

Abstract
The emerging complexity of large macromolecules has led to challenges in their full scale theoretical description and computer simulation. Multiscale multiphysics and multidomain models have been introduced to reduce the number of degrees of freedom while maintaining modeling accuracy and achieving computational efficiency. A total energy functional is constructed to put energies for polar and nonpolar solvation, chemical potential, fluid flow, molecular mechanics, and elastic dynamics on an equal footing. The variational principle is utilized to derive coupled governing equations for the above mentioned multiphysical descriptions. Among these governing equations is the Poisson-Boltzmann equation which describes continuum electrostatics with atomic charges. The present work introduces the theory of continuum elasticity with atomic rigidity (CEWAR). The essence of CEWAR is to formulate the shear modulus as a continuous function of atomic rigidity. As a result, the dynamics complexity of a macromolecular system is separated from its static complexity so that the more time-consuming dynamics is handled with continuum elasticity theory, while the less time-consuming static analysis is pursued with atomic approaches. We propose a simple method, flexibility-rigidity index (FRI), to analyze macromolecular flexibility and rigidity in atomic detail. The construction of FRI relies on the fundamental assumption that protein functions, such as flexibility, rigidity, and energy, are entirely determined by the structure of the protein and its environment, although the structure is in turn determined by all the interactions. As such, the FRI measures the topological connectivity of protein atoms or residues and characterizes the geometric compactness of the protein structure. As a consequence, the FRI does not resort to the interaction Hamiltonian and bypasses matrix diagonalization, which underpins most other flexibility analysis methods. FRI's computational complexity is of at most, where N is the number of atoms or residues, in contrast to for Hamiltonian based methods. We demonstrate that the proposed FRI gives rise to accurate prediction of protein B-Factor for a set of 263 proteins. We show that a parameter free FRI is able to achieve about 95% accuracy of the parameter optimized FRI. An interpolation algorithm is developed to construct continuous atomic flexibility functions for visualization and use with CEWAR.
INTRODUCTION
Proteins have a diverse range of structures and functions. The understanding of protein structure, function, and dynamics has grown rapidly in the past few decades. The conventional dogma of sequence-structure-function2 has been seriously challenged by the discovery that many intrinsically disordered proteins can also be functional.18, 41, 50, 65 The study of disordered proteins is of essential importance due to their connections to sporadic neurodegenerative diseases, such as mad cow disease, Alzheimer's disease, and Parkinson's disease.18, 59 Disordered proteins are traditionally assumed to be highly flexible. However, a frequently neglected fact is that well-folded proteins are flexible as well. Folded proteins experience everlasting intrinsic motions due to possible Brownian dynamics, rapid local motions of amino acid side chains, and spontaneous collective fluctuations. Therefore, flexibility, i.e., the ability to deform from the current conformation under external forces, is an intrinsic property of all proteins.
One of the major challenges in the biological sciences is the prediction of protein functions from protein structures. One key to protein function prediction is the protein flexibility which strongly correlates with enzymatic activity in proteins, such as allosteric transition, ligand binding and catalysis, as well as the stiffness and rigidity which is crucial to structural proteins. For instance, in enzymatic processes, protein flexibility enhances protein-protein interactions, which in turn reduce the activation energy barrier. Additionally, protein flexibility and motion amplify the probability of barrier crossing in enzymatic reactions. Therefore, the investigation of protein flexibility at a variety of energy spectra and time scales is vital to the understanding and prediction of protein functions. Currently, the most important technique for protein flexibility analysis is X-ray crystallography. Among the almost 100 000 structures in the protein data bank (PDB), more than 80% are collected by X-ray crystallography. The Debye-Waller factor, or B-factor, can be directly computed from X-ray diffraction or other diffraction data. Although atomic B-factors are directly associated with atomic flexibility, they can be influenced by the variations in atomic diffractive cross sections and chemical stability during the diffraction data collection. Therefore, only the B-factors for specific types of atoms, say Cα, can be directly interpreted as their relative flexibility without corrections. Another important method for accessing protein flexibility is nuclear magnetic resonance (NMR) which often provides structural flexibility information under physiological conditions. NMR spectroscopy allows the characterization of protein flexibility in diverse spatial dimensions and a large range of time scales. About 6% of structures in the PDB are determined by electron microscopy (EM) which does not directly offer the flexibility information at present.
In addition to experimental technologies, theoretical approaches play essential roles in biomolecular flexibility analysis and prediction. For example, molecular dynamics (MD) simulations have dramatically expanded our understanding of the conformational landscapes of proteins, particularly conformations that are not directly accessible via other techniques, i.e., protofibrils, amyloid-like fibrils, amyloids, intrinsically disordered proteins, and partially disordered proteins. However, the dynamics of large proteins typically occurs at time scales that are intractable to MD simulations. Alternative approaches have also been developed in the past few decades, including normal mode analysis (NMA),8, 25, 36, 55 elastic network model (ENM),58 Gaussian network model (GNM),5, 6, 22 and anisotropic network model (ANM).4 In fact, these methods can be regarded as time-independent molecular mechanics (MM) and are connected to MD methods via the time-harmonic approximation.44 Protein flexibility and B-factors can be approximated respectively from the first few eigenvectors and eigenvalues of the connection matrix. Such low-energy eigenvalues reflect the long-time behavior of the protein dynamics beyond the reach of MD simulations.6, 8, 36, 55, 58 These approaches have been improved in many aspects including crystal periodicity corrections28, 34, 35, 54 and density-cluster rotational-translational blocking.20 These methods are relatively inexpensive particularly in their coarse-grained settings. Their computational complexity is typically dominated by that of the diagonalization of the Hamiltonian matrix, i.e., , where N is the matrix dimension and k ≈ 3. These approaches give rise to quantitative predictions of biomolecular flexibility and their applications are discussed in many review papers.19, 38, 53, 68 However, for the structures of excessively large protein complexes which are typically obtained via cryo-EM, more efficient methods are required to analyze their flexibility. Although often called elastic models or elasticity analysis in the literature, the aforementioned methods are still microscopic in origin, and are fundamentally different from the stress and strain analysis of a truly continuum elasticity theory.
Recently, knowledge based methods, such as neural networks,47 support vector regression,69 and two-stage support vector regression,43 have also been developed for flexibility analysis. These approaches typically utilize large protein data sets as input training data. Computational accuracy, reliability, and complexity of these methods depend on the training data set. Jacobs et al.31 have utilized techniques from graph theory to analyze the bond networks in proteins. Their approach employs both geometric and energetic criteria to identify the flexible and rigid regions.
Another class of approaches for biomolecular flexibility analysis utilizes phenomenological theories and/or continuum mechanics. Linear and nonlinear elasticity models have been proposed for excessively large biomolecules, such as membranes, molecular motors, microtubules, and protein complexes.32, 51 One of phenomenological approaches is the Willmore flow energy functional66 which is in terms of the square of the difference between two principle curvatures. This model intends to minimize the deviation of a membrane surface from the local sphericity. As a generalization of the Willmore energy functional, Canham10 and independently, Helfrich,27 proposed an elasticity model for cellular membranes. The free energy functional of membrane bending consists of the Gaussian curvature of the membrane surface and the square of the difference between the mean curvature and the spontaneous curvature of the membrane. The minimization of the Helfrich energy functional leads to the equilibrium shape of the membrane.42 According to the Gauss-Bonnet theorem, the Gaussian curvature in the free energy functional will contribute to an unphysical jump in the free energy whenever there is a topology change in the membrane morphology. Computationally, phase field models can be used to simulate membrane curvature formation and evolution.21 In the past decade, membrane curvature has been a popular research topic partially due to the fact that except for the curvature, there is very little other quantitative information associated with membranes and membrane protein/DNA interactions. Indeed, protein membrane interaction39 and membrane curvature sensing have received much attention.3, 26, 45 So far, both theoretical modeling and numerical simulation in the field have been mostly phenomenological and qualitative,9 partially due to the lack of more quantitative experimental data. Another class of models directly utilizes continuum elasticity for biomolecular flexibility analysis. Recently, Zhou et al.73 have proposed an elasticity model which allows the electrostatic force of biomolecules to influence membrane stress.
The other class of theoretical methods for flexibility analysis has been developed via multiscale formulations. These methods combine elastic mechanics and molecular mechanics to significantly reduce the number of degrees of freedom of large biomolecular systems.9 For example, the classical theory of elasticity for DNA loops is combined with the MD description of protein for protein-DNA interaction complexes.60 Recently, the continuum elastic modeling of the Canham-Helfrich type of energy functional has been coupled with MD simulations to investigate the complex elastic behavior of Hepatitis B virus capsids.49 Multiscale based flexibility analysis has a wide range of technical variability. In the best scenario, multiscale methods can take the advantage of each scale to achieve excellent modeling accuracy and computational efficiency. However, multiscale methods are typically technically demanding and computationally complex. A major issue in the field is how to go beyond the phenomenological domain and make these approaches quantitative and predictive. Consistency analysis and validation with experimental data are indispensable procedures. There is a need to further develop and validate innovative approaches for the flexibility analysis of biomolecular systems.
