% oxygen over a 96-h time-period; 24 h (closed bars), 48 h (open bars), 72 h (cross-hatched bars), and 96 h (dotted bars). Neutrophil survival in untreated cells under 1% oxygen levels over the 24-, 48-, 72-, and 96-h time-points are 69.4%, 31.1%, 17.4%, and 8.8%, respectively (data not shown). (D) Representative flow cytometry plots of AnnV/PI binding at 24 h, 48 h, 72 h, and 96 h in culture with GM-CSF (20 ng/ml), with or without dexamethasone (100 nM) at 1% oxygen. Data represent n = 6 and are shown as mean ± sem. *P < 0.05; **P < 0.01.
Hypoxia-mediated neutrophil survival is propagated by inflammatory CM that GCs can impair
Inflammatory CM generated from LPS-treated MDMs increases neutrophil survival significantly, irrespective of oxygen concentration (Fig. 5A and B). Under standard culture conditions with 21% oxygen, dexamethasone does not alter CM-induced survival at 24 h or 48 h but causes a small, yet significant reduction in neutrophil survival at 72 h (Fig. 5A). Hypoxia and CM have an additive effect on the neutrophil lifespan inducing a significant and prolonged increase in survival (Fig. 5B). Dexamethasone has no effect on CM-mediated survival at 1% oxygen prior to 96 h but induces a significant reduction in neutrophil survival at this time-point (Fig. 5A). Importantly, whereas high concentrations of LPS (∼100 ng/ml) promote neutrophil survival [29], the 1-ng/ml used to stimulate the MDMs had no direct effect on neutrophil survival (Fig. 5C).
Figure 5. GCs attenuate CM-induced neutrophil survival.

(A) The percentage of neutrophil survival when cultured in inflammatory CM at 21% oxygen levels, with or without dexamethasone (100 nM). (B) The percentage of neutrophil survival cultured in inflammatory CM at 1% oxygen levels, with or without dexamethasone (100 nM); 24 h (closed bars), 48 h (open bars), 72 h (cross-hatched bars), and 96 h (dotted bars). (C) The direct impact of the 1-ng/ml LPS on neutrophil survival in standard media at 21% oxygen levels after 24 h. Data represent n = 6 and are shown as mean ± sem. *P < 0.05; ***P < 0.001. Control, Noninflammatory CM.
DISCUSSION
Here, we show that proinflammatory mediators, but not GCs, promote neutrophil survival in hypoxic conditions that would likely be found in inflamed tissues. Conversely, although GCs inhibit neutrophil survival at 21% oxygen, they are unable to promote neutrophil apoptosis in inflammatory environments in vitro.
The timing and manner of neutrophil cell death are fundamental for the controlled resolution of an innate inflammatory response [14]. In standard in vitro conditions (21% oxygen), GCs increase neutrophil survival by inhibition of apoptosis. In humans, GCs cause peripheral blood neutrophilia, and their ability to promote neutrophil survival has been suggested as a reason for the relatively limited efficacy of GCs in lung neutrophilic inflammation seen in diseases such as COPD [16, 18, 19]. This is in contrast to eosinophil-predominate diseases (such as asthma), where GCs are a highly effective therapy, in part, through their ability to induce rapid eosinophil apoptosis [30]. Therefore, the understanding of why GCs delay neutrophil apoptosis may allow the development of more effective anti-inflammatory drugs for GCunresponsive neutrophilic inflammation.
