

Abstract
We report the simultaneous mapping of multiple histone tail modifications on chromatin that has been confined to nanofluidic channels. In these channels, chromatin is elongated, and histone modification can be detected using fluorescently tagged monoclonal antibodies. Using reconstituted chromatin with three distinct histone sources and two histone tail modification probes (H3K4me3 and H3K9ac), we were able to distinguish chromatin from the different sources. Determined ratios of the two modifications were consistent with the bulk composition of histone mixtures. We determined that the major difficulty in transitioning the mapping method to site-specific profiling within single genomic molecules is the interference of naturally aggregating, off-the shelf antibodies with the internal structure of chromatin.
INTRODUCTION
As the availability of personal genomes is becoming widespread and genetic information is generally accessible, the question of how the raw information is utilized within each cell becomes central. Epigenetic factors are an important contributor to this process. The epigenetic individuality of cells within a heterogeneous tissue is particularly little understood, as it requires single-cell analysis.
In the nuclei of eukaryotic cells, genomic DNA is packaged as chromatin. The fundamental repeating unit of chromatin is the nucleosome, which is the complex formed when double-stranded DNA wraps around a histone octamer made of a central H3/H4 tetramer, sandwiched between two H2A/H2B dimers, around which 147 base pairs of DNA are wrapped.1 Each of these 4 types of core histone (H2A, H2B, H3, H4) has a flexible amino acid tail of 25–40 residues, known as a histone tail.2 Through the interaction of these histone tails and auxiliary proteins, a large variety of higher-order structures can be formed, which influence the transcriptional activity of chromatin.3 In its simplest form, the DNA–protein complex resembles a "beads on a string" fiber, where nucleosomes are located every 50 bp on a fiber that is approximately 10 nm in width.4, 5 Common histone tail covalent modifications, such as acetylation, mono-,di-, and tri-methylation, phosphorylation, and ubiquitination function to modulate the higher-order structure of chromatin. The correlation of modification profiles to transcriptional activity is an active field of research. The large number of combinations of histone modifications has led to a definition of the ‘histone code’ that extends the information content of the genome past the genetic (DNA) code.6 Some modifications can be inherited between cell generations, and are thus epigenetic marks. Because of the profound influence of histone modifications on cellular processes, we believe that single-cell analysis of the chromatin marks may give important insights for human health.
The dominant technique used in the analysis of histone modifications on chromatin is chromatin immunoprecipitation (ChIP),7 which can identify the DNA fragments that are bound to nucleosomes with a specific modification by sequencing or sequence-sensitive polymerase chain reaction (PCR). Within a cell population with epigenetic variability it would, in general, not be possible to retrieve information about correlations of the variability at distant genetic loci because data from multiple cells would become mixed.
Soloway and co-workers have overcome this problem with a nanofluidic platform in which specific nucleosomes from a single cell can be enriched, with the potential of resequencing the DNA from these selected nucleosomes.8, 9 However, for common modifications a large volume of data may be accumulated and needs to be analyzed if single cells are the target of a study.
An alternative pathway has been pursued in which DNA is not fragmented, and the genetic location of modification is inferred from a mapping of the spatial to genetic coordinates. In particular, Fluorescence in situ Hybridization (and its high-resolution variant fiber-FisH10, 11) can be extended to immunostaining for histone modification marks and chromatin-associated proteins to obtain large scale maps.12, 13 The resolution of such techniques is in general limited by the ability to stretch molecules gently and homogeneously. Any inhomogeneities in the molecule that are present at the time of tethering the molecule to the mapping surface are fixed, and cannot be removed through continued measurement.