Recently, we have introduced a new class of multiscale models, differential geometry based multiscale approaches, for biological and chemical systems.62, 63, 64 The essential idea is to use the differential geometry theory of surfaces and the geometric measure theory as a natural means to separate the solvent domain from the macromolecular domains. A number of physical phenomena, including polar and nonpolar solvation, molecular dynamics, quantum mechanics, fluid dynamics, electrokinetics, electrohydrodynamics, electrophoresis, and elastic dynamics are considered in our multiscale models via a total energy functional and a variational strategy. By using the Euler-Lagrange variation, the self-consistently coupled Laplace-Beltrami equation and Poisson-Boltzmann equation is obtained for solvation analysis. For charge and mass transport, additional generalized Poisson-Nernst-Planck equations and/or Navier-Stokes equations are incorporated. Multiscale (implicit solvent) MM is utilized to allow local conformational changes and elastic dynamics is considered for excessively large chemical and biological systems. Step by step, our differential geometry based multiscale models have been carefully validated in the past few years.11, 12, 14, 15, 16, 17, 64 The first series of validations was done for multiscale solvation models.14, 15, 16, 17, 29, 57, 72 The Eulerian formulation,14 Lagrangian formulation,15 and quantum formulation are constructed16 for the solvation analysis of hundreds of small and large molecules, including nonpolar ones.17 The quantum formulation is able to considerably improve model accuracy. The robustness of our differential geometry based solvation approaches comes from a significant reduction in the number of free parameters that users must “fit” or adjust in applications to real-world systems.57, 70 In fact, our differential geometry based nonpolar model offers some of the best predictions of nonpolar solvation energies for a large number of compounds.17
Another series of validations was done on charge transport in realistic ion channels.64, 71 Continuum descriptions are applied to the solvent domain while channel proteins are treated in molecular detail. In our energy functional, nonpolar energy, polar (electrostatic) energy, chemical potential, and possibly fluid energy are considered on an equal footing. Non-electrostatic van der Waals (VDW) interactions among all the ions, and between ions and proteins, including size (steric) effects are accounted for in our treatment.64 In this multiscale paradigm, the non-equilibrium multiscale transport theory reduces to the multiscale solvation model at equilibrium. Very good agreements between our model predictions and experimental measurements have been attained.64
The other series of validations of our multiscale models was on the proton transport through membrane proteins.11, 12 Proton transport plays an important role in the molecular mechanism of biological energy transduction, sensory systems, and reproduction of influenza A viruses.13 Due to the small mass and size of protons, proton permeation across membrane proteins involves significant quantum effects.40, 46 However, the quantum mechanical treatment of all individual protons can be computationally expensive. A new density functional theory based on the Boltzmann statistics rather than the Fermi-Dirac statistics has been developed to describe proton dynamics quantum mechanically while implicitly treating numerous solvent molecules as a dielectric continuum. To account for the gating effect, membrane proteins are described in atomistic detail. Densities of all other ions in the solvent are approximated by using the Boltzmann distributions, which were introduced in our earlier work71 and have been independently confirmed by using Monte Carlo simulations.33 Excellent predictions of experimental current-voltage curves have been observed.11, 12 Currently, the Poisson-Boltzmann model, or the Poisson model when there is no salt, has been proved to be a successful continuum model for biomolecular electrostatics at the quantitative level.1, 7, 24, 52 One of main reasons for its success is the continuum modeling which avoids the time consuming molecular dynamics description. While another reason for its success is the atomic detailed static charge description—the atomic point charges or charge distributions. In contrast, elasticity models are qualitative and phenomenological at moment. It is believed that elasticity analysis would play a much more important role in quantitative modeling and computation of biomolecular flexibility had atomic rigidity information been appropriately incorporated.
The objective of the present work is to develop differential geometry based multiscale, multiphysics, and multidomain models for biomolecular flexibility analysis. A major focus is to develop the theoretical model of continuum elasticity with atomic rigidity (CEWAR). Indeed, in our previous formulations, flexibility, rigidity, and elasticity have not been analyzed in detail, partially because of the fact that it is often more important and highly necessary to treat the macromolecular domain with atomistic descriptions. However, this situation has changed since the introduction of a multidomain formalism,63 which allows a biomolecular complex to be divided into multiple domains and simultaneously treated by multiple physics descriptions. As a result, it is advantageous to include elastic dynamics in certain domains. In our elastic treatment of biomolecular complexes, proteins are assumed to have atomic rigidity or shear modulus. Therefore, a robust method for the extraction of protein atomic flexibility and rigidity information is required. Due to dynamical nature of the CEWAR, the atomic flexibility and rigidity information needs to be extracted in a most efficient manner for both equilibrium and non-equilibrium structures. The present work presents one of the most efficient methods, called flexibility-rigidity index (FRI), for protein B-factor prediction and flexibility analysis. The basic assumption underlying the FRI is that protein functions are entirely determined by the structure of the protein complex. The FRI provides an accurate measure of geometric compactness and topological connectivity of a protein structure at each atom or residue. Physically, the FRI reflects the local interaction strength. As such, it gives rise to accurate prediction of protein B-factors. The proposed FRI method does not require a stringently minimized structure, harmonic assumption, interaction potential, or matrix decomposition, nor does it involve any training procedure as that used in the knowledge based approaches. Its computational complexity is at most of order .
The rest of this paper is organized as follows. A multiscale, multiphysics, and multidomain model that involves elastic dynamics, electrostatic interactions, molecular mechanics, and chemical potential effects is presented in Sec. 2 to facilitate the discussion on flexibility and rigidity. Equations for the elastic dynamics, elastostatics, and elastic vibration of macromolecular complexes are introduced. In particular, we show how the microscopic analysis of flexibility and rigidity is utilized in the macroscopic analysis of elasticity. A new model for protein flexibility evaluation is introduced in Sec. 3. We first define a diagonal-free correlation matrix to analyze the topological connectivity between protein atoms. Additionally, atomic flexibility and rigidity indices of each protein atom are deduced from the correlation matrix. Molecular rigidity index and averaged molecular rigidity index are proposed. The protein B-factors are directly associated with atomic flexibility indices which give rise to a practical method for B-factor prediction. Finally, a volumetric atomic rigidity function is constructed from the atomic rigidity index. A similar definition is also proposed for the flexibility. In Sec. 4, extensive numerical tests are carried out to validate the proposed method for protein flexibility analysis and B-factor prediction. Careful comparison with experimental data justifies our new approach. The proposed theory and formulation also offer new approaches for the visualization of biomolecular rigidity and flexibility. This paper ends with concluding remarks.
ELASTICITY IN MULTISCALE-MULTIPHYSICS-MULTIDOMAIN MODELING
We consider a multiscale, multiphysics, and multidomain model for biomolecular complexes in solvent where one domain is described by the continuum mechanics of elasticity. Some parts of the biomolecular system are described by using the molecular mechanics. The solvent consists of various charged species and water. Fluid mechanics is used to describe possible fluid motion of the solvent. Electrostatic interactions are considered in the whole computational domain. We first provide a simplified description of the action functional, followed by the derivation of governing equations. Special attention is given to elastic analysis.
The action functionals
Elastic energies
Experimental measurements indicate that protein, DNA, and other biomolecular systems exhibit elasticity, i.e., they are able to deform under prescribed external forces and restore to their original states when the external forces are no longer applied. The amount of deformation under a given external force is determined by internal forces that oppose the deformation. The resistance to the internal force or the stiffness is measured by various elastic moduli, such as Young's modulus, the bulk modulus, and the shear modulus in elasticity theory. For a protein or biomolecule, the internal force or resistance is not uniform and is position dependent. Certain parts of the protein are highly flexible while other parts are highly rigid, as indicated by the variation in protein B-factors.
To analyze the elasticity of arbitrarily shaped biomolecules, we consider the displacement w of a point r to its new position in
| (1) |
The difference between the squares of infinitesimal changes is
| (2) |
where the Einstein summation notation is used to simplify tensorial quantities and σij is the strain tensor
| (3) |
The strain tensor describes the change of a point between before and after the elastic deformation. For relatively small deformations, one omits the term that is nonlinear in w and obtains a linear strain tensor
| (4) |
Obviously, the linear strain has computational advantages.
We denote the elastic potential energy density in the Einstein notation as
| (5) |
where λE is the lame parameter, describing the compressibility of the elastic macromolecule, and μE is the shear modulus, or rigidity, describing the stiffness of the elastic macromolecule under external force. In the present work, an atomistic description of μE = μE(r) will be provided.
The kinetic energy density of the elastic system is given by where ρE is the mass density of the elastic macromolecule and denotes the velocity of the displacement. In general, the difference between the kinetic energy density and potential energy density gives rise to the Lagrangian, a functional for variational derivation of governing equations.