Here, we have shown that dexamethasone is able to mediate an additional neutrophil prosurvival effect at 10% and 5% oxygen levels at 24 h, suggesting that GCs may be able to promote neutrophil survival at the higher oxygen levels found in the circulation, thus potentially contributing to GC-induced neutrophilia [1]. Although we have also confirmed that reduction in oxygen tension induces neutrophil survival in a concentration-dependent fashion, the ability of GCs to induce an additional neutrophil prosurvival effect is lost at 1% oxygen. This is despite hypoxia and GCs eliciting neutrophil survival through distinct signaling pathways, as illustrated by the involvement of PI3K/Akt signaling in the GC but not the hypoxic-mediated survival effect [7, 9]. However, GC and hypoxia increased the expression of NF-κB, the major mediator of hypoxia-HIF-1α in neutrophils [9], and consistent with the literature, the expression of the NF-κB regulated prosurvival factor Mcl-1 [8, 31]. Therefore, their respective signaling pathways may eventually converge on a common mechanism that could explain the lack of an additive effect. Hypoxia itself increased the expression of the GR, but this increase in expression did not translate into an increase in GC-mediated survival. This observation suggests that although GCs may promote neutrophil survival in the circulation, they are not likely to augment the hypoxia-mediated delay in apoptosis at an inflamed site where severe hypoxia may occur. An assessment of neutrophil apoptosis after dexamethasone treatment at the site of inflammation in vivo would be required to confirm this observation.
The observed GC-mediated neutrophil survival at 21% oxygen is, however, time-dependent, being maximal at 24 h, diminishing by ∼50% at 48 h, and lost by 72 h. A similar time-dependent pattern is also observed from the initial induction of survival, mediated by hypoxia alone or with GM-CSF alone. This suggests that a single survival signal alone is adequate to induce the prolonged neutrophil survival that is seen at an inflammatory site. Both GM-CSF and the CM containing the inflammatory milieu from LPS-stimulated MDMs promote neutrophil survival. Similar to GCs and hypoxia, the neutrophil survival mediated by GM-CSF or CM alone was not sustained and diminished over time. However, unlike GCs, the increase in neutrophil survival mediated by GM-CSF and CM was increased at 1% oxygen, resulting in a significant reduction in the rate of neutrophil death over time. This prolongation of neutrophil survival with combinations of prosurvival factors is likely to represent multiple points of control. This ensures that no one signal can affect neutrophil survival inappropriately, thus permitting neutrophils to carry out their inflammatory functions on an appropriate time-scale.
Interestingly, although dexamethasone had little effect on neutrophil survival mediated by GM-CSF at 21% oxygen, under hypoxic conditions (1% oxygen), dexamethasone reduced GM-CSF-mediated survival significantly from 48 h onward. Similarly, dexamethasone was able to attenuate the neutrophil survival induced by CM under hypoxia (1% oxygen) at later time-points (72 h). Therefore, although GCs exert a prosurvival effect on resting neutrophils at 21% oxygen, this effect is ameliorated (in the case of hypoxia alone) or reversed by the presence of an inflammatory environment. The finding that GCs can be prosurvival or proapoptotic, depending on the time-scale and microenvironment, is similar to findings with TNF-α that can promote neutrophil survival or apoptosis, depending on the time-scale of exposure and the state of NF-κB activation [5, 32]. Therefore, GCs may only exert a proapoptotic effect, with combinations of hypoxia and inflammatory mediators seen at sites of neutrophilic inflammation. Again, an assessment of neutrophil apoptosis after dexamethasone treatment at sites of inflammation in vivo would be required to confirm this observation.
In summary, our data show that the promotion of neutrophil survival by GCs is lost in the severe hypoxia found within inflamed tissues. Inflammatory mediators but not GCs prolong survival during hypoxia. In fact, GCs inhibit proinflammatory, mediator-induced neutrophil survival at low oxygen levels. These data suggest that the relative ineffectiveness of GC to control lung neutrophilic inflammation is not a result of a GC-dependent promotion of neutrophil survival at sites of inflammation.
ACKNOWLEDGMENTS
This study was funded by a project grant from Medical Research Scotland (318FRG) awarded to J.A.M. The authors acknowledge funding from the Medical Research Council (G0601481 and MR/K013386/1) and Asthma UK (01/042). D.A.D. and C.D.L. are funded by the Wellcome Trust (WT096497 and WT094415).
