

Abstract
Genetic predisposition and environmental factors contribute to an individual's susceptibility to develop hepatosteatosis. In a systematic, comparative survey we focused on genotype-dependent and -independent adaptations early in the pathogenesis of hepatosteatosis by characterizing C3HeB/FeJ, C57BL/6NTac, C57BL/6J, and 129P2/OlaHsd mice after 7, 14, or 21 days high-fat-diet exposure. Strain-specific metabolic responses during diet challenge and liver transcript signatures in mild hepatosteatosis outline the suitability of particular strains for investigating the relationship between hepatocellular lipid content and inflammation, glucose homeostasis, insulin action, or organelle physiology. Genetic background-independent transcriptional adaptations in liver paralleling hepatosteatosis suggest an early increase in the organ's vulnerability to oxidative stress damage what could advance hepatosteatosis to steatohepatitis. “Universal” adaptations in transcript signatures and transcription factor regulation in liver link insulin resistance, type 2 diabetes mellitus, cancer, and thyroid hormone metabolism with hepatosteatosis, hence, facilitating the search for novel molecular mechanisms potentially implicated in the pathogenesis of human non-alcoholic-fatty-liver-disease.
Abbreviations: NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic hepatosteatitis; T2D, type 2 diabetes mellitus; IR, insulin resistance; VLDL, very low density lipoprotein; C3H, C3HeB/FeJ; B6N, C57BL/6NTac; B6J, C57BL/6J; 129, 129P2/OlaHsd; HFD, high-fat diet; LFD, low fat rodent laboratory diet; TAG, triacylglycerol; WAT, white adipose tissue; ALT, alanine aminotransferase; HDL, high-density lipoprotein; LDL, low-density lipoprotein
Keywords: Non-alcoholic fatty liver disease, Inflammation, Oxidative stress, Thyroid metabolism, Insulin resistance, Cancer
1. Introduction
In the pathogenesis of non-alcoholic fatty liver disease (NAFLD) moderate pathophysiological changes in initial stages of hepatosteatosis may be critical in mediating hepatocellular injury and progression to non-alcoholic hepatosteatitis (NASH), the latter associated with severe metabolic conditions [1–3]. It is still an unresolved matter why not all patients with risk factors for developing NAFLD progress from benign steatosis to inflammation (steatohepatitis), fibrosis, cirrhosis, and hepatocellular cancer [4]. From a number of studies published over the recent years ethnic variations outline a role for genetic factors as well as a link between hepatic steatosis and hepatic inflammation, and insulin resistance or type 2 diabetes mellitus (T2D) in combination with obesity [5,6]. Hepatic steatosis usually originates from an increase in dietary fat intake, lipolysis in adipose tissue, de novo lipogenesis in hepatocytes, a decrease in hepatic very-low-density lipoprotein secretion and/or lipid breakdown. Excessive lipid accumulation in liver is frequently accompanied by endoplasmic reticulum and oxidative stress responses as well as alterations in the production and release of liver cell-derived cytokines that might mediate inflammatory responses in the organ. Taken together, the complex interplay of genetic risk factors with environmental, hormonal, and metabolic alterations in liver might promote the development of a chronic pro-inflammatory state, leading to hepatic cell injury, apoptosis, fibrosis, and progression to NASH [4,7].
Ethnic differences suggest that NAFLD-susceptibility is modulated by the way in which organs store fat. Even though associated risk factors such as T2D, insulin resistance, and obesity are similar in Hispanics and African-Americans, the latter are less prevalent to develop NAFLD, less likely advance to later NAFLD stages and are less prone to hypertriglyceridemia and abdominal fat mass gain [8,9]. Obese African-Americans appear to store fat to a larger extent subcutaneously whereas obese Hispanics tend to deposit fat in liver and visceral adipose tissue depots. Liver fat has been suggested to contribute to the risk of developing metabolic diseases rather than visceral fat and moderate increases in the hepatocellular lipid content have been associated with multiple metabolic phenotypes that precede NAFLD [8].
Simultaneous exposure of genetically heterogeneous mouse strains to a defined environmental (dietary) challenge allows determination of genetic and exogenous factors involved in development of HFD-induced hepatosteatosis. For this reason we subjected the inbred mouse strains C3HeB/FeJ, C57BL/6NTac, C57BL/6J, and 129P2/OlaHsd, which are frequently used in biomedical research, to a HFD challenge and systematically compared early physiological responses on the organism level as well as accompanying transcriptional alterations in modestly steatotic livers. Besides strain specific responses we identified genetic background-independent alterations in gene expression signatures pointing out novel candidates that potentially link the development of human NAFLD with T2D, cancer, and thyroid hormone metabolism.