Action functional of a multiscale multiphysics and multidomain model
Let us label the continuum solvent, molecular mechanics, and elasticity descriptions, respectively, by S, M, and E so that SI (I = S, M, and E) are the characteristic functions of the solvent, molecular, and elastic domains. Similarly, pI, γI, εI, and ϱI are, respectively, pressures, surface tensions, dielectric constants, and charge densities associated with I = S, M, and E. The total action functional of our multiscale multiphysics and multidomain model is given by Ref. 63:
| (6) |
where US includes all the non-electrostatic (or nonpolar) interactions involving the solvent,63 Φ is the electrostatic potential, and ρα, ρα0, qα, and μα0 are, respectively, the density, bulk density, charge, and relative reference chemical potential of αth component of the solvent. Here, ρ = ∑αρα is the total solvent mass density, v is the flow stream velocity, and μf is the viscosity of the fluid. The Einstein notation is used in the fluid potential energy. Here, ρj = mjδ(zj − xj) is the mass density of the jth atom in a molecular dynamics description, with mj and xj being the mass and the macroscopic position of the jth atom, respectively. Here, UM(z) and are, respectively, the potential and kinetic energy densities of the jth atom with . We use the short-hand notations and with Na the total number of atoms in the domain of molecular dynamics description. We assume that the potential interactions UM(z) include all bonding and non-bonding components as used in implicit MD calculations.23, 37 The integration is over the macroscopic variable r, microscopic variable z, and time t.
Physically, in Eq. 6, the first is the nonpolar solvation free energy, followed by the electrostatic free energy in the second row, the chemical potential related energy in the third row, the Lagrangian of the fluid dynamics in the fourth row, the Lagrangian of the molecular dynamics in the fifth row, and finally the Lagrangian of the elastic dynamics in the last row.
Governing equations
It has become a standard procedure to derive governing equations by a total variation.62, 63 We briefly discuss these equations below.
Generalized Laplace-Beltrami equation
Using the Euler-Lagrange variation, we derive two generalized Laplace-Beltrami equations, respectively, for characteristic functions of molecular mechanics domain and the elastic domain
| (7) |
where driven terms VM and VE are, respectively, given by
| (8) |
and
| (9) |
Surface of the solvent is determined via relation SS = 1 − SM − SE.
Generalized Poisson equation
The electrostatic potential (Φ) is determined by the generalized Poisson equation
| (10) |
where ε(S) = SSεS + SMεM + SEεE is the generalized permittivity function.
Generalized Nernst-Planck equation
The derivation of the generalized Nernst-Planck involves the variation with respect to solvent species ρα and the generalized Fick's law62, 63, 64
| (11) |
where is the density production of α species per unit volume in the jth chemical reaction.62 Equation 11 describes generalized mass conservation law in which the rate of change of each mass density is balanced by convection, diffusion, and reactions.
Generalized Navier-Stokes equation
The total variation of functional 6 gives rise to the generalized Navier-Stokes equation
| (12) |
where flow stress tensor T can be expressed as
| (13) |
where symbol T denotes the transpose. The force FE has the form
| (14) |
In the inner solvent domain (i.e., SM = SE = 0), Eq. 14 reduces to the standard Navier-Stokes equation for incompressible flows except for an extra force term which is due to solvent-solvent interactions. In fact, away from the flow boundary, becomes negligible too.
Generalized Newton equation
As discussed in our earlier work,62 the variation with respect to δzj leads to Newton's equation for the molecular mechanics
| (15) |
where the force term is given by
| (16) |
| (17) |
| (18) |
| (19) |
Here, , and are, respectively, solvent-solute interaction force, reaction field (RF) force, and potential interaction force due to atomic interactions.
Elastic dynamics
Note that in the present work, the rigidity μE = μE(r) is continuous function with atomic rigidity information. Considering such a position dependence, the governing equation for the elastic dynamics of the macromolecule can also be derived by variation
| (20) |
where fE is the total force
| (21) |
where
| (22) |
| (23) |
| (24) |
Here, is the fluid-structure interaction (FSI) force and is the RF force which is due to the charge distributions of biomolecules in the molecular dynamics domain and the elastic domain. Term is the heterogeneous (HG) force due to the inhomogeneity of the biomolecules. If we assume that λ is independent of position (∇wλ = 0), or the biomolecule is incompressible (∇ · w = 0), we can drop the first part of the HG force. Additionally, in the elastic domain, one has SE = 1.
To simplify Eq. 20, we make use of the stress tensor of the elastic molecule62
| (25) |
Obviously, the stress tensor is symmetric with respect to labels i and j. By means of the stress tensor, the elastic dynamics Eq. 20 is
| (26) |
Clearly, Eq. 26 is a generalization of the classical elastic dynamics. It is essentially the Newton's equation of motion for elastic molecules and is parallel to the Newton's equation for atomistic molecular dynamics. The product of mass and acceleration is balanced by the internal friction , external forces , and internal forces ( − Φ∇wϱE, (∇ · w)2∇wλ, and ). The dependent variable in Eq. 26 is a vector in , while the spatial dimension of molecular dynamics is 3Na.
The elastostatic state is given by
| (27) |
Here, Eq. 27 describes the shape of elastic biomolecule at the balance of internal friction and external force. It is meaningful for the equilibrium state but may also be used for a non-equilibrium conformation.
Assume that the elastic dynamics of biomolecules admits a time-harmonic solution, i.e., its time dependence is proportional to eiωt, we have the following eigenvalue equation:
| (28) |
Therefore, the diagonalization of the operator in Eq. 28 for biomolecules produces eigenvalues and eigenvectors. The latter can be used to analyze and visualize the vibration of macromolecules. The low order vibrational modes reflect protein collective motions.
In the present multiscale, multiphysics, and multidomain theory, the elastic dynamics 26 is coupled to the generalized Laplace-Beltrami equation 7, Poisson equation 10, Nernst-Planck equation 11, Navier-Stokes equation 12, and Newton's equations 15. There are many ways to simplify this coupled system. For example, one can omit the molecular dynamics by assuming a static conformation in the MD domain, neglect fluid dynamics when there is no flow velocity and disregard the Nernst-Planck equation when there is no ion permeation. In fact, one can also utilize sharp interface and dielectric approximation to skip the Laplace-Beltrami equation. However, more detailed discussion along this line is beyond the scope of the present work and will be carried out in our future work.
FLEXIBILITY AND RIGIDITY
In this section, we present a microscopic theory for molecular rigidity and flexibility in light of CEWAR. We propose a FRI method for protein flexibility analysis and B-factor prediction. The FRI is a structure, or geometry, based method, and does not involve the interaction Hamiltonian used in energy based approaches.
Rigidity
The behavior of the stress tensor 25 determines the dynamics and elastostatics of the biomolecular elasticity. Currently, elastic moduli λE and μE are taken to be constants in most theoretical modeling and computational experiments.51 The lame parameter λE is typically relatively small for biomolecules because of the incompressibility of macromolecules under physiological condition. The shear modulus μE, i.e., rigidity, should vary from position to position so that the continuum elasticity with atomic rigidity can play an important role in the elastic analysis of excessively large macromolecules.
Consider a macromolecule of N particles (or atoms) with a conformation vector , where is the position of jth particle or atom. Let us denote ‖ri − rj‖ the Euclidean distance between particles ri and rj. We assume that the correlation between particles ri and rj has the form
| (29) |
where ηij are characteristic distances between particles and Φ(‖ri − rj‖; ηij) is a correlation kernel, also called an unnormalized density estimator.61
In general, the correlation kernel is a real-valued, smooth, and monotonically decreasing function. It has properties
| (30) |
| (31) |
Many decaying radial basis functions can be used for correlation kernels. Typical examples include generalized exponential function
| (32) |
and generalized Lorentz function61
| (33) |
Certainly, many other alternative choices, such as delta sequence kernels of the positive type discussed in an earlier reference61 can be employed as well. For example, one can use the product of exponential and Lorentz functions
We shall not exploit all alternatives at present because our goal is to establish a theoretical framework. The basic idea is that the correlation between any two particles should decay according to their distance.
We construct a N × N symmetric correlation map C = {Cij}. The correlation map contains topological connectivity between atoms and thus, is also called connectivity. The behavior of the correlation map C is explored in Sec. 4.
For the shear modulus in the continuum elasticity analysis, we need a continuous function defined in the elastic domain (ΩE = {r|SE(r) ≠ 0}). To this end, we define the correlation from an arbitrary point r to the jth particle
| (34) |
where r is in the proximity of ith particle. We define an atomic rigidity function μ(r) as
| (35) |
where wj(r) are particle-type related weights. The atomic rigidity function μ(r) measures the local rigidity or local stiffness at point r.
The average rigidity (or averaged rigidity index function) can be calculated by
| (36) |
Therefore, parameter wj in Eq. 35 can be determined by a comparison of with its experimental value, the shear modulus, for a given macromolecule. This procedure can help the parametrization of wj in Eq. 35, though wj should also reflect the difference in different types of atoms in a macromolecule.
It is important to have a discrete representation of rigidity on a set of atoms or particles. To this end, we define an atomic rigidity index as
| (37) |
where wij = wj(ri). It is convenient to further define the molecular rigidity index as a summation of all the atomic ones
| (38) |
Obviously, the molecular rigidity index of a given macromolecule is a direct measure of its total interaction strength in a general sense.
For the purpose of comparison among different molecules, we further define an averaged molecular rigidity index
| (39) |
Similar to the Wiener index, both the molecular rigidity index and the averaged molecular rigidity index must strongly correlate with many physical properties, such as molecular thermal stability, density (compactness), boiling points of isomers, the ratio of surface area over volume, surface tension, bulk modulus, etc. However, a thorough investigation of these aspects is beyond the scope of the present work and is left for future work.