Footnotes
- AnnV
- Annexin V
- CM
- conditioned media
- COPD
- chronic obstructive pulmonary disease
- DHR
- dihydrorhodamine 123
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- HIF-1α
- hypoxia-inducible factor 1α
- Mcl-1
- myeloid cell leukemia sequence 1
AUTHORSHIP
J.A.M. and A.G.R. conceived of the study. J.A.M., K.O.J., T.A.S., S.F., C.W., and J.M. conducted the experiments. J.A.M., D.A.D., C.D.L., and A.G.R. provided intellectual input and data analyses and wrote the manuscript. R.D., M.B., N.H., I.D., C.H., and A.G.R. provided guidance and critical review of methods.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Athens J. W., Haab O. P., Raab S. O., Mauer A. M., Ashenbrucker H., Cartwright G. E., Wintrobe M. M. (1961) Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J. Clin. Invest. 40, 989–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffin R., Leitch A. E., Fox S., Haslett C., Rossi A. G. (2010) Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol. Rev. 236, 28–40 [DOI] [PubMed] [Google Scholar]
- 3. Haslett C., Guthrie L. A., Kopaniak M. M., Johnston R. B., Jr., Henson P. M. (1985) Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am. J. Pathol. 119, 101–110 [PMC free article] [PubMed] [Google Scholar]
- 4. Haslett C., Lee A., Savill J.S., Meagher L., Whyte M. K. (1991) Apoptosis (programmed cell death) and functional changes in aging neutrophils. Modulation by inflammatory mediators. Chest 99, 6S. [PubMed] [Google Scholar]
- 5. Ward C., Chilvers E. R., Lawson M. F., Pryde J. G., Fujihara S., Farrow S. N., Haslett C., Rossi A. G. (1999) NF-κB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 274, 4309–4318 [DOI] [PubMed] [Google Scholar]
- 6. Cross A., Barnes T., Bucknall R. C., Edwards S. W., Moots R. J. (2006) Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J. Leukoc. Biol. 80, 521–528 [DOI] [PubMed] [Google Scholar]
- 7. Hannah S., Mecklenburgh K., Rahman I., Bellingan G. J., Greening A., Haslett C., Chilvers E. R. (1995) Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 372, 233–237 [DOI] [PubMed] [Google Scholar]
- 8. Leuenroth S. J., Grutkoski P. S., Ayala A., Simms H. H. (2000) Suppression of PMN apoptosis by hypoxia is dependent on Mcl-1 and MAPK activity. Surgery 128, 171–177 [DOI] [PubMed] [Google Scholar]
- 9. Walmsley S. R., Print C., Farahi N., Peyssonnaux C., Johnson R. S., Cramer T., Sobolewski A., Condliffe A. M., Cowburn A. S., Johnson N., Chilvers E. R. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J. Exp. Med. 201, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein J. B., Rane M. J., Scherzer J. A., Coxon P. Y., Kettritz R., Mathiesen J. M., Buridi A., McLeish K. R. (2000) Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 164, 4286–4291 [DOI] [PubMed] [Google Scholar]
- 11. Moulding D. A., Akgul C., Derouet M., White M. R., Edwards S. W. (2001) BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol. 70, 783–792 [PubMed] [Google Scholar]
- 12. Wardle D. J., Burgon J., Sabroe I., Bingle C. D., Whyte M. K., Renshaw S. A. (2011) Effective caspase inhibition blocks neutrophil apoptosis and reveals Mcl-1 as both a regulator and a target of neutrophil caspase activation. PLoS One 6, e15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Savill J. S., Wyllie A. H., Henson J. E., Walport M. J., Henson P. M., Haslett C. (1989) Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83, 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Serhan C. N., Savill J. (2005) Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Cousin J. M., Hughes J., Van Damme J., Seckl J. R., Haslett C., Dransfield I., Savill J., Rossi A. G. (1999) Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J. Immunol. 162, 3639–3646 [PubMed] [Google Scholar]
- 16. Meagher L. C., Cousin J. M., Seckl J. R., Haslett C. (1996) Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 156, 4422–4428 [PubMed] [Google Scholar]