Here, we utilize nanofluidic stretching to perform a measurement that is conceptually similar to fiber-FisH, but without the step of immobilizing DNA on a surface. Nanofluidic confinement stretches DNA in a equilibrium process through introduction into a nanochannel that has an approximately square cross-section about 100 nm wide and a length of hundreds of microns.14 Because the stretching is achieved for a free molecule in equilibrium without any hydrodynamic or mechanical stresses, the molecule can fluctuate around its equilibrium point and assume a large variety of conformations, which can be meaningfully averaged.15, 16, 17 We have previously shown that chromatin can be stretched in a very similar fashion to DNA,18 and that fluorescent beacons can be used to map epigenetic marks on nanochannel-stretched DNA,19 in particular 5-methyl cytosine modifications.20 Reference 20 established that artificially imprinted methylation patterns could be faithfully recovered using labeled methyl-binding domain (MBD) protein fragments. Matsuoka and coworkers showed that histone-specific antibodies could be used to visualize chromatin in conceptually similar devices made from flexible polydimethylsiloxane (PDMS).21
In this publication, we show the simultaneous use of multiple antibodies to interrogate and identify heterogeneous reconstituted chromatin from multiple sources. Specifically, we seek to distinguish between the histone modifications in chromatin reconstituted from calf thymus, HeLa core, and chicken erythrocyte histones, via fluorescently labeled anti-trimethyl-H3K4 and anti-acetyl-H3K9 monoclonal antibodies (Fig. 1). The ability to distinctly identify each individual histone groups opens the door for building of epigenetic marker maps on a single molecule level. We could test such mapping of profiles within single molecules because of the lacking ability to reconstitute chromatin with deterministic spatial modification profiles.
Figure 1.
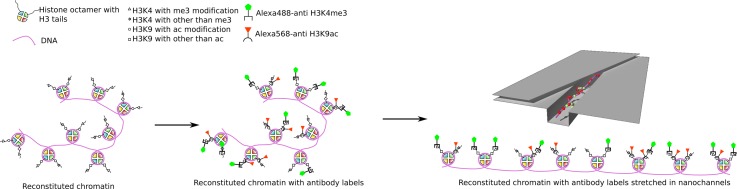
Schematic of experiment. Chromatin is reconstituted with histone mixtures from different sources, and will in general display a heterogeneous modification profile. It is double-labeled using antibodies for two distinct histone tail modifications that are not mutually exclusive, and introduced into a device that elongates the chromatin molecule. The pattern of binding yields a modification profile.
METHODS AND MATERIALS
We used bacteriophage T4 GT7 DNA (∼166 kbp) at 330 μg/ml stock concentration (Nippon Gene). Chromatin was reconstituted from calf thymus (Sigma H9250), HeLa core (Activ Motif 53501), and chicken erythrocyte (Genway Biotech, Inc 11-511-248355) histones. 6 μl of T4 GT7 DNA was dispersed in a buffer containing 20 μl of 10× NEBuffer2 (New England Biolabs), 44.5 μl of 0.1 M Na EDTA, 236.8 μl of 4 M NaCl, 10 μl of 20 mg/ml bovine serum albumin (BSA) in 152 μl of ultrapure water to give a final NaCl concentration of 2 M. For the reconstitution of calf thymus, HeLa core, and chicken erythrocyte chromatin, the histones of calf thymus, HeLa core, and chicken erythrocytes, were added at 2 μg, 6 μg, and 10 μg, respectively. The different histone concentrations were necessary to obtain mechanically similar chromatin as determined by nanochannel stretching and molecular combing assays that we performed in exploratory stages of this work.
The salt concentration was decreased by adding HEPES buffer (50 mM HEPES, 1 mM Na EDTA, 5 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), pH 7.5) in the following order: 156 μl, 327.6 μl, 234 μl, 171.6 μl, 234 μl, 327.6 μl, 483.6 μl, 1450.8 μl, and 967.2 μl. At each dilution step, we equilibrated at 30 °C for 15 min. At the end of the series of HEPES dilutions, the NaCl concentration was ∼200 mM. The NaCl concentration was finally reduced to 100 mM by addition of 4826 μl of Nonidet P-40 based buffer (10 mM Tris HCl, 1 mM Na EDTA, 0.1% v/v Nonidet P-40, 5% v/v glycerol, 0.1 mg/ml BSA, 5 mM DTT, 0.5 mM PMSF, pH 7.5). The chromatin is concentrated to roughly 3 μg/ml by placement into a dialysis tube (50 kDa cutoff) in contact with a G-Biosciences protein concentrator powder. The resulting chromatin is stored at 4 °C in the dark until further use.
