

Abstract
It is evident that there is a relationship between the brain's serotonin system and obesity. Although it is clear that drugs affecting the serotonin system regulate appetite and food intake, it is unclear whether changes in the serotonin system are cause or consequence of obesity. To determine whether obesogenic eating habits result in reduced serotonin transporter (SERT)-binding in the human hypothalamic region, we included 25 lean, male subjects who followed a 6-week-hypercaloric diet, which were high-fat-high-sugar (HFHS) or high-sugar (HS) with increased meal size or -frequency (=snacking pattern). We measured SERT-binding in the hypothalamic region with SPECT. All hypercaloric diets significantly increased body weight by 3–3.5%. Although there were no differences in total calories consumed between the diets, only a hypercaloric HFHS-snacking diet decreased SERT-binding significantly by 30%. We here show for the first time in humans that snacking may change the serotonergic system increasing the risk to develop obesity.
Keywords: Serotonin transporters, Meal pattern, Hypercaloric diet, Sugar, Fat, Human imaging
1. Introduction
Obesity is a worldwide health problem of epidemic proportions and is a consequence of an ongoing dysbalance in energy intake and energy expenditure. Why so many people are not capable to balance their energy metabolism is still poorly understood. To target the obesity epidemic, it is important to understand the mechanisms underlying the chronic intake of energy surplus. The hypothalamus is viewed as the centre of feeding regulation [1] and it has been shown that brain circuits involved in food intake and appetite control are disturbed in rodent models of obesity. Besides the hypothalamic control of food intake, nuclei in the brainstem and the mesolimbic cortex are also involved in this complex regulatory system [2].
The neurotransmitter serotonin innervates most of these brain structures and is therefore a likely candidate to coordinate hunger and satiety signalling to and from the periphery. Indeed, serotonin acts as an anorexigenic signal: food intake increases both extracellular serotonin levels and serotonin turnover in the brain while serotonin inhibits food intake and promotes satiety [3,4]. In addition, neurochemical depletion of brain serotonin, as well as genetic and pharmacological manipulations of several parts of the serotonin system in rodents, results in obesity and hyperphagia [5–7].
Serotonin transporters (SERTs) are determinants of serotonin bioavailability in the synaptic cleft and are important regulators of serotonergic transmission by facilitating reuptake of serotonin from the synaptic cleft into the presynaptic neuron. After reuptake, serotonin can be released into the cleft again and stimulate signal transmission [8]. Lower cerebral SERT binding was reported in an obese rat model [9] while in humans cerebral SERT binding measured with PET was inversely related to body mass index (BMI) [10]. In line with these findings, a SERT promoter polymorphism has been identified as risk factor for obesity [11]. Finally, methylation of the SERT promoter correlates with obesity measures in monozygotic twins [12]. Interestingly, drugs that increase extracellular serotonin via inhibition of SERT reduce food intake and result in a decrease in body weight in both animals [13] and in humans [4]. However, long-term use of selective serotonin reuptake inhibitors (SSRIs) is associated with obesity [14,15]. The mechanisms underlying this difference remain unclear.
Thus, lower SERT is clearly associated with obesity; however whether this is cause or consequence, and whether this is explained by BMI per se or excessive food intake remains to be determined. As obesity may result from either change in meal pattern and/or meal composition we here studied the relationship between cerebral SERT and different patterns of excessive food intake. We therefore measured cerebral SERT with SPECT before and after a 6 weeks hypercaloric diet intervention in lean men.
2. Methods
2.1. Subjects
We included 39 healthy lean men (age 22.6±3.5 years; BMI 22.4±1.5 kg/m2). All subjects had normal insulin sensitivity defined as HOMA-IR <2.5. Subjects had no history of neuropsychiatric or eating disorders and did not use any medication. They were not known with substance abuse and did not perform shift work. All subjects had healthy eating behaviour assessed via an online diet journal (eetmeter.voedingscentrum.nl) defined as a eucaloric diet containing ~45–50% carbohydrates, ~30–35% fat and ~15–20% protein. The study was approved by the medical ethics committee of the AMC Amsterdam. Written informed consent was received from all participants prior to the start of study participation.