- 17. Ito K., Herbert C., Siegle J. S., Vuppusetty C., Hansbro N., Thomas P. S., Foster P. S., Barnes P. J., Kumar R. K. (2008) Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 39, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barnes P. J. (2007) New molecular targets for the treatment of neutrophilic diseases. J. Allergy Clin. Immunol. 119, 1055–1062 [DOI] [PubMed] [Google Scholar]
- 19. Barnes P. J., Adcock I. M. (2009) Glucocorticoid resistance in inflammatory diseases. Lancet 373, 1905–1917 [DOI] [PubMed] [Google Scholar]
- 20. Sitkovsky M., Lukashev D. (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1 α and adenosine receptors. Nat. Rev. Immunol. 5, 712–721 [DOI] [PubMed] [Google Scholar]
- 21. Caldwell C. C., Kojima H., Lukashev D., Armstrong J., Farber M., Apasov S. G., Sitkovsky M. V. (2001) Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J. Immunol. 167, 6140–6149 [DOI] [PubMed] [Google Scholar]
- 22. Eltzschig H. K., Carmeliet P. (2011) Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walmsley S. R., Chilvers E. R., Thompson A. A., Vaughan K., Marriott H. M., Parker L. C., Shaw G., Parmar S., Schneider M., Sabroe I., Dockrell D. H., Milo M., Taylor C. T., Johnson R. S., Pugh C. W., Ratcliffe P. J., Maxwell P. H., Carmeliet P., Whyte M. K. (2011) Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Invest. 121, 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charron C. E., Chou P. C., Coutts D. J., Kumar V., To M., Akashi K., Pinhu L., Griffiths M., Adcock I. M., Barnes P. J., Ito K. (2009) Hypoxia-inducible factor 1α induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J. Biol. Chem. 284, 36047–36054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossi A. G., Sawatzky D. A., Walker A., Ward C., Sheldrake T. A., Riley N. A., Caldicott A., Martinez-Losa M., Walker T. R., Duffin R., Gray M., Crescenzi E., Martin M. C., Brady H. J., Savill J. S., Dransfield I., Haslett C. (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 12, 1056–1064 [DOI] [PubMed] [Google Scholar]
- 26. Dorward D. A., Lucas C. D., Alessandri A. L., Marwick J. A., Dransfield I., Haslett C., Dhaliwal K., Rossi A. G. (2009) Technical Advance: Autofluorescence-based sorting: rapid and nonperturbing isolation of ultrapure neutrophils to determine cytokine production. J. Leukoc. Biol. 94, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michlewska S., Dransfield I., Megson I. L., Rossi A. G. (2009) Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-α. FASEB J. 23, 844–854 [DOI] [PubMed] [Google Scholar]
- 28. Taylor E. L., Megson I. L., Haslett C., Rossi A. G. (2001) Dissociation of DNA fragmentation from other hallmarks of apoptosis in nitric oxide-treated neutrophils: differences between individual nitric oxide donor drugs. Biochem. Biophys. Res. Commun. 289, 1229–1236 [DOI] [PubMed] [Google Scholar]
- 29. Sabroe I., Prince L. R., Jones E. C., Horsburgh M. J., Foster S. J., Vogel S. N., Dower S. K., Whyte M. K. (2003) Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J. Immunol. 170, 5268–5275 [DOI] [PubMed] [Google Scholar]
- 30. Woolley K. L., Gibson P. G., Carty K., Wilson A. J., Twaddell S. H., Woolley M. J. (1996) Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 154, 237–243 [DOI] [PubMed] [Google Scholar]
- 31. Saffar A.S., Dragon S., Ezzati P., Shan L., Gounni A. S. (2008) Phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase regulate induction of Mcl-1 and survival in glucocorticoid-treated human neutrophils. J. Allergy Clin. Immunol. 121, 492.e10–498.e10 [DOI] [PubMed] [Google Scholar]
- 32. Murray J., Barbara J. A., Dunkley S. A., Lopez A. F., Van Ostade X., Condliffe A. M., Dransfield I., Haslett C., Chilvers E. R. (1997) Regulation of neutrophil apoptosis by tumor necrosis factor-α: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood 90, 2772–2783 [PubMed] [Google Scholar]