2. Materials and methods
2.1. Mouse strains and in vivo metabolic experiments
C3HeB/FeJ (C3H; strain does not carry mouse mammary tumor virus or abnormal allele at Tlr4 locus but is homozygous for retinal degradation allele Pde6brd1; The Jackson Laboratory, Maine, U.S.A.), C57BL/6NTac (B6N; Taconic, Ry, Denmark), C57BL/6J (B6J; The Jackson Laboratory, Maine, U.S.A.), and 129P2/OlaHsd (129; Harlan Laboratories, Indianapolis, U.S.A.) were bred and housed under standard vivarium conditions (12:12 light–dark-cycle). At an age of 14 weeks male mice of each strain were single housed, litter-matched and allocated to six groups. Three groups per strain were switched to a high-fat-diet (custom-made HFD, Ssniff, Soest, Germany) and three continued on low-fat-diet (LFD, Diet#1310, Altromin, Lage, Germany) for 7, 14, or 21 days (Table 1). Body mass and composition, the latter determined by in vivo 3H NMR-spectroscopy (MiniSpecLF60, Bruker Optics, Ettlingen, Germany), were measured 1 day prior to the start and end of the experiment. Between 9 and 11 a.m. at the terminal study-endpoint random-fed mice were killed with isoflurane. V. cava blood samples were obtained, immediately centrifuged at 4 °C, and plasma aliquots frozen in liquid nitrogen. Liver and epididymal white adipose tissues were dissected, weighted, and quickly freeze-clamped in liquid nitrogen or immersed in paraformaldehyde. All animals received humane care and study-protocols complied with the institution's guidelines. Animal experiments were approved by the Upper-Bavarian district government (Regierung von Oberbayern, Gz.55.2-1-54-2532-4-11).
Table 1.
Composition of low-fat diet (LFD: heat of combustion 17.0 KJ/g) and high-fat diet (HFD: heat of combustion 24.3 KJ/g).
| Macronutrients | Low-fat diet (content; g/kg) | High-fat diet (content, g/kg) |
|---|---|---|
| Protein | 27% calories from corn (17), wheat (63), soybean (145) | 15% calories from casein (200) |
| Carbohydrate | 60% calories from corn (34), wheat (223), soybean (148) | 27% calories from cornstarch (227), maltose dextrin (132), cellulose (50) |
| Fat | 13% calories from corn (7), wheat (16), soybean (7), soybean oil (20) | 58% calories from soybean oil (70), safflower oil (270) |
2.2. Plasma analysis
Plasma immunoreactive insulin concentrations were determined via ELISA (Insulin Mouse Elisa, Mercodia, Uppsala, Sweden) and all other plasma parameters with an AU400 autoanalyzer (Olympus Germany, Hamburg, Germany) using adapted reagents of Beckman Coulter (Krefeld, Germany), Wako Chemicals GmbH (Neuss, Germany), and Randox Laboratories GmbH (Crumlin Co. Antrim, Northern Ireland, United Kingdom).
2.3. Liver triacylglycerol (TAG) analysis
Approximately 50 mg ground liver aliquots were homogenized (TissueLyserII, Qiagen, Hilden, Germany) with 1 ml 5% Triton-X100, samples were heated to 80–100 °C, cooled to room temperature, and heated once more. After centrifugation supernatants were diluted (HFD: 20-fold, LFD 10-fold) and TAG concentrations quantified enzymatically (Biocat, Heidelberg, Germany).
2.4. Liver histochemistry
For Oil-Red-O histochemistry 8 μm cryosections were fixed in paraformaldehyde, rinsed with 60% isopropanol, sections 10 min stained with freshly prepared Oil-Red-O working solution, and rinsed with 60% isopropanol. Nuclei were counterstained with Mayer's haematoxylin, rinsed with tap water and mounted in glycerin jelly. Images were acquired with a Hamamatsu NanoZoomer 2.0HT (Shizuoka, Japan).