Flexibility
For polyatomic molecules, we should have μi > 0. Therefore, we can define an atomic flexibility index as
| (40) |
Atomic flexibility indices {fi} of a macromolecule must be proportional to its B-factor {Bi}
| (41) |
where are theoretically predicted B-factors. Here, constants a and b do not depend on index i and can be determined by a simple linear regression. The procedure outlined above is perhaps the simplest one for B-factor prediction. Unlike ENM, GNM, ANM, and many other methods, the proposed approach by-passes the matrix diagonalization (or decomposition) procedure in conventional B-factor prediction and flexibility analysis. It is well known that the computational complexity of matrix diagonalization is asymptotically close to , while that of a two-parameter linear regression given in Eq. 41 is asymptotically of . The construction of the correlation map C can be made linear in complexity with appropriate spatial index techniques, although the construction of a spatial database may be of in complexity. Nevertheless, our FRI based B-factor prediction gives rise to a dramatic reduction in the computational complexity compared with conventional approaches. We expect that the proposed method will outperform other methods in computational efficiency and be potentially useful for the flexibility analysis of excessively large macromolecules.
Let us define a molecular flexibility index as a sum of atomic indices
| (42) |
and an averaged molecular flexibility index
| (43) |
Similar to the Wiener index, the molecular flexibility index or the averaged molecular flexibility index reflects the atomic geometric irrelevance and topological disconnectivity in a molecule, and must strongly correlate with energy and disorderliness. These aspects will be explored in our future work.
Finally, atomic flexibility functions can be defined in two ways. For example, one simply defines
| (44) |
It is reasonable to assume that the atomic rigidity function is non-singular in the domain ΩE. On the other hand, it is convenient to define the atomic flexibility function by using a set of B-factors {Bj}
| (45) |
where Ψ(‖r − rj‖) is a general interpolation kernel. The B-factors can be obtained either from experimental data or from theoretical predictions. When there are different types of atoms, the experimental B-factors should be corrected according to diffraction cross sections before they are interpreted as atomic flexibility. Such a correction can be by-passed when only the same type of atoms, i.e., Cα, is involved.
NUMERICAL EXPERIMENTS
In this section, we validate the concepts, demonstrate the usefulness, and explore the efficiency of the proposed theory and algorithm for flexibility and rigidity analysis. We first analyze the correlation map, which reveals the topological connectivity among atoms in a macromolecule. The prediction of B-factor based on the proposed atomic flexibility index is demonstrated. Finally, we illustrate the use of atomic rigidity function and atomic flexibility function in protein visualization.
In the present experiments, we employ a coarse-grained representation with amino acid residues and consider only Cα atoms. We can set weights wij = 1 and assign a common value to characteristic length parameter ηij = η in Eq. 37
| (46) |
We consider only the generalized exponential kernel
| (47) |
and the generalized Lorentz kernel in the present numerical experiment
| (48) |
By appropriate selection of power υ, κ, and η, we actually end up with a parameter-free atomic flexibility index
| (49) |
In general, Eqs. 46, 47, 48 are used for computing correlation maps and their combination with Eqs. 40, 41 provides the FRI scheme for B-factor predictions.
To quantitatively assess the performance of the proposed FRI method for the B-factor prediction, we consider the correlation coefficient
| (50) |
where are a set of predicted B-factors by using the proposed method and are a set of experimental B-factors downloaded from the Protein Data Bank (PDB). Here, and are the statistical averages of theoretical and experimental B-factors, respectively.
Correlation map
Similar to the cross correlations of the GNM and other methods, FRI correlation maps computed using Eq. 46 qualitatively reflect the three-dimensional structure of a protein. As a consequence, distinct secondary structures such as α helices and β-sheets exhibit characteristic patterns. After some studying of the patterns, it is possible to approximate a protein's secondary and tertiary structures from the patterns of the correlation map alone. However, unlike the cross correlations of the GNM, the FRI correlation maps are able to further offer quantitative structural information. In fact, since the kernel used to generate the map is known, the distances between all atoms can be calculated and the three-dimensional structure can be reconstructed from the correlation map. Figure 1 displays four examples of correlation maps next to their corresponding three-dimensional structure. The scale-bars of the correlation maps include distance values to emphasize the preservation of the 3D structural information.
Figure 1.

Correlation maps (left column) and secondary structure representations (right column) for proteins 1C26, 1BK2, 1PGA, and 1NH9, from top to bottom. Correlation maps are generated using Eq. 48 with υ = 2.5 and η = 1.0 Å. Secondary structure visualizations are generated with VMD.30 Colors represent distance and correlation values for each pair of atoms. Red represents nearby atoms with high correlation values and blue represents distant atoms with low correlation values. The residue numbers for each Cα are listed along the x- and y-axes. The protein are displayed in VMD's “new cartoon” representation and colored by secondary structure determined by STRIDE. The color scheme for secondary structure is: Purple – α helix, blue – 3(10) helix, yellow – β-sheet, cyan – turn, white – coil. The correlation map of an α helix gives rise to a widened diagonal pattern (see the first row). In contrast, the correlation map of paired beta sheets has two patterns. One is a line that is perpendicular to the diagonal with distances around 5-10 Å for a pair of anti-parallel beta sheets (see the first and second beta sheets in the third row). The other is a line that is parallel to the diagonal with distances around 5-10 Å for a pair of parallel beta sheets (see the first and fourth beta sheets in the third row).
As stated previously, each secondary structure exhibits a distinct pattern in our correlation maps. The pattern for an α helix is shown in the first row of Fig. 1. The α helix creates a band of high correlation extending about 4 amino acids in either direction from the diagonal. The correlation has a local maximum at the third neighbor residue, due to the structure of the α helix (3.6 amino acid residues per turn). Therefore, the peak at the third residue serves as another signature of an α helix in the FRI correlation map. An increase in correlation between two such neighboring atoms compared to other neighboring pairs indicates the interaction of the α helix and another component. For example, in the third row of Fig. 1, the correlation strength between 29th Cα and 32th Cα is higher, due to interaction of 29th Cα with the third and fourth beta sheets. This is an example of how this type of correlation kernels reflects tertiary structure information.
Other folds such as β-sheets are also easily identified by distinct patterns. One can easily distinguish parallel β-sheets from anti-parallel β-sheets by their patterns with this method. The second row of Fig. 1 is a good example of the pattern generated by anti-parallel β-sheets. Anti-parallel β-sheets appear as lines that are perpendicular to the diagonal of the map and the intersection of the two lines of high correlation are the turns between each β strand. Parallel β-sheets appear as lines parallel to the diagonal. In the third row of Fig. 1, an anti-parallel β-sheet is formed by the first and last ten amino acids resulting in a line in the top left and bottom right of the correlation matrix.
The last two rows of Fig. 1 both display complex patterns which reflect not only secondary structure information but also the three-dimensional arrangement of the secondary structure features. Clearly, from the last correlation map, the first β-sheet interacts strongly with the first α helix and the second β-sheet in a parallel manner. It also interacts to a lesser degree with the second α helix and with the last β-sheet in an anti-parallel manner. These patterns and the stabilizing forces from the interactions they represent are lost if one uses a contact or Kirchoff matrix based method instead of a monotonically decreasing radial basis function based correlation map.
FRI based B-factor prediction
To further validate our FRI method, we compare the B-factor predictions with the experimental B-factors from protein X-ray crystallography experiments as shown in Eq. 50. A set of 263 proteins was collected from the PDB with preference for high resolution (1.5 Å) protein-only structures that lack structural co-factors. The impact of co-factors on protein stability requires an all atom model and is a topic that will be explored in our future work. The set of 263 proteins was converted to a Cα only format and when atoms have multiple coordinates with occupancy <1.0 the highest occupancy coordinate was kept and all others were discarded. This is a potential source of error in the B-factor predictions. However, some proteins with multiple coordinates for atoms were among the highest scoring which suggests that the impact in most cases is small.
The correlation coefficients of B-factor prediction are displayed in Fig. 2 for both exponential (expo) and Lorentz kernels. Each protein was tested with both the exponential and Lorentz correlation kernels across a range of parameter values of κ and η for the exponential kernel and υ and η for the Lorentz kernel. Correlation coefficient scores for B-factor predictions below 0.5 account for just 19 out of 263 proteins for the Lorentz kernel based FRI and 14 out of 263 for the exponential kernel based FRI and are not shown in Fig. 2. The reasons for these low scores are the subject of future research and are likely related to the influence of crystal packing effects, structural ligands and side-chain effects that are not approximated well by the Cα course grained model. The accuracy of B-factor prediction is also dependent upon the quality of the experimental data. If multiple coordinates are reported for an atom along with multiple B-factors, then we do not have high confidence in the B-factor and thus the prediction will appear to be less accurate.
Figure 2.