2.2. Diets
Subjects were randomised into one of 4 hypercaloric diet groups or a control group. Subjects in the control group (N=5) underwent all the measurements but did not follow a diet.
All hypercaloric diets were based on a 40% caloric surplus on top of the ad libitum diet. The diet was followed for 6 consecutive weeks. Subjects were contacted weekly. Randomisation is displayed in Figure 1. The hypercaloric diet groups were:
-
1.
High-fat high-sugar (HFHS) diet using a liquid meal (Nutridrink Compact, Nutricia® Advanced Medical Nutrition; Zoetermeer; The Netherlands) 3 times a day to be consumed together with the 3 daily main meals. This group represents the HFHS-increased meal size (HFHS-S) group.
-
2.
High-fat high-sugar (HFHS) diet using a liquid meal (Nutridrink Compact, Nutricia®) 3 times a day in between the 3 daily main meals (2–3 h after each meal). This group represents the HFHS-increased meal frequency (HFHS-F) group.
-
3.
High-sugar (HS) diet using commercially available sugar-sweetened beverages 3 times a day to be consumed together with the 3 daily main meals. This group represents the HS-increased meal size (HS-S) group.
-
4.
High-sugar (HS) diet using commercially available sugar-sweetened beverages 3 times a day in between the 3 daily main meals (2–3 hours after each meal). This group represents the HS-increased meal frequency (HS-F) group.
Figure 1.
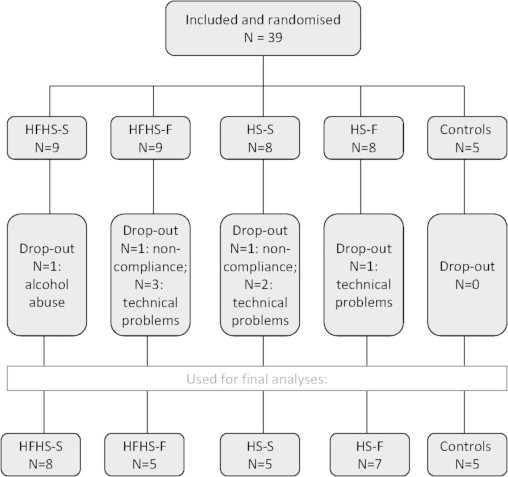
Randomisation flowchart. Number of subjects initially included and randomised; drop-out and reason for drop-out and final number of subjects on which data analysis was performed. HFHS=high-fat-high-sugar; HS=high-sugar. S=increased meal size; F=increased meal frequency.
The nutritive value of Nutridrink Compact® was as follows: 240 kcal/100 ml; 16 En% protein (mainly casein), 49 En% carbohydrates (mainly maltose and polysaccharides) and 35 En% fat (mainly mono- and poly-unsaturated fat). As high-sugar liquids, the subjects consumed commercially available soft drinks. A list of drinks with comparable nutritive value of which subjects could choose their favourite beverage was used. The soft drinks contained no fat or protein and were mainly sweetened with sucrose. The ad libitum diet was monitored online. Subjects registered their daily food consumption on a website (eetmeter.voedingscentrum.nl). When caloric intake was lower than caloric need, assessed by measured resting energy expenditure (REE), subjects were instructed to increase their healthy ad libitum diet.
2.3. Resting energy expenditure (REE)
REE was measured by use of indirect calorimetry. VO2 and VCO2 were measured in the supine position during 30 min using a ventilated hood system (Sensor Medics, Vmax Encore 29 N, Anaheim, CA). REE and respiratory exchange ratio (RER) were calculated as described previously.[16] The abbreviated Weir equation was used to calculate the 24-h energy expenditure.