2.5. Microarray analysis
Total RNA was isolated from frozen liver homogenates using the miRNeasy Minikit (Qiagen) including DNAse treatment. RNA quality was assessed with an Agilent bioanalyzer kit and total RNA (200 ng, RIN>5) was amplified using the GeneChip Whole Transcript (WT) Sense Target Labeling Assay (Affymetrix). Amplified cDNA was hybridized on Affymetrix Mouse Gene ST 1.0 arrays. Expression console (v.1.1, Affymetrix) was used for quality control and to obtain annotated normalized RMA gene-level data for each mouse strain separately. Data were filtered for significantly regulated genes defined at a fold-change >1.3 for HFD treated mice compared to LFD fed mice of the same genetic background. Under this condition a False Discovery Rate (FDR) below 10 was determined in all 4 strains (Supplemental Table 2). Statistical analyses were performed in CARMAweb [10]. For genewise testing for differential expression limma t-test and Benjamini–Hochberg multiple testing correction were employed. Hierarchical Cluster analysis [11] was employed for identification of similar expression patterns between strains. As distance the average-linkage-method and as distance-metric the Euclidean Distance was applied. Array data are available in the GEO database under accession number GSE43106 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=dlolzewsasoeile&acc=GSE43106). The overlap between strains and strain-specific differences of regulated genes were determined by and represented in Venn-diagrams [12]. The STRING database v9.1 was employed for literature-based analyses of overrepresented GO terms and KEGG pathways of significantly differentially expressed genes as well as graphical representation of regulated genes in network diagrams [13]. Transcription factor binding site analysis was conducted with P-scan. Groups of co-expressed genes were scanned for over-represented (p<0.01) motifs predicting the binding of transcription factors [14].
2.5.1. Quantitative real-time-PCR-based (qRT-PCR)
QRT-PCR analysis was performed with samples used for microarray analysis (n=5–6/group). After 1 µg total RNA was reverse transcribed (Invitrogen, Darmstadt, Germany) with random primers, PCR was performed with an ABI Prism 7900HT Sequence detection system (Applied Biosciences, Warrington, UK) using Power Sybr Green (Applied Biosciences, Warrington, UK). The following forward (f) and reverse (r) primers were specific for the indicated genes: Decr1_f ATTTGACGGTGGAGAGGAAGTATT and Decr1_r GATTATATCCCACTCCTCCTTGGT; Decr2_f ACTGTCTTCCTGAATACCACCATC and Decr2_r CAGAACCACCACCTGTGATAAAG; Dio1_f GAGAGGCTCTACGTGATACAGGAG and Dio1_r GACTTCCTCAGGATTGTAGTTCCA; Gst3_f AGTATCGTATGTTTGAGCCCAAGT and Gst3_r ATGTAGGCAGAGATCTTCTTGAGG; Gsta2_f TCTCTATGTTGAAGAGCTTGATGC and Gsta2_r CTGCCAGGATGTAGGAACTTCT; Gsta4_f TCCTAGAAGCCATTTTGATGGT and Gsta4_r GTAGGAATGTTGCTGATTCTTGTC; Gst1_f TGACATTCTTGACCAGTACCGTAT and Gst1_r ATGTAGGCAGAGATCTTCTTGAGG; Gstp2_f AATGGTAAGAATGACTACGTGAA and Gstp2_r AAGTTGTAATCGGCAAAGGAGA; Ppia_f ACTTCATCCTAAAGCATACAGGTC and Ppia_r CACAATGTTCATGCCTTCTTT; Ywhaz_f GCCCTCAACTTCTCTGTGTTCTAT and Ywhaz_r TAACTGCATTATTAGCGTGCTGTC; B2m_f GTCTTTCTGGTGCTTGTCTCACT and B2m_r TTACGTAGCAGTTCAGTATGTTCG. Each sample was measured in triplicate and ΔCT values normalized to peptidylprolyl isomerase A (Ppia; cyclophilin A) but comparable results observed after normalization with the alternative housekeeping genes 14-3-3 protein zeta/delta (Ywhaz) and beta-2 microglobulin (B2m). Via the comparative method linear fold-changes of qRT-PCR products in livers of individual HFD-treated mice to the mean of LFD-fed mice of the same strain were calculated.