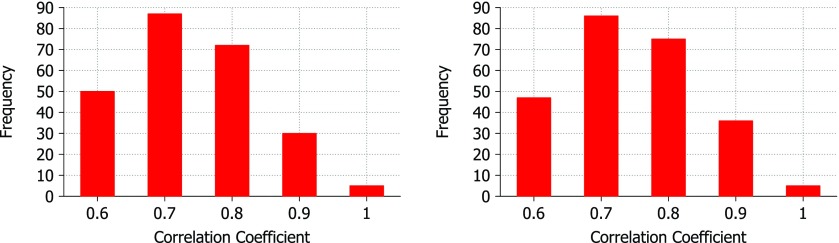
Correlation coefficients for experimental vs predicted B-factors using the Lorentz kernel (left) and exponential (right) kernel. The test set consists of 263 Cα only PDB files. Scores below 0.5 are not shown. For the Lorentz kernel, υ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 1.0 Å to 40.0 Å at an interval of 1.0 Å. For the exponential kernel, κ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 0.5 Å to 20.0 Å at an interval of 0.5 Å.
A comparison of the experimental vs predicted B-factors for two proteins, 1DF4 and 2Y7L, is shown in Fig. 3 to demonstrate the accuracy of our FRI method. These two proteins were in the top five highest correlation coefficients for B-factor predictions using the exponential (2Y7L: 0.928, 1DF4: 0.909) and Lorentz (2Y7L: 0.928, 1DF4: 0.917) kernels. It can be seen from the correlation scores and Figs. 23 that both correlation kernels give similar results, especially for these highly accurate predictions.
Figure 3.

Experimental B-factors (black) vs predicted B-factors (red) using the Lorentz (top) and exponential (bottom) correlation kernels. The structures used for comparison are 1DF4 (left) and 2Y7L (right). For these comparisons, the optimal parameters were used for υ, κ, and η based on the parameter searches for each correlation kernel. For the Lorentz kernel, υ = 1.5 and η = 2.0 Å are the parameters used for 1DF4 and υ = 1.5 and η = 19 Å are used for 2Y7L. For the exponential kernel, κ = 0.5 and η = 1.0 Å are employed for 1DF4 and κ = 0.5 and η = 2.5 Å for 2Y7L.
B-factor prediction was calculated for each protein at a range of parameter values in each kernel. The Lorentz kernel requires parameters, υ and η, while the exponential kernel requires κ and η. The aim is to find values for these parameters that are suitable for most or all proteins so that the method may be made parameter free. The parameters which result in the highest correlation coefficient for each protein are displayed in Figs. 46 for the Lorentz and exponential kernels, respectively.
Figure 4.
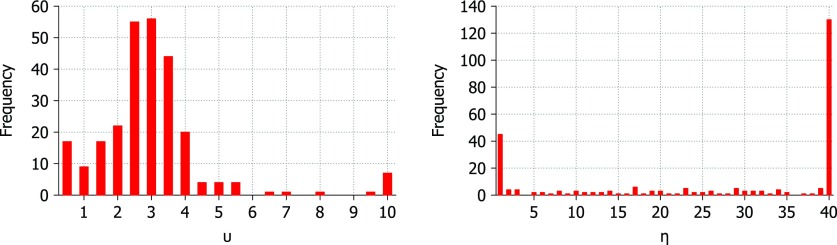
Optimal υ parameter value for 263 proteins using the Lorentz correlation kernel. B-factor prediction was calculated for υ values ranging from 0.5 to 10 at an interval of 0.5 and η values ranging from 1.0 Å to 40.0 Å at an interval of 1.0 Å.
Figure 6.
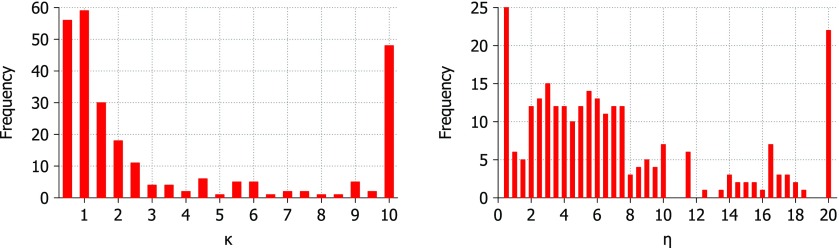
Optimal parameters for 263 structures using the exponential correlation kernel. Here, κ values range from 0.5 to 10.0 at an interval of 0.5. η values range from 0.5 Å to 20.0 Å at an interval of 0.5 Å.
The optimal value for υ in the Lorentz kernel is found to be near 2.5 for most proteins in the test set. The optimal value for η is typically the highest or lowest tested. The results of the parameter search for υ and η are shown in Fig. 4. This result is a close match to the findings of Yang et al.67 and their parameter free ENM (pfENM) model. In the pfENM, spring constants are scaled by an inverse power. Yang et al.67 tested powers 1-10 and found second and third inverse power relationships were the most accurate for B-factor predictions.67 In our study, we also test non-integer powers over the range 0.5-10.0 and come to a similar conclusion. The optimal value for υ is plotted against the optimal value for η and colored by the size of protein in Fig. 5. There is no clear pattern based on protein size except that some smaller proteins (under 100 atoms) prefer very high values of υ which may be due to a lack of long range interactions.
Figure 5.

Phase diagram for Lorentz kernel optimal parameter values υ and η colored by the size of structure and with shapes corresponding to correlation coefficient. Diamond − 0.5, downward triangle − 0.6, upward triangle − 0.7, square − 0.8 , circle − 0.9. υ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 1.0 Å to 40 Å at an interval of 1.0 Å.
For the exponential kernel, the optimal κ value for most proteins is between 0.5 and 1 while the optimal η values are more spread out with the majority of proteins having optimal η values from 0.5 Å to 8 Å. This ambiguity in the optimal parameter value makes the choice of parameters for a parameter free version difficult, however, the testing of the parameter free exponential kernel method shows that it performs as well as the parameter free Lorentz kernel methods. The optimal values for κ and η for all proteins in the test set are shown in Fig. 6. Optimal values for κ are 0.5 or 1.0 in most cases with a significant peak at κ = 10 which is the highest value tested. Optimal values for η are more varied and there is no clear choice for a parameter free version. There is a large peak at the highest η value tested (η = 20 Å) as there was for κ, however, these two peaks do not correspond to the same set of proteins. This point is illustrated in Fig. 7 which compares κ and η values. Figure 7 also shows that there is no relationship between number of atoms or correlation coefficient and the parameters κ and η. To further inform our choice of parameters for the parameter free exponential method, we look at the patterns of correlation scores for every κ and η value combination in Fig. 8. The parameter maps show that for most proteins the choice of κ is most important and that when κ ⩽ 1 there are many choices for η that result in very similar correlation coefficients.
Figure 7.

Phase diagram for exponential kernel optimal parameter values κ and η colored by the size of structure and with shapes corresponding to correlation coefficient. Diamond − 0.5, downward triangle − 0.6, upward triangle − 0.7, square − 0.8, circle − 0.9. κ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 0.5 Å to 20 Å at an interval of 0.5 Å.
Figure 8.

Complete results of optimal parameter searches using the exponential correlation kernel for structures 1DF4 (top left), 2Y7L (top right), 2Y9F (bottom left), and 3LAA (bottom right). Structures 1DF4 and 2Y7L (top) represent the high scoring structures, those with scores near 0.9. Structures 2Y7L and 3LAA (bottom) show the typical pattern for correlation scores based on parameter values for the majority of proteins. κ values range from 0.5 to 20.0 at an interval of 0.5 and η values range from 0.5 Å to 20 Å at an interval of 0.5 Å.
To test parameter free versions of the FRI method, we chose υ = 2.5 and η = 1.0 Å for the Lorentz kernel and κ = 1.5 and η = 5.0 Å for the exponential kernel. These choices were made based on the parameter searches and limited tests of various parameter values. In Fig. 9, we compare the exponential and Lorentz kernel performance based on correlation coefficients from B-factor prediction. The correlation coefficients were highest overall when using the exponential kernel with optimized parameters. The average correlation coefficient of B-factor prediction using the exponential kernel is 0.681 using optimal parameters and 0.627 using the parameter free version. The average correlation coefficient of B-factor prediction using the Lorentz kernel is 0.668 using optimal parameters and 0.627 using the parameter free version. The difference between the exponential and Lorentz kernels is small when using optimized parameters with an average deviation of just 0.0182. The parameter free versions of the kernels also produce very similar correlation coefficients with an average deviation of 0.0365.
Figure 9.

Comparison of correlation coefficients calculated using optimal parameters for both Lorentz and exponential correlation kernels. Average deviation = 0.0182 (left) and 0.0365 (right). For the Lorentz kernel optimal parameter search, υ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 1.0 Å to 40.0 Å at an interval of 1.0 Å. For the exponential kernel parameter search, κ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 0.5 Å to 20.0 Å at an interval of 0.5 Å. The parameter free Lorentz kernel uses υ = 2.5 and η = 1.0 Å and the parameter free exponential kernel uses κ = 1.5 and η = 5.0 Å.