2.4. SPECT imaging
After an overnight fast subjects underwent a single photon emission computed tomography (SPECT) scan 2 h after intravenous administration of 115 MBq 123I-FP-CIT (range 110–120 MBq; specific activity >750 MBq/nmol; radiochemical purity >98%, produced according to GMP criteria at GE Healthcare, Eindhoven, The Netherlands). In a previous study, we showed that this is the optimal time-point to measure SERT with 123I-FP-CIT SPECT [17]. Each participant was pre-treated with potassium iodide to block thyroid uptake of free radioactive iodide. SPECT imaging was performed using a 12-detector, single slice brain-dedicated scanner (Neurofocus), using an acquisition protocol as described earlier with slight modifications (interslice distance 5 mm, acquisition time 210 s per slice) [18]. All scans were reconstructed in 3D and corrected for attenuation.
2.5. Image analysis
For quantification, a ROI analysis was performed to determine specific binding activity in the diencephalon, which includes the hypothalamus and thalamus, as described previously [18]. Briefly, the 4 consecutive slices with the highest diencephalic binding were selected to assess binding to SERT. Activity in the cerebellum (3 consecutive slices) was assumed to represent non-displaceable activity (nonspecific binding and free radioactivity). Finally, a specific-to-nonspecific binding ratio was calculated as activity in ROI minus cerebellar binding/cerebellar binding [19]. This ratio was used as the outcome measure.
2.6. Statistics
For body weight and food intake data we used a 1-way ANOVA to determine overall effects. If a significant result was found, posthoc analysis (Students t-test) was performed with Bonferroni correction to compare data between the different groups. For analysis of SERT binding changes within the hypercaloric diet groups, we used 2 way ANOVA's to determine overall effects. If significant effects were determined, we used paired student t-tests to compare the data before and after the diet intervention within the different hypercaloric diet group.
3. Results
3.1. Randomisation and Body weight
39 subjects were randomised and completed the 6-week diet period. Two subjects were excluded because compliance with the diet was uncertain and one subject was excluded because of excessive alcohol consumption during the days prior to the second study day. In 6 subjects the SPECT images could not be used because of technical failure. Randomisation and drop-out rate is displayed in Figure 1. The final data analyses were performed on 30 subjects with a mean age of 22.1±2.5 years and a mean BMI of 22.5±1.3 kg/m2. Baseline characteristics are described in Table 1. Subjects in all 4 diet groups significantly gained body weight compared to the control group (Figure 2A; Table 2), on average 3–3.5% of total body weight, yet BMI after the diet remained within the normal range (20–25 kg/m2). There was no statistical significant difference in percentage weight gain between the diet intervention groups. Subjects in the control group (n=5) did not change significantly in body weight (Figure 2A).
Table 1.
Baseline characteristics.
| HFHS-S | HFHS-F | HS-S | HS-F | CP | ANOVA | |
|---|---|---|---|---|---|---|
| N | 8 | 5 | 5 | 7 | 5 | |
| Age (yr) | 22.6±2.7 | 21.0±1.4 | 21.8±1.6 | 21.7±2.9 | 23.0±3.1 | ns |
| BMI (kg/m2) | 22.2±0.9 | 22.6±1.7 | 22.3±0.8 | 23.0±1.3 | 22.3±2.1 | ns |
| Body weight (kg) | 78.0±5.6 | 80.6±9.6 | 79.8±7.8 | 82.5±8.4 | 76.6±7.7 | ns |
| Resting Energy Expenditure (REE) | 1926±91 | 1975±179 | 1965±130 | 2002±152 | 1893±110 | ns |
| Caloric intake (kCal) | 2566±317 | 2692±341 | 2631±165 | 2426±301 | 2490±262 | ns |
Data are displayed as mean±SD; ns=not significant.
Figure 2.