Monoclonal antibodies specific to trimethylated H3K4 (Abcam ab1012) and acetylated H3K9 (Abcam ab12179) were fluorescently labeled with Alexa 488 carboxylic acid, 2,3,5,6 tetrafluorophenyl ester (Invitrogen A30005) and Alexa 568 carboxylic acid, succinimidyl ester (Invitrogen A20003) dyes, respectively, according to recommendations by the manufacturer. Anti-H3K4me3 antibodies were labeled with Alexa 488 (green), while the anti-H3K9ac antibodies are labeled with Alexa 568 (red).
About 1 μg of chromatin was incubated with 4 μg of dye labeled antibody, with 10 mg/ml of BSA and 0.5 mM of PMSF, at 4 °C overnight in the dark. Excess antibodies were removed by dialysis into 1× TBE (pH 8.4, with 3 mM beta-mercaptoethanol, 0.1% v/v Tween20), followed by concentration to 3 μg/ml. Prior to imaging, the chromatin concentration was lowered to 1 μg/ml in a 1.5× Tris Borate EDTA (TBE) buffer at pH 8.4 that contained 10% polyvinylpyrrolidone (PVP), 5 mM DTT, 0.5 mM PMSF, and 0.02% Tween 20. Fluorescent staining of DNA with YOYO-1 and YOYO-3 (both Invitrogen) was performed at a molar ratio of 1:10 (dye:basepairs) for chromatin only and 1:50 for chromatin decorated with one set of antibodies. Chromatin with two sets of antibodies was not stained using a DNA-specific dye.
Integrated nano/microfluidic channels were fabricated in fused silica using methods described elsewhere.22 The device layout is illustrated in that publication, following a design principle demonstrated by Reisner et al.,23 in which DNA is localized in the field of view through the combination of nanogrooves and a thin shunt layer that allows liquid to escape but traps DNA within the grooves. Note that the actual device is an array of such channels. The effective channel cross-section was 200 nm × 200 nm, and the shunt channel was 50 nm deep. Channel dimensions were confirmed using SEM after the device had been used in experiments.
Molecules were observed using an inverted fluorescence microscope with 1.35 N.A. oil immersion objective (Nikon) coupled to an emCCD camera (Andor). Green and red images were separated by an image splitter (collection at 620/60 and 520/40). Green and red images were obtained under illumination from 473 nm and 561 nm DPSS lasers, respectively. There was no detectable emission in the green channel by Alexa568/ YOYO-3 and red channel by Alexa488/YOYO-1 under 473 nm and 561nm illumination, respectively.
In experiments using only Alexa568 and Alexa488 labels, the ratio of the number densities of both dyes can be determined if both red and green fluorescence are collected under illumination at 473 nm. For this one needs to consider for each dye: (i) the molar extinction coefficient at 473 nm, calculated from the molar extinction at the absorbance maximum and the absorption spectrum of the dye, (ii) the fraction of fluorescence collected by the optical system in the red and green channel from the transmission spectra of filters and normalized emission spectrum of each dye, (iii) the quantum yield of each dye.
DNA was driven through both micro- and nano-fluidic channels using a pressure differential of about 30 psi. Once molecules had been localized in the nanochannel, the pressure was removed to recover an equilibrium configuration that is independent of liquid flow. After observation and recording, the channels were flushed using one-sided pressure application.
RESULTS AND DISCUSSION
We first verified the chromatin reconstitution process using T4 GT7 DNA and the histone mixture from calf thymus. After reconstituting, chromatin was stained using YOYO-1 and introduced into nanochannels, where an elongation of single molecules was observed (Fig. 2a). Molecules were stretched to a length of approximately 4 μm or 41 kbp/μm. Using a deGennes model24 with 200 × 200 nm2 channel cross-section, the known extension of dsDNA in channels of that diameter,15 and the estimated chromatin parameters which we described in Ref. 18, we find a predicted extension for our experiments of 42 kbp/μm, which is a remarkably good agreement. The stretching is about half that reported in high-resolution fiber-chromatin experiments.13
Figure 2.

Fluorescence images of chromatin reconstituted from calf-thymus histones. (a) Visualized using only YOYO-1. Chromatin in (b) is stained with Alexa488 anti H3K4-me3 (green) and YOYO-3 (red), in (c) it was stained using Alexa 568 anti H3K4-me3 (red) and YOYO-1 (green), and in (d) it was stained using Alex 568 anti H3K9-ac (red) and YOYO-1 (green). In panels (b)–(d) we present the combined image (center), and show the color channels above (green) and below (red) for clarity. In all panels the scale bar is 5 μm.