The parameter free Lorentz and exponential kernels appear to have similar performance and these results do not indicate a clear advantage in using either kernel. In Fig. 10, we compare the correlation coefficients from the parameter free and optimized versions of the method for both correlation kernels. In each case, the optimized method outperforms the parameter free method no matter which kernel is used. Again this suggests that neither kernel has an advantage over the other for this method. The maximal average deviation among these methods is 0.0549, meaning that the parameter free exponential kernel captures 94% of the best results generated by optimized Lorentz kernel for this set of proteins. Similarly, the parameter free exponential kernel captures 94% of the best results from the optimized exponential kernel. It is worthwhile to note that the parameter free Lorentz kernel (υ = 2.5 and η = 1.0 Å) is able to capture 95% of the best results generated by either the optimized exponential or Lorentz kernel for this set of proteins. Therefore, it appears that the both parameter free kernels are very robust for practical applications.
Figure 10.

Comparison of correlation coefficients calculated using optimal parameters and parameter free versions of the FRI. The optimized correlation coefficients are the highest scoring from a parameter search. For the Lorentz kernel optimal parameter search, υ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 1.0 Å to 40.0 Å at an interval of 1.0 Å. For the exponential kernel parameter search, κ values range from 0.5 to 10.0 at an interval of 0.5 and η values range from 0.5 Å to 20.0 Å at an interval of 0.5 Å. The parameter free Lorentz kernel uses υ = 2.5 and η = 1.0 Å and the parameter free exponential kernel uses κ = 1.5 and η = 5.0 Å. The line y = x is shown for reference. Points on the line indicate little or no difference between optimized parameters and the parameter free results. Average deviations are 0.0410, 0.0549, 0.0463, and 0.0540 (from left to right and from top to bottom).
Rigidity and flexibility visualization
From the above analysis, the rigidity and flexibility indices can be obtained at coordinates of Cα atoms in the protein. Such values can be utilized directly for visualization. For the purpose of visualization, it is sufficient to plot either rigidity or flexibility. A large value of the flexibility index can be represented by a large atomic radius in the visualization while a small flexibility index corresponds a small atomic radius. Therefore, we scale atomic van der Waals radii by their flexibility indices as shown in Fig. 11 for 1QD9. Clearly, Cαs located near molecular boundary are more flexible.
Figure 11.

Cα atoms of 1QD9 in VDW representation scaled by predicted B-factor (both images) and colored with electrostatics (right). Larger VDW radii represent more flexible atoms such as those near the surface of this soluble protein. Smaller VDW radii represent more rigid atoms such as those in the core of the protein. On the right, atoms are colored by electrostatics revealing two charged domains. First, the flexible outer amino acids have some areas of positive charge that interact with the bulk solvent. Second, a highly negatively charged portion of the protein core is highlighted in red. These charges are stabilized by internal water molecules.
Additionally, the flexibility index can be visualized together with electrostatic potential. Specifically, the flexibility is represented by the atomic size while the electrostatics is illustrated by color as shown in the right chart of Fig. 11. There is a correlation between flexibility and partial charge at the protein outer surface—charged residues are more rigid. From these figures we see the image of a typical soluble protein with flexible, partially charged residues on the solvent-solute boundary and a less flexible, rigid core. It is well-known that the partially charged flexible outer protein surface is responsible for many protein functions in enzymes, cell signaling, and ligand binding. Interestingly, this soluble protein has a highly charged core made up of many negatively charged residues interacting with a network of water molecules. This results in a negatively charged, rigid core which is represented by small, red VDW spheres.
Furthermore, in order to study the elastic dynamics, elastostatics, and collective motion of a macromolecule, the continuous atomic rigidity and flexibility functions are required in our multiscale multiphysics multiphysics and multidomain models. The spatially scattered information at each Cα coordinate needs to be interpolated into continuous atomic rigidity and flexibility functions. In this work, we employ the modified Shepard's method to interpolate rigidity and flexibility values at Cα coordinates to build their continuous functions.48, 56 The essence of Shepard's method is to blend local interpolants with locally supported weight functions. For example, the atomic flexibility function can be expressed as
| (51) |
where the locally supported weight function is defined as
| (52) |
| (53) |
Here, Ri > 0 is a constant radius with ith Cα as its center. Its value varies with i so as to include different numbers of points into its influence domain when it is necessary.56
Our input data are a set atomic flexibility indices {fi} or the predicted B-factors located at Cαs. We denote r = (x, y, z), r ∈ SE a general position inside the elastic domain of a macromolecule, and the local interpolant is a nodal function defined as
| (54) |
where aij are coefficients and Qi(r) is a quadratic polynomial function which interpolates the predicted B-factors at neighboring set of Cα locations, namely,
| (55) |
where δij is the Kronecker delta function. For a given ith Cα, Eq. 55 is repeatedly employed on all Cαs within the given sphere of radius Ri and results in a number of algebraic equations. The algebraic equations are solved by using the weighted least square method, which determines coefficients aij. For sufficiently large data, we can choose 32 surrounding atomic flexibility indices to fit coefficients.56 Note that the atomic rigidity function (μ(r)) can be constructed in the same manner by replacing with μj.
In Fig. 12, we compare an atomistic and a continuous representation for flexibility of protein 1QD9. The molecular surface on the left is colored by X-ray B-factors, while the molecular surface on the right is colored by the interpolated flexibility values. Overall, the interpolated values mimic the B-factor pattern closely. However, the predicted flexibility at the inner ring of the structure is higher than that given by X-ray B-factors due to the fact water molecules fill part of the inner core in the full structure. The B-factor color map is discontinuous. In contrast, the flexibility map generated with the FRI method has the advantage of being continuous both on the surface and in the interior of the protein. The atomic rigidity function and atomic flexibility function constructed in the present work will be utilized to study macromolecular elastic dynamics, elastostatics, and elastic vibration in our future work.
Figure 12.

The molecular surface of Protein 1QD9 colored by B-factor (left) and continuous FRI representation (right). The flexibility index is calculated using the Lorentz method with υ = 2.5 and η = 1.0 Å. Images generated by VMD using BWR color bar and scale 10-50 for B-factors and 0.75–0.90 for the flexibility index. In both images, blue regions indicate low flexibility and red regions indicate high flexibility. On the left, B-factor is an atomistic representation of flexibility. On the right, FRI is used to predict flexibility and the continuum representation is mapped to the protein surface. The continuum prediction matches the experimental flexibility pattern closely except for near the core of the protein which contains some structural water not included in our model.
CONCLUSION
This work puts forward a multiscale, multiphysics, and multidomain model for the theoretical description and computer simulation of large macromolecular complexes. The multidomain setting enables the simultaneous multiscale and multiphysical treatment of biomolecular systems. In a special example, we consider a few physical descriptions, including nonpolar solvation, electrostatic interaction, multiple charged species, fluid flow, molecular mechanics, and elasticity. Of these descriptions, molecular mechanics is the only one defined at a microscopic level, while the rest are described using macroscopic theories. The interfaces between various domains are characterized by Laplace-Beltrami flows. The total energy functional is utilized to assemble various physical descriptions on an equal footing. The Euler-Lagrange variation is utilized to derive coupled governing equations for various physical descriptions. Apart from Laplace-Beltrami equations for interfaces, the Poisson-Boltzmann equation, the Navier-Stokes equation, Nernst-Planck equations, the elastic equation, and Newton's equations are obtained, respectively, for electrostatics, fluid flow, ion densities, elastic dynamics, and molecular mechanics in the model. A distinguishing feature of the present theory is that the elasticity theory is made non-uniform. Unlike the usual continuum elasticity analysis which utilizes a uniform shear modulus or rigidity, the present work introduces non-uniform shear modulus based on flexibility and rigidity analysis of macromolecules. This approach, called CEWAR, incorporates microscopic rigidity information in continuum elasticity analysis. The essential idea is to decouple the dynamics complexity of macromolecular system from its static complexity, such that the time-consuming molecular dynamics of the macromolecular system is replaced with a continuum elastic dynamics, while the relatively low-cost static analysis is computed with atomic rigidity.
We propose a FRI to estimate the static property of macromolecules. We utilize monotonically decreasing functions, including delta sequence of positive type,61 to measure the geometric compactness of a protein and quantify the topological connectivity of atoms or residues in the protein. Physically, the FRI characterizes the total interaction strength at each atom or residue, and thus reflects the atomic rigidity and flexibility. Additionally, we define the total rigidity of a molecule by a summation of atomic rigidities. Furthermore, the spatial varying shear modulus is obtained by an interpolation using atomic rigidities. A practical validation of the proposed FRI is the prediction of B-factors, or temperature factors of proteins, measured by X-ray crystallography. We employ a set of 263 proteins to examine the validity, explore the reliability, and demonstrate the robustness of the proposed FRI method for B-factor prediction. We analyze the performance of two classes of correlation kernels, i.e., the exponential type and the Lorentz type, for the B-factor prediction. The exponential type of correlation kernel involves two parameters, exponential order and characteristic length. The Lorentz type of correlation kernel also involves two parameters, power order and characteristic length. By searching the parameter space for optimal predictions, parameter-free correlation kernels are obtained. It is found that the parameter-free correlation kernel of the Lorentz type is able to retain about 95% accuracy compared to the optimized results.
A basic assumption of the present FRI theory is that the geometry or structure of a given protein together with its specific environment, namely, solvent, assembly, or crystal lattice, completely determines the biological function and properties including flexibility, rigidity, and energy. As such, the present approach bypasses the construction of the Hamiltonian and interaction potentials. A possible drawback of the present method is that the full geometric and topological information of a protein complex is usually not available, which contributes to modeling errors.