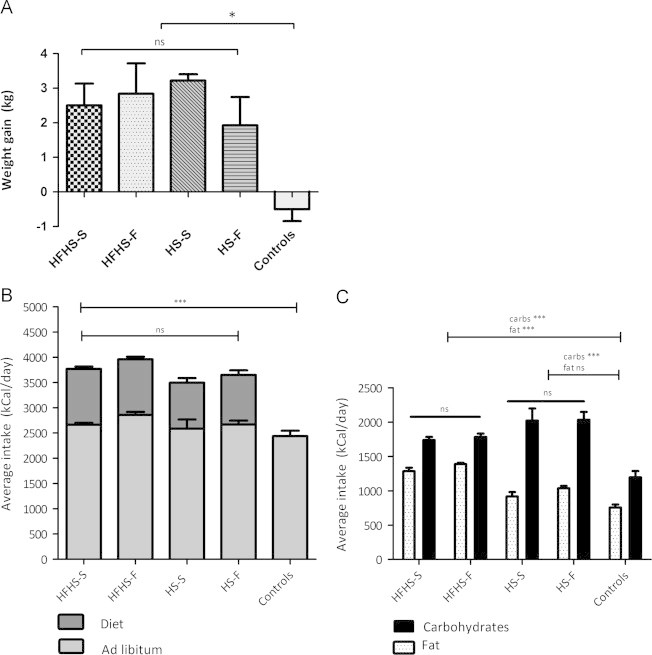
Body weight gain and food intake. HFHS=high-fat-high-sugar; HS=high-sugar. S=increased meal size; F=increased meal frequency.(A) All hypercaloric diet groups showed significantly increased body weight compared to the control group. There were no differences in body weight gain between the hypercaloric diet groups (data are depicted in mean ±SEM; *p<0.01). (B) Caloric intake in kCal/day during the intervention period: Ad libitum intake is depicted in light grey and added HFHS or HS calories are depicted in dark grey. Total intake, and also ad lib and diet intake were similar in all hypercaloric diet groups, whereas intake for all hypercaloric diet groups were increased compared to the control group (data depicted as mean±±SEM; ***p<0.0001). (C) Carbohydrate- and fat intake in kCal/day during the intervention period displayed as mean and SEM. Subjects on HFHS consumed more fat and carbohydrate calories compared to the controls, whereas subject on HS consumed more carbohydrate calories, but equal fat calories, compared to controls. No significant differences were detected between HFHS-S and HFHS-F, and between HS-S and HS-F (data depicted as mean±±SEM; ***p<0.0001).
Table 2.
BMI and food intake before- and after a hypercaloric diet.
|
Control |
HFHS |
HS |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | T=6 weeks | Paired t-test | Baseline | Hypercaloric | Paired t-test | Baseline | Hypercaloric | Paired t-test | |
| BMI (kg/m2) | 22.3±2.1 | 22.2±2.2 | ns | 22.4±1.2 | 23.1±1.2 | ⁎⁎⁎ | 22.7±1.1 | 23.4±1.2 | ⁎⁎⁎ |
| Caloric intake (kCal/24 h) | 2490±262 | 2502±271 | ns | 2615±319 | 3862±251 | ⁎⁎⁎ | 2512±266 | 3954±257 | ⁎⁎⁎ |
| Carbohydrate (g) | 322±68 | 299±51 | ns | 297±42 | 441±37 | ⁎⁎⁎ | 272±46 | 546±61 | ⁎⁎⁎ |
| Carbohydrate (En%) | 51±8 | 46±9 | ns | 46±5 | 46±3 | ns | 43±4 | 57±3 | ⁎⁎⁎ |
| Fat (g) | 79±9 | 84±11 | ns | 94±19 | 147±14 | ⁎⁎⁎ | 98±20 | 112±13 | ⁎⁎ |
| Fat (En%) | 29±4 | 34±5 | ns | 32±4 | 35±3 | ⁎⁎ | 35±6 | 27±3 | ⁎⁎⁎ |
| Protein (g) | 92±17 | 89±14 | ns | 94±12 | 146±11 | ⁎⁎⁎ | 103±16 | 112±16 | ⁎ |
| Protein (En%) | 15±3 | 15±3 | ns | 15±2 | 15±1 | ns | 16±2 | 12±2 | ⁎⁎⁎ |
Data are displayed as mean±SD. HFHS=hypercaloric high-fat-high-sugar diets (N=13); HS=hypercaloric high-sugar diets (N=12). Within the HFHS- and the HS-group there were no significant differences (see Figure 2) and for legibility the groups are combined in this table. Paired t-tests: ns=not significant.
p<0.01.
p<0.001.
p<0.0001.