We chose such wide channels and moderate stretching because of the tendency of antibodies to form aggregates and prevent DNA entry into nanochannels. Antibody quality is discussed later. Narrower channels can be used to stretch chromatin, but increasing care has to be taken not to strip nucleosomes off by the increased driving pressure. Note that chromatin reconstituted with histones from different sources in general showed moderately different extensions in nanochannels (∼15%), which is an indication of the different quality of histone sources.
In Fig. 2b we present calf thymus-derived chromatin that was labeled with Alexa488 anti-H3K4-me3 monoclonal antibody (green) and co-stained with YOYO-3 dye (red). A continuous staining of DNA using the YOYO-3 dye along the entire length of the chromatin was observed. The antibody signal was also homogeneous, except at the ends where antibodies and histones appeared to be stripped off the molecule in some cases. Extremely compact molecules were observed in isolation.
For epigenetic profiling, there is the need to determine correlations between patterns of different types of modification by multiplexing labels of different color. Thus we decided to utilize our proven antibody (H3K4-me3), and add a different fluorescent label. Fig. 2c shows anti-H3K4-me3 labeled with Alexa568 (red) bound to calf thymus-derived chromatin that was co-stained with YOYO-1 (green). The binding pattern was more uneven, likely due to the switch of chromophore. It is generally accepted that Alexa568 has a higher tendency towards aggregation than Alexa488. We believe that the discontinuous histone labeling is due to chromatin disruption at the time of nanochannel insertion if the interaction between aggregated, genetically distant nucleosomes becomes stronger than the binding between DNA and histone octamer. Consequently, histones may be mechanically stripped due to tension during nanochannel insertion. The aggregation of antibodies appears to be the major impediment towards a quantitative use of the proposed method in an intra-molecular profiling application. While the development of low-aggregating antibodies is in general possible, we do not currently have the resources to do so. Dense binding of antibodies will in general modify the effective polymer parameters in the de Gennes model, and thus a change in stretch is anticipated. We conclude that mapping would be best either for sparse of dense antibody binding, with intermediate binding leading to the highest heterogeneity in stretch.
The final step in developing a system capable of differentiating between different sets of histone modification marks on chromatin was choosing a second set of monoclonal antibodies, that to the epigenetically active H3K9 acetylation mark, which we tagged with Alexa 568. The H3K4me3 and H3K9ac marks are both associated with transcriptionally active chromatin, but are not directly correlated or anticorrelated.25 In Fig. 2d we show calf thymus-derived chromatin labeled with this antibody, and co-stained using YOYO-1 dye. Point-wise binding of antibodies was observed.
We tested the ability of our assay to discriminate chromatin from different origins by comparing chromatin reconstituted with histone mixtures from calf thymus, chicken erythrocyte, and HeLa cells in a dual-labeling experiment with both Alexa488-labeled anti-H3K4me3 and Alexa568-labeled anti-H3K9ac. In Fig. 3a, we show calf thymus-derived chromatin: both antibodies were bound and could be observed, with some clustering of red and green fluorescence. In Fig. 3b, we demonstrate for chromatin reconstituted with HeLa histones, the abundance of anti-H3K9ac is greater than that of anti-H3K4me3. In Fig. 3c we show that chromatin reconstituted with histones from chicken erythrocytes carries both H3K4me3 and H3K9ac markers, with a strong enrichment in H3K4me3.
Figure 3.

Fluorescence images of double-stained chromatin with Alexa488-anti H3K4me3 (green channel and top panel) and Alex568-anti H3K9ac (red channel and bottom panel). Chromatin was reconstituted with histone extracted from (a) calf thymus, (b) HeLa cells, and (c) chicken erythrocytes. The scale bar is 5 μm.