2.6. Statistical analysis
All data, except microarray data, are expressed as means±SEM. Analysis of Variance (ANOVA, Bonferroni correction) or Student's T-tests for pairwise comparisons of genes regulated in livers of HFD-treated versus LFD treated mice in each mouse strain (qRT-PCR) were performed. Spearman's rank correlation coefficient (Bonferroni correction) was used to correlate the expression level of individual genes in liver with individual physiological parameters. Statistical information on functional enrichments of significantly regulated genes (fold-change >1.3, Bonferroni correction) in networks were provided by the STRING database v9.1.
3. Results
3.1. Mouse strains
In this survey we compared the four inbred mouse strains C3HeB/FeJ (C3H), C57BL/6N (B6N), C57BL/6J (B6J), and 129P2/OlaHsd (129). Criteria for selection were the highest frequency of these genetic backgrounds for systemic primary phenotyping in the German Mouse Clinic [15]. Furthermore, B6N is a standard inbred strain and its embryonic stem cell lines are facilitated by the International Knockout Mouse Consortium [16]. The C3H strain is a resource in the Munich ENU Mouse Mutagenesis Project [17] and the UK ENU Mutagenesis program [18]. Due to their global use in mouse genetics and metabolic research all strains serve as controls for primary phenotyping in the European Mouse Disease Clinic [19] and B6N in the International Mouse Phenotyping Consortium [20].
3.2. Strain-dependent and -independent phenotypic characteristics during LFD exposure
Upon single housing and continuation on our animal facilities' LFD, 129 males progressively gained body mass and whole body fat mass (Figure 1A and B open bars). Males of all other strains decreased body mass and whole body fat mass between study start and day 7 of the experiment but, except for B6J males, recovered initial body mass within 14 or 21 days (Figure 1A and B open bars). Hepatic TAG concentrations (three time-point average) ranged between ~2.5% (129), ~2.2% (C3H, B6N), and ~1.5% (B6J).
Figure 1.
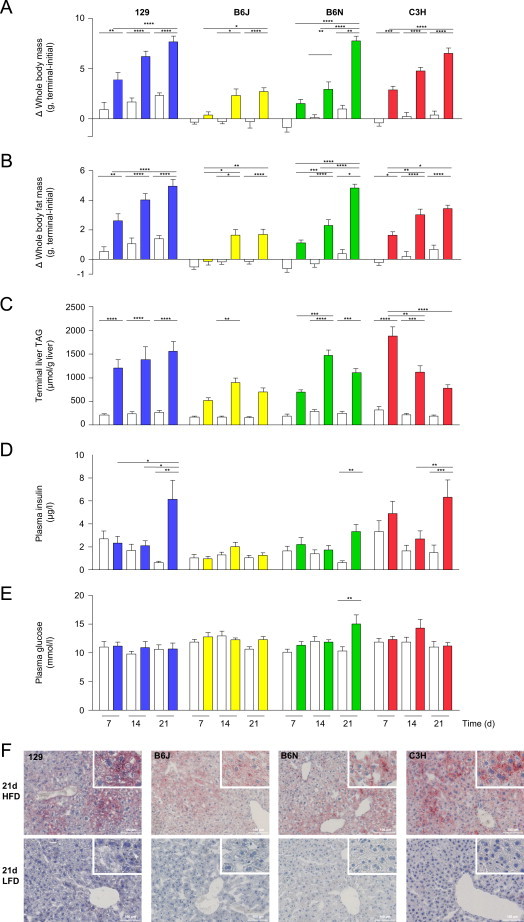
Phenotypic comparison of 129, B6N, B6J, and C3H males fed a HFD (colored bars) or LFD (open bars) for 7, 14, or 21 days. During the survey (A) whole body mass changes and (B) whole body fat mass changes (initial versus days 7, 14, or 21), as well as (C) liver TAG concentrations, (D) plasma insulin, and (E) plasma glucose concentrations following 7, 14, or 21 days HFD intervention were determined. Data are means±SEM (n=8–12/group, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; Bonferroni correction). (F) Representative Oil-Red-O stained liver-section images depict neutral lipid (red) after 21 days HFD (upper panel) or LFD (lower panel) exposure. Nuclei (blue) are visualized with hematoxilin (20× and 100× magnifications). Abbreviations: HFD, high-fat diet; LFD, low-fat diet; TAG, triacylglycerol.