3.2. Caloric intake
Ad libitum nutrient intake was similar between the diet groups. Diet intervention data are displayed in Table 2 and Figure 2B and C. The subjects in the 4 diet groups all consumed a hypercaloric diet compared to the control group (p<0.0001). Control subjects consumed a eucaloric diet of ~2400 kCal/day consisting for ~45–50% of carbohydrates, ~30–35% of fat and for ~15% of protein. As expected, the subjects in the HS groups consumed excessive carbohydrates (~60% of total caloric intake; p=<0.0001 compared to baseline diet), which was significantly higher compared to the control and HFHS group (p<0.0001). Absolute intake of protein (~103 gram per day at baseline versus ~112 gram per day during the diet, p=0.02), and fat (98 gram per day at baseline versus 112 gram per day during the diet, p=0.001), were higher during the HS diet. However, due to excessive carbohydrate intake, relative intake of protein (~16% at baseline versus ~12% during the diet , p<0.0001) and fat (~35% at baseline versus ~27% during the diet p<0.0001) were decreased. There was no difference in carbohydrate intake between both HS groups (p=0.33). Subjects in the HFHS groups consumed both excessive fat (~94 gram per day at baseline versus ~147 gram per day during the diet, p<0.0001) and carbohydrates (~297 gram per day at baseline versus ~441 gram per day during the diet, p<0.0001) with no difference between the HFHS groups (p=0.35). Both HFHS groups had a higher absolute intake of protein (~98 gram per day before versus ~146 gram per day during the diet; p<0.0001). Relative diet composition remained stable at ~45% carbohydrate, ~35% fat and ~15% protein. Subjects in both HFHS-groups consumed similar amounts of Nutridrink®.
3.3. SERT in the hypothalamic region
A 2 way ANOVA detected an overall effect of size versus frequency on SERT specific-to-nonspecific binding ratio (SNS-BR) in the hypothalamic region (p=0.01), with lower SERT binding when surplus calories were consumed in between the meals (frequency) compared to surplus calories consumed with the meals (size) (Figure 3A). When analysing the changes in SERT binding within the specific hypercaloric diet groups and within the control group, SERT binding activity in the HFHS-F group was 30% reduced after the diet (0.65±0.15 vs. 0.46±0.13; p=0.004) (Figure 3B). In the other 3 hypercaloric diet groups and in the control group, there was no significant change in SERT binding over the 6 weeks of treatment (data not shown).
Figure 3.
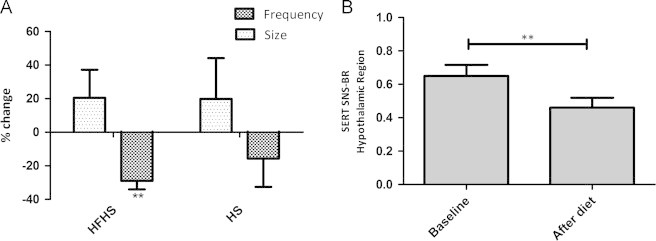
SERT binding. HFHS=high-fat-high-sugar; HS=high-sugar. S=increased meal size; F=increased meal frequency. (A) SERT binding activity in diencephalon in the 4 hypercaloric diet groups displayed as % change. A hypercaloric diet with surplus calories in between the meals (frequency) showed lower SERT binding activity compared to hypercaloric diet with the surplus given with the meals (size) (overall effect of meal pattern (freq vs. size): p=0.01). (B) SERT binding activity decreased 30% after 6 weeks in a HFHS-F diet (paired t-test”: **p=0.004). There was no significant change in SERT binding activity in the other diet groups or the control group (not shown).
4. Discussion
We here show for the first time that in lean healthy male subjects SERT binding in the diencephalon, which includes the hypothalamus, decreases after a hypercaloric HFHS diet. This decrease is only observed when the caloric surplus is consumed in between meals but not when caloric surplus is consumed together with the meals. This suggests a selective effect of hypercaloric snacking on SERT in the hypothalamic region that is absent following increased meal size. Our data are in line with, and extend, previous findings showing a negative correlation between SERT and BMI [10,12], and could imply that a hypercaloric snacking diet, and not so much obesity itself, underlies these changes in SERT in obese individuals. Thus, overeating by high-fat high-sugar snacking might contribute to a disturbed appetite control with a risk of obesity through modulation of cerebral serotonin metabolism.