The generalization of the present work is underway on a few fronts. First, a comparison of the present FRI and two other state of the art approaches, namely, the GNM and the coarse-grained normal mode analysis (cgNMA) will be carried out for B-factor prediction. Unlike GNM and cgNMA, the FRI does not require matrix diagonalization or decomposition. Its computational complexity is at most . The performances of FRI, GNM, and cgNMA in terms of accuracy, reliability, and computational efficiency will be examined with a large number of proteins. Additionally, the performance of the present FRI will be improved by the consideration of co-factors, crystal periodicity, and X-ray diffraction cross-section. Moreover, the collective motions of proteins will be studied by solving the eigenvalue problem of the elasticity equation with atomic rigidity. Furthermore, the interaction between elastic domains and other domains will be studied by using the elastostatic equation. Finally, elastic dynamics will be simulated using the theory developed in the present work.
ACKNOWLEDGMENTS
G.W.W. was supported in part by National Science Foundation (NSF) Grant Nos. DMS-1160352 and IIS-1302285, and National Institutes of Health (NIH) Grant No. R01GM-090208. The authors acknowledge the Mathematical Biosciences Institute for hosting valuable workshops.
References
- Alexov E. and Gunner M. R., “Calculated protein and proton motions coupled to electron transfer: Electron transfer from to QB in bacterial photosynthetic reaction centers,” Biochemistry 38, 8253–8270 (1999). 10.1021/bi982700a [DOI] [PubMed] [Google Scholar]
- Anfinsen C. B., “Einfluss der configuration auf die wirkung den,” Science 181, 223–230 (1973). 10.1126/science.181.4096.223 [DOI] [PubMed] [Google Scholar]
- Antonny B., “Mechanisms of Membrane Curvature Sensing,” Ann. Rev. Biochem. 80, 101–123 (2011). 10.1146/annurev-biochem-052809-155121 [DOI] [PubMed] [Google Scholar]
- Atilgan A., Durrell S., Jernigan R., Demirel M. C., Keskin O., and Bahar I., “Anisotropy of fluctuation dynamics of proteins with an elastic network model,” Biophys. J. 80, 505–515 (2001). 10.1016/S0006-3495(01)76033-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I., Atilgan A., Demirel M., and Erman B., “Vibrational dynamics of proteins: Significance of slow and fast modes in relation to function and stability,” Phys. Rev. Lett. 80, 2733–2736 (1998). 10.1103/PhysRevLett.80.2733 [DOI] [Google Scholar]
- Bahar I., Atilgan A., and Erman B., “Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential,” Folding Des. 2, 173–181 (1997). 10.1016/S1359-0278(97)00024-2 [DOI] [PubMed] [Google Scholar]
- Baker N. A., in Biomolecular Applications of Poisson-Boltzmann Methods, Reviews in Computational Chemistry Vol. 21, edited by Lipkowitz K. B., Larter R., and Cundari T. R. (John Wiley and Sons, Hoboken, NJ, 2005). [Google Scholar]
- Brooks B. R., Bruccoleri R. E., Olafson B. D., States D., Swaminathan S., and Karplus M., “Charmm: A program for macromolecular energy, minimization, and dynamics calculations,” J. Comput. Chem. 4, 187–217 (1983). 10.1002/jcc.540040211 [DOI] [Google Scholar]
- Brown M. F., “Curvature forces in membrane lipid-protein interactions,” Biochemistry 51(49), 9782–9795 (2012). 10.1021/bi301332v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canham P. B., “The minimum energy of bending as a possible explanation of the biconcave shape of the human red blood cell,” J. Theor. Biol. 26, 61–81 (1970). 10.1016/S0022-5193(70)80032-7 [DOI] [PubMed] [Google Scholar]
- Chen D., Chen Z., and Wei G. W., “Quantum dynamics in continuum for proton transport II: Variational solvent-solute interface,” Int. J. Numer. Methods Biomed. Eng. 28, 25–51 (2012). 10.1002/cnm.1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. and Wei G. W., “Quantum dynamics in continuum for proton transport—Generalized correlation,” J. Chem. Phys. 136, 134109 (2012). 10.1063/1.3698598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. N., Wu Y. J., and Voth G. A., “Proton transport behavior through the influenza A M2 channel: Insights from molecular simulation,” Biophys. J. 93, 3470–3479 (2007). 10.1529/biophysj.107.105742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Baker N. A., and Wei G. W., “Differential geometry based solvation models I: Eulerian formulation,” J. Comput. Phys. 229, 8231–8258 (2010). 10.1016/j.jcp.2010.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Baker N. A., and Wei G. W., “Differential geometry based solvation models II: Lagrangian formulation,” J. Math. Biol. 63, 1139–1200 (2011). 10.1007/s00285-011-0402-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. and Wei G. W., “Differential geometry based solvation models III: Quantum formulation,” J. Chem. Phys. 135, 194108 (2011). 10.1063/1.3660212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Zhao S., Chun J., Thomas D. G., Baker N. A., Bates P. B., and Wei G. W., “Variational approach for nonpolar solvation analysis,” J. Chem. Phys. 137, 084101 (2012). 10.1063/1.4745084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F. and Dobson C. M., “Protein misfolding, functional amyloid, and human disease,” Annu. Rev. Biochem. 75, 333–366 (2006). 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- Cui Q. and Bahar I., Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems (Chapman and Hall, 2010). [Google Scholar]
- Demerdash O. N. A. and Mitchell J. C., “Density-cluster NMA: A new protein decomposition technique for coarse-grained normal mode analysis,” Proteins: Struct., Funct., Bioinf. 80(7), 1766–1779 (2012). 10.1002/prot.24072 [DOI] [PubMed] [Google Scholar]
- Du Q., Liu C., and Wang X. Q., “A phase field approach in the numerical study of the elastic bending energy for vesicle membranes,” J. Comput. Phys. 198, 450–468 (2004). 10.1016/j.jcp.2004.01.029 [DOI] [Google Scholar]
- Flory P., “Statistical thermodynamics of random networks,” Proc. R. Soc. London, Ser. A 351, 351–378 (1976). 10.1098/rspa.1976.0146 [DOI] [Google Scholar]
- Geng W. and Wei G. W., “Multiscale molecular dynamics using the matched interface and boundary method,” J. Comput. Phys. 230(2), 435–457 (2011). 10.1016/j.jcp.2010.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K., Davis M. E., Luty B. A., and McCammon J. A., “Computation of electrostatic forces on solvated molecules using the Poisson-Boltzmann equation,” J. Phys. Chem. 97(14), 3591–3600 (1993). 10.1021/j100116a025 [DOI] [PubMed] [Google Scholar]
- Go N., Noguti T., and Nishikawa T., “Dynamics of a small globular protein in terms of low-frequency vibrational modes,” Proc. Natl. Acad. Sci. U.S.A. 80, 3690–3700 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann B., “The BAR-domain family of proteins: A case of bending and binding? The membrane bending and GTPase-binding functions of proteins from the BAR-domain family,” EMBO Rep. 5(3), 250–255 (2004). 10.1038/sj.embor.7400105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich W., “Elastic properties of lipid bilayers: Theory and possible experiments,” Z. Naturforsch. C 28, 693–703 (1973). [DOI] [PubMed] [Google Scholar]
- Hinsen K., “Structural flexibility in proteins: Impact of the crystal environment,” Bioinformatics 24, 521–528 (2008). 10.1093/bioinformatics/btm625 [DOI] [PubMed] [Google Scholar]
- Hu L. H. and Wei G. W., “Nonlinear Poisson equation for heterogeneous media,” Biophys. J. 103, 758–766 (2012). 10.1016/j.bpj.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., and Schulten K., “VMD – visual molecular dynamics,” J. Mol. Graphics 14(1), 33–38 (1996). 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Jacobs D., Rader A., Kuhn L., and Thorpe M., “Protein flexibility predictions using graph theory,” Proteins: Struct., Funct., Genet. 44(2), 150–165 (2001). 10.1002/prot.1081 [DOI] [PubMed] [Google Scholar]
- Janosi I., Chretien D., and Flyvbjerg H., “Modeling elastic properties of microtubule tips and walls,” Eur. Biophys. J. Biophys. Lett. 27(5), 501–513 (1998). 10.1007/s002490050160 [DOI] [PubMed] [Google Scholar]
- Kiselev Y. V., Leda M., Lobanov A. I., Marenduzzo D., and Goryachev A. B., “Lateral dynamics of charged lipids and peripheral proteins in spatially heterogeneous membranes: Comparison of continuous and Monte Carlo approaches,” J. Chem. Phys. 135, 155103 (2011). 10.1063/1.3652958 [DOI] [PubMed] [Google Scholar]