3.3. General, strain-independent phenotypic alterations during HFD-induced hepatosteatosis
In all strains HFD exposure, at least at one time-point, increased body mass, whole body fat mass, epididymal white adipose tissue mass, as well as plasma HDL and total cholesterol concentrations compared to littermates fed LFD (Figure 1A and B; Supplemental Table 1A–D). Development of moderate obesity caused by HFD intervention was paralleled by an expansion of hepatic lipid stores (Figure 1C and F). In all mouse strains HFD exposure raised hepatic triacylglycerol concentrations between approximately 3- and 7-fold during the study time course compared to the respective genotype control (Figure 1C).
3.4. Strain-specific phenotypic alterations during HFD-induced hepatosteatosis
Solely 129 males progressively gained body mass and whole body fat mass, increased epididymal adipose tissue mass, paralleled by declining plasma non-esterified fatty acid concentrations during 21 days of HFD feeding compared to LFD-fed littermates (Figure 1A and B; Supplemental Table 1A). In contrast to B6J, B6N exhibited a pronounced body mass and whole body fat mass gain between day 14 and 21 of HFD-feeding compared to LFD-fed littermates (Figure 1A and B; Supplemental Table 1B and C). Noteworthy, among all strains HFD-fed B6J displayed the lowest absolute whole body fat mass and epididymal fat pad mass at any time point during HFD-intervention (Figure 1B; Supplemental Table 1B). Only C3H progressively gained body mass paralleled by a blunted whole body fat gain between day 14 and 21 of HFD-intervention (Figure 1A and B; Supplemental Table 1D).
HFD-induced hepatic lipid deposition followed a strain-characteristic, temporal pattern. Highest absolute levels were reached in C3H, followed by 129, B6N, and B6J males (Figure 1C). Exclusively 129 males progressively increased hepatic TAG concentrations during HFD-feeding (Figure 1C). In livers of B6N and B6J, TAG concentrations reached an intermediary peak at day 14 of HFD feeding whereas in those of C3H earlier at day 7 of HFD intervention (Figure 1C). All strains developed mild hepatosteatosis within 21 days of HFD-exposure outlined by an increase in hepatic lipid deposition (Figure 1F). Visual observation suggested the development of macrovesicular steatosis in livers of most 129 and C3H males, and rather microvesicular steatosis in B6N and B6J.
In the majority of patients NAFLD is associated with insulin resistance, considered a key pathogenic factor in its development [3,5,7,21]. Suggesting development of insulin resistance, 21 days of HFD-intervention were paralleled by a marked increase in random-fed plasma insulin concentrations in 129, B6N, and C3H mice compared to the respective LFD-fed littermates (Figure 1D). In addition to hyperinsulinemia the B6N strain displayed a statistically significant increase in random fed plasma glucose concentrations following 21 days HFD-intervention compared to LFD-fed littermates (Figure 1E). Over the study time course only HFD-fed B6J maintained euglycemia and -insulinemia compared to LFD-fed age-matched littermates (Figure 1D and E).
3.5. Liver transcriptome profiles associated with HFD-induced hepatosteatosis
Next we explored genetic background-specific and -independent transcriptional alterations in steatotic livers after 21 of HFD exposure compared to the respective genotype-control group fed LFD. Heat maps of all significantly differentially expressed genes (fold-change <1.3) in livers are provided in the supplement section for all strains (Supplemental Table 3A–D). We employed cluster, literature-based network, functional classification, and transcription factor binding site analyses. In total 209 probe sets exhibiting significantly differential signals were identified in livers of 129 mice, 314 in B6J mice, 440 in B6N mice, and 230 in C3H mice after 21 days HFD intervention (Figure 2A and B). Despite pronounced phenotypic variations during induction of HFD-induced hepatosteatosis, hierarchical cluster analysis outlined similarities in hepatic gene expression patterns in response to a HFD-challenge in the four mouse strains (Figure 2C). Noteworthy, 44 ‘common’ probesets were significantly regulated in a genetic background-independent fashion (Figure 2A) and a subset of 43 probe sets out of the 44 was consistently increased or decreased (Figure 2D) in all four strains. QRT-PCR data from selected ‘common’ genes were largely concordant with expression array data, although the magnitude of some of the changes detected varied between the two methods (Figure 2E). An extended list of significantly differentially regulated probesets in livers of two or three HFD-fed strains compared to LFD controls is included as Supplemental Figure 1.
Figure 2.