Understanding the role of SERT in the hypothalamus in the regulation of food intake and body weight regulation is difficult. The interaction between SERT, extracellular serotonin and serotonin signal transmission is dynamic and complex [20]. Moreover, serotonin is produced not only in the brain but also in the enteric nerve system. Total SERT depleted animals have increased extracellular serotonin and display an obese insulin resistant phenotype without an increase in food intake [21] while the short-term use of SERT inhibitors is associated with lower food intake and body weight [4]. On the other hand, chronic use of SSRI's might increase the risk for obesity in humans [14,22]. It could well be that there is a difference between acute and chronic SERT inhibition on extracellular serotonin levels and thereby on food intake and body weight control. The change in SERT binding in our subjects might be a physiological response to reduce food intake through increasing extracellular serotonin. However, one would then expect a decrease in all hypercaloric diet groups, but we found a decrease only in the HFHS-F group. Therefore it seems more likely that a decrease in SERT results in a decrease in extracellular serotonin, also explaining the failure of SSRIs for long-term weight loss maintenance. A decrease in extracellular serotonin might result in less inhibition of food intake predisposing especially snacking individuals to obesity. However, this remains to be investigated.
The HFHS groups consumed more protein, which was inevitable since the liquid meal (Nutridrink®) contained protein. Dietary proteins are involved in brain satiation mechanisms: they induce gut peptide secretion (e.g. GLP-1, PPY) and production of hormones such as insulin and ghrelin. These neuropeptides and hormones can signal to brain regions involved in hunger and satiety including the hypothalamus [23]. There is currently no evidence for the presence of a relationship between SERT and dietary proteins. A few rodent studies have investigated the possibility of a relationship between dietary protein and serotonin in the brain but did not report a consistent association [24,25]. One study showed activation of the hypothalamic serotonin system after ingestion of animal proteins in rats [26]. However, in the present study the protein surplus is casein and not animal proteins. Moreover, the reduction in SERT binding was found only in the HFHS-F group while protein intake did not significantly differ from the HFHS-S group making it unlikely that the effect on the brain is explained by higher protein intake.
In the present study we investigated SERT binding in the diencephalon. The diencephalon includes the hypothalamus, which is a key regulatory brain centre for energy balance [1]. Future studies will need to establish in which specific nucleus or nuclei SERT is decreased after similar diet interventions. A hypercaloric snacking high-fat high-sugar diet during 6 weeks in lean men decreases SERT binding in the hypothalamic region while increasing glucose intake or increasing meal size did not affect SERT binding. Interestingly, in today's society there is increasing snacking behaviour and it has been shown that snacking is associated with obesity [27–29]. We propose that a decrease SERT in the hypothalamic region caused by high-fat-high-sugar snacking might be considered as a mechanism for disturbed appetite control resulting in hyperphagia and obesity.
Conflict of interest
None declared.