In order to quantify the binding profiles, we mapped the ratios of occupancy for both markers on each chromatin species in a 2-dimensional histogram of instantaneous pixel brightness in the red and green spectral channel (Fig. 4). In order to avoid problems of relative intensity variations of the two lasers, all data are derived solely under 473 nm illumination. The drawback is that some Alexa488 emission will bleed through into the red spectral channels, which detects all Alex568. In all panels of Fig. 4, we find a circular feature at low intensity that is indicative of mostly empty nanochannels and camera electronics with Gaussian noise. From this circular feature, approximately linear “jets” emerge. All panels appear to contain a predominantly green jet, which is mainly horizontal. We attribute this jet to nearly pure Alexa488 emission since Alexa488 is visible in both the green and red collection channel. In the chicken erythrocyte-derived chromatin histogram (Fig. 4c), only the green jet is visible, while for calf thymus-derived chromatin (Fig. 4a) and HeLa cell-derived chromatin (Fig. 4b) both the green jet and a second more red jet are apparent. Visual inspection of the raw data reveals that in both the calf and HeLa chromatin sets some free Alexa488-labelled H3K4me3 antibodies flow through the nanochannels, which accounts for the green component. Red fluorescence, attributed to both Alex568 and Alexa488, was overwhelmingly located within channel stretches associated with chromatin. We thus believe that the jets with higher red content are the true chromatin-associated signal. The green signal due to free antibodies could be suppressed using autocorrelation techniques,16, 17 but we decided to keep the raw data for this publication.
Figure 4.
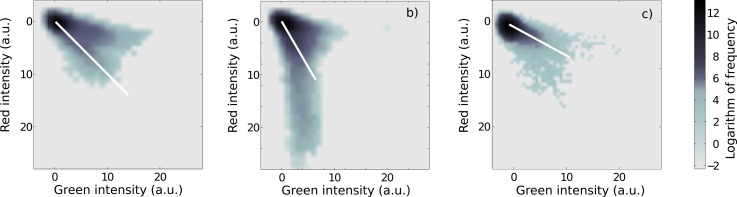
Histograms of pixel brightness within channel. Chromatin was reconstituted from (a) calf thymus histones, (b)HeLa cells, and (c) chicken erythrocytes. The density scale is logarithmic, and the white lines indicate the slope from which the ratio of H3K4me3 to H3K9ac was derived.
The existence of defined jet directions in Fig. 4 is indication that each chromatin source has a well-defined ratio of the two modifications. Indeed, that would be the expectation for randomly assembled chromatin from diverse sources. The fact that we observe jets instead of isolated patches in Fig. 4 must be attributed to the tendency of aggregation, and the fact that aggregate sizes have a disappointingly wide range. By measuring the asymptotic angle of jets and using the known spectra for the fluorophores, we can find different characteristic ratios of red to green emission for the different histone source.
We conclude that chicken blood chromatin has the highest ratio of H3K4me3 to H3K9ac (0.19), followed by calf chromatin (0.11), and lowest ratio for HeLa Chromatin (0.09). The error of these values is given by the uncertainty of measurement of the angle of the asymptotic line (±5°), which leads to an error of all three values is about ±30% of the above number. These ratios were obtained using spectral properties provided by the manufacturer for free dye, and not the conjugated form. Thus, all ratios could be substantially biased if spectra have shifted, although the trend would remain intact. Furthermore, we assume similar dye conjugation efficiencies for both sets of antibodies. While we have observed that chromatin reconstitution required different quantities of core histones for different sources, we believe that the relative abundance of histone modifications incorporated into chromatin is representative of the histones before chromatin reconstitution. We thus tested histone mixes using a total histone content kit (EpiQuik Global Acetyl H3-K9 Quantification Kit, P-4011-48, and Global Trimethyl H3-K4 Quantification Kit, P-3027-48), and found a result with the same ordering of relative histone modification ratios as in our nanochannel mapping. It thus appears that the fluorescence mapping method in nanochannels reproduced the bulk measurement. It could hence potentially be used to obtain spatially ordered maps if such a spatially ordered substrate could be prepared.