- Kondrashov D. A., Van Wynsberghe A. W., Bannen R. M., Cui Q., and Phillips J. G. N., “Protein structural variation in computational models and crystallographic data structure,” Structure 15, 169–177 (2007). 10.1016/j.str.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S., Melton J. S., Sorensen D. C., and Phillips J. G. N., “Dynamics of proteins in crystals: Comparison of experiment with simple models,” Biophys. J. 83, 723–732 (2002). 10.1016/S0006-3495(02)75203-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M., Sander C., and Stern P. S., “Protein normal-mode dynamics: Trypsin inhibitor, crambin, ribonuclease and lysozyme,” J. Mol. Biol. 181(3), 423–447 (1985). 10.1016/0022-2836(85)90230-X [DOI] [PubMed] [Google Scholar]
- Lu Q. and Luo R., “A Poisson-Boltzmann dynamics method with nonperiodic boundary condition,” J. Chem. Phys. 119(21), 11035–11047 (2003). 10.1063/1.1622376 [DOI] [Google Scholar]
- Ma J., “Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes,” Structure 13, 373–180 (2005). 10.1016/j.str.2005.02.002 [DOI] [PubMed] [Google Scholar]
- McMahon H. and Gallop J., “Membrane curvature and mechanisms of dynamic cell membrane remodelling,” Nature (London) 438(7068), 590–596 (2005). 10.1038/nature04396 [DOI] [PubMed] [Google Scholar]
- Nagle J. F. and Morowitz H. J., “Molecular mechanisms for proton transport in membranes,” Proc. Natl. Acad. Sci. U.S.A 75(1), 298–302 (1978). 10.1073/pnas.75.1.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic J. N., Luthey-Schulten Z., and Wolynes P. G., “Theory of protein folding: The energy landscape perspective,” Annu. Rev. Biochem. 48, 545–600 (1997). 10.1146/annurev.physchem.48.1.545 [DOI] [PubMed] [Google Scholar]
- Ou-Yang Z. C. and Helfrich W., “Bending energy of vesicle membranes: General expressions for the first, second, and third variation of the shape energy and applications to spheres and cylinders,” Phys. Rev. A 39, 5280–5288 (1989). 10.1103/PhysRevA.39.5280 [DOI] [PubMed] [Google Scholar]
- Pan X.-Y. and Shen H.-B., “Robust prediction of B-factor profile from sequence using two-stage SVR based on random forest feature selection,” Protein Pept. Lett. 16(12), 1447–1454 (2009). 10.2174/092986609789839250 [DOI] [PubMed] [Google Scholar]
- Park J. K., Jernigan R., and Wu Z., “Coarse grained normal mode analysis vs. refined Gaussian network model for protein residue-level structural fluctuations,” Bull. Math. Biol. 75, 124–160 (2013). 10.1007/s11538-012-9797-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B., Kent H., Mills I., Vallis Y., Butler P., Evans P., and McMahon H., “BAR domains as sensors of membrane curvature: The amphiphysin BAR structure,” Science 303(5657), 495–499 (2004). 10.1126/science.1092586 [DOI] [PubMed] [Google Scholar]
- Pomes R. and Roux B., “Structure and dynamics of a proton wire: A theoretical study of H+ translocation along the single-file water chain in the gramicidin A channel,” Biophys. J. 71, 19–39 (2002). 10.1016/S0006-3495(96)79211-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P., Obradovic Z., Smith D. K., Zhu G., Vucetic S., Brown C. J., Lawson J. D., and Dunker A. K., “Protein flexibility and intrinsic disorder,” Protein Sci. 13, 71–80 (2004). 10.1110/ps.03128904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renka R. J., “Multivariate interpolation of large sets of scattered data,” ACM Trans. Math. Softw. 14(2), 139–148 (1988). 10.1145/45054.45055 [DOI] [Google Scholar]
- Roos W. H., Gibbons M. M., Arkhipov A., Uetrecht C., Watts N. R., Wingfield P. T., Steven A. C., Heck A. J. R., Schulten K., Klug W. S., and Wuite G. J. L., “Squeezing protein shells: How continuum elastic models, molecular dynamics simulations, and experiments coalesce at the nanoscale,” Biophys. J. 99(4), 1175–1181 (2010). 10.1016/j.bpj.2010.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M. and Kaufman R. J., “The mammalian unfolded protein response,” Annu. Rev. Biochem. 74, 739–789 (2005). 10.1146/annurev.biochem.73.011303.074134 [DOI] [PubMed] [Google Scholar]
- Sept D. and MacKintosh F. C., “Microtubule elasticity: Connecting all-atom simulations with continuum mechanics,” Phys. Rev. Lett. 104, 018101 (2010). 10.1103/PhysRevLett.104.018101 [DOI] [PubMed] [Google Scholar]
- Sharp K. A. and Honig B., “Calculating total electrostatic energies with the nonlinear Poisson-Boltzmann equation,” J. Phys. Chem. 94, 7684–7692 (1990). 10.1021/j100382a068 [DOI] [Google Scholar]
- Skjaerven L., Hollup S. M., and Reuter N., “Normal mode analysis for proteins,” J. Mol. Struct.: THEOCHEM 898, 42–48 (2009). 10.1016/j.theochem.2008.09.024 [DOI] [Google Scholar]
- Song G. and Jernigan R. L., “vGNM: A better model for understanding the dynamics of proteins in crystals,” J. Mol. Biol. 369(3), 880–893 (2007). 10.1016/j.jmb.2007.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasumi M., Takenchi H., Ataka S., Dwidedi A. M., and Krimm S., “Normal vibrations of proteins: Glucagon,” Biopolymers 21, 711–714 (1982). 10.1002/bip.360210318 [DOI] [PubMed] [Google Scholar]
- Thacker W. I., Zhang J., Watson L. T., Birch J. B., Iyer M. A., and Berry M. W., “Algorithm 905: SHEPPACK: Modified Shepard algorithm for interpolation of scattered multivariate data,” ACM Trans. Math. Softw. 37(3) (2010). 10.1145/1824801.1824812 [DOI] [Google Scholar]
- Thomas D., Chun J., Chen Z., Wei G. W., and Baker N. A., “Parameterization of a geometric flow implicit solvation model,” J. Comput. Chem. 34, 687–695 (2013). 10.1002/jcc.23181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirion M., “Large amplitude elastic motions in proteins from a single-parameter, atomic analysis,” Phys. Rev. Lett. 77, 1905–1908 (1996). 10.1103/PhysRevLett.77.1905 [DOI] [PubMed] [Google Scholar]
- Uversky V. and Dunker A. K., “Controlled chaos,” Science 322, 1340–1341 (2008). 10.1126/science.1167453 [DOI] [PubMed] [Google Scholar]
- Villa E., Balaeff A., Mahadevan L., and Schulten K., “Multiscale method for simulating protein-DNA complexes,” Multiscale Model. Simul. 2(4), 527–553 (2004). 10.1137/040604789 [DOI] [Google Scholar]
- Wei G. W., “Wavelets generated by using discrete singular convolution kernels,” J. Phys. A 33, 8577–8596 (2000). 10.1088/0305-4470/33/47/317 [DOI] [Google Scholar]
- Wei G. W., “Differential geometry based multiscale models,” Bull. Math. Biol. 72, 1562–1622 (2010). 10.1007/s11538-010-9511-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G.-W., “Multiscale, multiphysics and multidomain models I: Basic theory,” J. Theor. Computat. Chem. 12(8), 1341006 (2013). 10.1142/S021963361341006X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G.-W., Zheng Q., Chen Z., and Xia K., “Variational multiscale models for charge transport,” SIAM Rev. 54(4), 699–754 (2012). 10.1137/110845690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. H. and Wimley W. C., “Membrane protein folding and stability: Physical principles,” Annu. Rev. Biophys. Biomol. Struct. 28, 319–365 (1999). 10.1146/annurev.biophys.28.1.319 [DOI] [PubMed] [Google Scholar]
- Willmore T. J., Riemannian Geometry (Oxford University Press, USA, 1997). [Google Scholar]
- Yang L., Song G., and Jernigan R. L., “Protein elastic network models and the ranges of cooperativity,” Proc. Natl. Acad. Sci. U.S.A. 106(30), 12347–12352 (2009). 10.1073/pnas.0902159106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. W. and Chng C. P., “Coarse-grained models reveal functional dynamics–I. Elastic network models–theories, comparisons and perspectives,” Bioinf. Biol. Insights 2, 25–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z., Bailey T., and Teasdale R., “Prediction of protein B-factor profiles,” Proteins: Struct., Funct., Bioinf. 58(4), 905–912 (2005). 10.1002/prot.20375 [DOI] [PubMed] [Google Scholar]
- Zhao S., “Pseudo-time-coupled nonlinear models for biomolecular surface representation and solvation analysis,” Int. J. Numer. Methods Biomed. Eng. 27, 1964–1981 (2011). 10.1002/cnm.1450 [DOI] [Google Scholar]
- Zheng Q. and Wei G. W., “Poisson-Boltzmann-Nernst-Planck model,” J. Chem. Phys. 134, 194101 (2011). 10.1063/1.3581031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Yang S. Y., and Wei G. W., “Molecular surface generation using PDE transform,” Int. J. Numer. Methods Biomed. Eng. 28, 291–316 (2012). 10.1002/cnm.1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. C., Holst M. J., and McCammon J. A., “A nonlinear elasticity model of macromolecular conformational change induced by electrostatic forces,” J. Math. Anal. Appl. 340, 135–164 (2008). 10.1016/j.jmaa.2007.07.084 [DOI] [PMC free article] [PubMed] [Google Scholar]