Acknowledgements
Author JB is a consultant at GE Healthcare. This study was funded by a PhD fellowship grant awarded by the AMC Executive Board. There were no other sources of funding.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Horvath T.L. The hardship of obesity: a soft-wired hypothalamus. Nature Neuroscience. 2005;8:561–565. doi: 10.1038/nn1453. [DOI] [PubMed] [Google Scholar]
- 2.Morrison C.D., Berthoud H.R. Neurobiology of nutrition and obesity. Nutrition Reviews. 2007;65:517–534. doi: 10.1301/nr.2007.dec.517-534. [DOI] [PubMed] [Google Scholar]
- 3.Lam D.D., Garfield A.S., Marston O.J., Shaw J., Heisler L.K. Brain serotonin system in the coordination of food intake and body weight. Pharmacology Biochemistry & Behavior. 2010;97:84–91. doi: 10.1016/j.pbb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Halford J.C., Harrold J.A., Boyland E.J., Lawton C.L., Blundell J.E. Serotonergic drugs: effects on appetite expression and use for the treatment of obesity. Drugs. 2007;67:27–55. doi: 10.2165/00003495-200767010-00004. [DOI] [PubMed] [Google Scholar]
- 5.Breisch S.T., Zemlan F.P., Hoebel B.G. Hyperphagia and obesity following serotonin depletion by intraventricular p-chlorophenylalanine. Science. 1976;192:382–385. doi: 10.1126/science.130678. [DOI] [PubMed] [Google Scholar]
- 6.Saller C.F., Stricker E.M. Hyperphagia and increased growth in rats after intraventricular injection of 5,7-dihydroxytryptamine. Science. 1976;192:385–387. doi: 10.1126/science.1257774. [DOI] [PubMed] [Google Scholar]
- 7.Waldbillig R.J., Bartness T.J., Stanley B.G. Increased food intake, body weight, and adiposity in rats after regional neurochemical depletion of serotonin. Journal of Comparative and Physiological Psychology. 1981;95:391–405. doi: 10.1037/h0077790. [DOI] [PubMed] [Google Scholar]
- 8.Squire L.R., Berg D., Bloom F.E., du Lac S., Ghosh A., Spitzer N.C. Fundamental Neuroscience. 2008;3:143–144. [Google Scholar]
- 9.Ratner C., Ettrup A., Bueter M., Haahr M.E., Compan V., le Roux C.W., Levin B., Hansen H.H., Knudsen G.M. Cerebral markers of the serotonergic system in rat models of obesity and after Roux-en-Y gastric bypass. Obesity (Silver Spring) 2012;20:2133–2141. doi: 10.1038/oby.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erritzoe D., Frokjaer V.G., Haahr M.T., Kalbitzer J., Svarer C., Holst K.K., Hansen D.L., Jernigan T.L., Lehel S., Knudsen G.M. Cerebral serotonin transporter binding is inversely related to body mass index. Neuroimage. 2010;52:284–289. doi: 10.1016/j.neuroimage.2010.03.086. [DOI] [PubMed] [Google Scholar]
- 11.Sookoian S., Gemma C., Garcia S.I., Gianotti T.F., Dieuzeide G., Roussos A., Tonietti M., Trifone L., Kanevsky D., Gonzalez C.D., Pirola C.J. Short allele of serotonin transporter gene promoter is a risk factor for obesity in adolescents. Obesity (Silver Spring) 2007;15:271–276. doi: 10.1038/oby.2007.519. [DOI] [PubMed] [Google Scholar]
- 12.Zhao J., Goldberg J., Vaccarino V. Promoter methylation of serotonin transporter gene is associated with obesity measures: a monozygotic twin study. International Journal of Obesity (London) 2012;37(1):140–145. doi: 10.1038/ijo.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simansky K.J. Serotonergic control of the organization of feeding and satiety. Behavioural Brain Research. 1996;73:37–42. doi: 10.1016/0166-4328(96)00066-6. [DOI] [PubMed] [Google Scholar]
- 14.Raeder M.B., Bjelland I., Emil V.S., Steen V.M. Obesity, dyslipidemia, and diabetes with selective serotonin reuptake inhibitors: the Hordaland Health Study. Journal of Clinical Psychiatry. 2006;(67):1974–1982. doi: 10.4088/jcp.v67n1219. [DOI] [PubMed] [Google Scholar]
- 15.Fava M., Judge R., Hoog S.L., Nilsson M.E., Koke S.C. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. Journal of Clinical Psychiatry. 2000;61:863–867. doi: 10.4088/jcp.v61n1109. [DOI] [PubMed] [Google Scholar]