Throughout our experiments, we observed changes in chromatin length as the molecule was modified through antibody binding. We observed that antibody binding itself could result in more compact configurations than as-reconstituted chromatin. However, upon introduction into nanochannels, nucleosomes can be stripped off the DNA. We believe that both bare antibodies, as well as the fluorescent tags attached to the antibodies contribute to this modification through aggregation and bridging of multiple nucleosomes. These aggregates stabilize loops of DNA, which in effect shorten the DNA. If the interaction between nucleosomes and antibodies is stronger than the interaction between nucleosomes and DNA, then nucleosomes can dissociate from DNA under the stress of nanochannel insertion, and large stretches of chromatin convert to bare DNA. This is particularly apparent in Fig. 2c. Commercial antibodies, at least those used by us, in general have a strong tendency to aggregate, but lower aggregation has been achieved for antibodies used in therapeutics, for instance. We also observe that the antibodies are stickier than the chromatin, and tend to adhere to channel walls unless mitigated by a PVP additive. Nonetheless, we are able to flush most of the residual adhered antibodies out and reuse chips 3 to 5 times.
CONCLUSIONS
Our results on the detection of trimethylated H3K4 and acetylated H3K9 histone markers on the three different reconstituted chromatin in our nanochannels demonstrate the success of our technique in being able to quantify the relative histone makeup at a single molecule scale. We analyzed large molecules (160 kbp), without the need to fragment them. There is a clear pathway for scaling up to even larger molecules, and we envision detection of histone markers on longer and larger stretched chromatin with our technique, with slight modifications to nanochannel design, to ensure that the entire chromatin can be imaged in the field of view. While we focused on histone tail marks in this publication, the general concept of detecting proteins bound to DNA using nanochannel stretching and antibodies should hold for a wide variety of other DNA-binding proteins.
ACKNOWLEDGMENTS
We acknowledge support from the National Cancer Institute (CA132075) and the National Institute of Child Health and Human Development (HD065222). A portion of this research was conducted at the Center for Nanophase Materials Sciences, which was sponsored at Oak Ridge National Laboratory by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. This work was performed in part at the Georgia Tech Nanotechnology Research Center and the Cornell NanoScale Facility, members of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation (Grant No. ECCS-0335765).
References
- Felsenfeld G. and Groudine M., “ Controlling the double helix,” Nature 421(6921), 448–453 (2003). 10.1038/nature01411 [DOI] [PubMed] [Google Scholar]
- Luger K., Mäder A. W., Richmond R. K., Sargent D. F., and Richmond T. J., “ Crystal structure of the nucleosome core particle at 2.8 A resolution,” Nature 389(6648), 251–260 (1997). 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- Kouzarides T., “ Chromatin modifications and their function,” Cell 128(4), 693–705 (2007). 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- McGhee J. D. and Felsenfeld G., “ Nucleosome structure,” Annu Rev Biochem. 49, 1115–1156 (1980). 10.1146/annurev.bi.49.070180.005343 [DOI] [PubMed] [Google Scholar]
- Wolffe A. P., Chromatin: Structure and Function, 3rd ed. (Academic Press, Waltham, MA, 1999). [Google Scholar]
- Strahl B. D. and Allis C. D., “ The language of covalent histone modifications,” Nature 403(6765), 41–45 (2000). 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- Collas P., “ The current state of chromatin immunoprecipitation,” Mol Biotechnol. 45(1), 87–100 (2010). 10.1007/s12033-009-9239-8 [DOI] [PubMed] [Google Scholar]
- Cipriany B. R., Zhao R., Murphy P. J., Levy S. L., Tan C. P., Craighead H. G., and Soloway P. D., “ Single molecule epigenetic analysis in a nanofluidic channel,” Anal. Chem. 82(6), 2480–2487 (2010). 10.1021/ac9028642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P. J., Cipriany B. R., Wallin C. B., Ju C. Y., Szeto K., Hagarman J. A., Benitez J. J., Craighead H. G., and Soloway P. D., “ Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells,” Proc. Natl. Acad. Sci. U. S. A. 110(19), 7772–7777 (2013). 10.1073/pnas.1218495110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter P., Tang C. J., Call K., Hermanson G., Evans G. A., Housman D., and Ward D. C., “ High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones,” Science 247(4938), 64–69 (1990). 10.1126/science.2294592 [DOI] [PubMed] [Google Scholar]