- 16.Frayn K.N. Calculation of substrate oxidation rates in vivo from gaseous exchange. Journal of Applied Physiology. 1983;55:628–634. doi: 10.1152/jappl.1983.55.2.628. [DOI] [PubMed] [Google Scholar]
- 17.Koopman K.E., la Fleur S.E., Fliers E., Serlie M.J., Booij J. Assessing the optimal time point for the measurement of extrastriatal serotonin transporter binding with 123I-FP-CIT SPECT in healthy, male subjects. Journal of Nuclear Medicine. 2012;53:1087–1090. doi: 10.2967/jnumed.111.102277. [DOI] [PubMed] [Google Scholar]
- 18.Booij J., de J.J., de B.K., Knol R., de Win M.M., van Eck-Smit B.L. Quantification of striatal dopamine transporters with 123I-FP-CIT SPECT is influenced by the selective serotonin reuptake inhibitor paroxetine: a double-blind, placebo-controlled, crossover study in healthy control subjects. Journal of Nuclear Medicine. 2007;48:359–366. [PubMed] [Google Scholar]
- 19.Innis R.B., Cunningham V.J., Delforge J., Fujita M., Gjedde A., Gunn R.N., Holden J., Houle S., Huang S.C., Ichise M., Iida H., Ito H., Kimura Y., Koeppe R.A., Knudsen G.M., Knuuti J., Lammertsma A.A., Laruelle M., Logan J., Maguire R.P., Mintun M.A., Morris E.D., Parsey R., Price J.C., Slifstein M., Sossi V., Suhara T., Votaw J.R., Wong D.F., Carson R.E. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. Journal of Cerebral Blood Flow & Metabolism. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 20.Daws L.C. Unfaithful neurotransmitter transporters: focus on serotonin uptake and implications for antidepressant therapy. Pharmacology & Therapeutics. 2009;121:89–99. doi: 10.1016/j.pharmthera.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X., Margolis K.J., Gershon M.D., Schwartz G.J., Sze J.Y. Reduced serotonin reuptake transporter (SERT) function causes insulin resistance and hepatic steatosis independent of food intake. PLoS One. 2012;7:e32511. doi: 10.1371/journal.pone.0032511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serretti A., Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. Journal of Clinical Psychiatry. 2010;71:1259–1272. doi: 10.4088/JCP.09r05346blu. [DOI] [PubMed] [Google Scholar]
- 23.Journel M., Chaumontet C., Darcel N., Fromentin G., Tome D. Brain responses to high-protein diets. Advances in Nutrition. 2012;3:322–329. doi: 10.3945/an.112.002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi S., Disilvio B., Fernstrom M.H., Fernstrom J.D. Meal ingestion, amino acids and brain neurotransmitters: effects of dietary protein source on serotonin and catecholamine synthesis rates. Physiology and Behavior. 2009;98:156–162. doi: 10.1016/j.physbeh.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Peters J.C., Harper A.E. Acute effects of dietary protein on food intake, tissue amino acids, and brain serotonin. American Journal of Physiology. 1987;252:R902–R914. doi: 10.1152/ajpregu.1987.252.5.R902. [DOI] [PubMed] [Google Scholar]
- 26.Nagasawa M., Murakami T., Sato M., Takahata Y., Morimatsu F., Furuse M. Dietary animal proteins alter monoamine metabolism in the brain. Animal Science Journal. 2012;83:493–498. doi: 10.1111/j.1740-0929.2011.00987.x. [DOI] [PubMed] [Google Scholar]
- 27.Jahns L., Siega-Riz A.M., Popkin B.M. The increasing prevalence of snacking among US children from 1977 to 1996. Journal of Pediatrics. 2001;138:493–498. doi: 10.1067/mpd.2001.112162. [DOI] [PubMed] [Google Scholar]
- 28.Mesas A.E., Munoz-Pareja M., Lopez-Garcia E., Rodriguez-Artalejo F. Selected eating behaviours and excess body weight: a systematic review. Obesity Reviews. 2012;13:106–135. doi: 10.1111/j.1467-789X.2011.00936.x. [DOI] [PubMed] [Google Scholar]
- 29.Zizza C., Siega-Riz A.M., Popkin B.M. Significant increase in young adults' snacking between 1977–1978 and 1994–1996 represents a cause for concern! Preventive Medicine. 2001;32:303–310. doi: 10.1006/pmed.2000.0817. [DOI] [PubMed] [Google Scholar]