- Heng H. H. Q., Squire J., and Tsui L. C., “ High-resolution mapping of mammalian genes by in situ hybridization to free chromatin,” Proc. Natl. Acad. Sci. U. S. A. 89(20), 9509–9513 (1992). 10.1073/pnas.89.20.9509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower M. D., Sullivan B. A., and Karpen G. H., “ Conserved organization of centromeric chromatin in flies and humans,” Dev. Cell 2(3), 319–330 (2002). 10.1016/S1534-5807(02)00135-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. M., Chastain P. D., Cordeiro-Stone M., and Kaufman D. G., “ DNA replication and the GINS complex: Localization on extended chromatin fibers,” Epigenetics Chromatin 2(1), 6 (2009). 10.1186/1756-8935-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegenfeldt J. O., Prinz C., Cao H., Chou S., Reisner W. W., Riehn R., Wang Y. M., Cox E. C., Sturm J. C., Silberzan P., and Austin R. H., “ The dynamics of genomic-length DNA molecules in 100-nm channels,” Proc. Natl. Acad. Sci. U.S.A. 101(30), 10979–10983 (2004). 10.1073/pnas.0403849101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner W., Morton K. J., Riehn R., Wang Y. M., Yu Z. N., Rosen M., Sturm J. C., Chou S. Y., Frey E., and Austin R. H., “ Statics and dynamics of single DNA molecules confined in nanochannels,” Phys. Rev. Lett. 94(19), 196101 (2005). 10.1103/PhysRevLett.94.196101 [DOI] [PubMed] [Google Scholar]
- Carpenter J. H., Karpusenko A., Pan J., Lim S. F., and Riehn R., “ Density fluctuations dispersion relationship for a polymer confined to a nanotube,” Appl. Phys. Lett. 98(25), 253704 (2011). 10.1063/1.3602922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpusenko A., Carpenter J. H., Zhou C., Lim S. F., Pan J., and Riehn R., “ Fluctuation modes of nanoconfined DNA,” J. Appl. Phys.111(2), 24701–247018 (2012). 10.1063/1.3675207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streng D. E., Lim S. F., Pan J. H., Karpusenka A., and Riehn R., “ Stretching chromatin through confinement,” Lab Chip 9(19), 2772–2774 (2009). 10.1039/b909217j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. M., Tegenfeldt J. O., Reisner W., Riehn R., Guan X. J., Guo L., Golding I., Cox E. C., Sturm J., and Austin R. H., “ Single-molecule studies of repressor-DNA interactions show long-range interactions,” Proc. Natl. Acad. Sci. U.S.A. 102(28), 9796–9801 (2005). 10.1073/pnas.0502917102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. F., Karpusenko A., Sakon J. J., Hook J. A., Lamar T. A., and Riehn R., “ DNA methylation profiling in nanochannels,” Biomicrofluidics 5(3), 034106 (2011). 10.1063/1.3613671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T., Kim B. C., Huang J., Douville N. J., Thouless M. D., and Takayama S., “ Nanoscale squeezing in elastomeric nanochannels for single chromatin linearization,” Nano Lett. 12(12), 6480–6484 (2012). 10.1021/nl304063f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehn R., Reisner W., Tegenfeldt J. O., Wang Y. M., Tung C.-K., Lim S. F., Cox E. C., Sturm J., and Austin R. H., “ Nanochannels for genomic DNA analysis: The long and short of it,” in Integrated Biochips for DNA Analysis, 1st ed., edited by Liu R. H. and Lee A. P. (Landes Bioscience, Austin, Texas, 2007), pp. 151–186. [Google Scholar]
- Reisner W., Larsen N. B., Silahtaroglu A., Kristensen A., Tommerup N., Tegenfeldt J. O., and Flyvbjerg H., “ Single-molecule denaturation mapping of DNA in nanofluidic channels,” Proc. Natl. Acad. Sci. 107(30), 13294–13299 (2010). 10.1073/pnas.1007081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gennes P. G., Scaling Concepts in Polymer Physics (Cornell University Press, Ithaca, NY, 1979). [Google Scholar]
- Crump N. T., Hazzalin C. A., Bowers E. M., Alani R. M., Cole P. A., and Mahadevan L. C., “ Dynamic acetylation of all lysine-4 trimethylated histone H3 is evolutionarily conserved and mediated by p300/CBP,” Proc. Natl. Acad. Sci. U.S.A. 108(19), 7814–7819 (2011). 10.1073/pnas.1100099108 [DOI] [PMC free article] [PubMed] [Google Scholar]