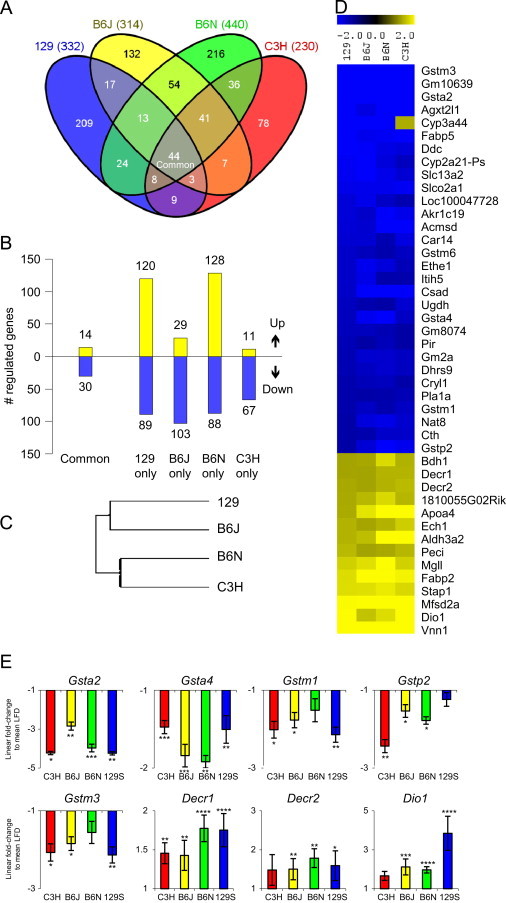
HFD-induced alterations in gene expression patterns in livers of 129, B6N, B6J, and C3H mice. (A) Venn-diagram depicts regulated probesets following 21 days HFD exposure in each strain. (B) Outlines the number of significantly induced (yellow) and repressed (blue) probesets in liver after 21 days HFD intervention in each strain as well as in all strains (Common). (C) Hierarchical cluster analysis indicates similarities between all regulated genes in the four mouse strains. (D) Heat map outlines the 44 probesets (rows) significantly differentially expressed in all four strains (columns). Data points represent mean ratios in 21 days HFD mice compared to LFD-fed littermates. Expression value intensities (blue down-, yellow up-regulated) are depicted by the color-indicator ranging from −2 and 2 on a log2 scale. (E) Validation of selected ‘common’ genes by qRT-PCR in livers of 21 days HFD treated mice compared to LFD-fed littermates. Data are means±SEM (n=5–6/group, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
3.6. Classification and network analysis of genes in liver associated with HFD-induced hepatosteatosis
For classification of regulated strain-specific and -independent genes in HFD-mediated steatotic livers we analyzed over-represented GO terms and KEGG pathways (Supplemental Table 3E). Analysis of the significantly upregulated genes in livers of HFD-fed 129 mice suggest a marked association with immune system activation and responses to stress or inflammation (Supplemental Table 4A). Exclusively in livers of HFD-fed B6J mice significantly downregulated genes were enriched in processes and pathways related to the metabolism of pyrimidine, purine, nicotinate, and nicotinamide (Supplemental Table 4B). In B6N autoimmune-associated diseases (e.g. graft-versus-host disease, type I diabetes, autoimmune thyroid disease, etc.), maturity onset diabetes of the young, or ascorbate/aldarate, carbohydrate, retinol, and steroid hormone metabolism were overrepresented. Significantly induced genes in HFD-fed B6N livers were associated with mitochondria, mitochondrial (and peroxisomal) lipid metabolism (e.g. fatty acid β-oxidation), ketone body metabolism, and lipid transport (Supplemental Table 4C). Specifically in steatotic livers of C3H males significantly downregulated genes were related to glycerolipid metabolism and the ribosomal compartment whereas significantly upregulated genes to acyl-CoA, thioester, and cellular lipid metabolic processes (Supplemental Table 4D).
Special focus of this study was to identify genes in steatotic livers regulated in a genetic background-independent fashion in mice and putatively implicated in the development of human hepatosteatosis. Within the ‘common’ murine gene set, upregulated transcripts in steatotic livers were related to the cytosolic, peroxisomal, or mitochondrial compartment and to lipid, fatty acid, and ketone metabolism whereas downregulated genes to glutathione, cellular modified amino acid, nitrogen, and ketone metabolism (Supplemental Table 4E). Predicted and evidence based interactions of the whole set of ‘common’ genes or their products are visualized in a network (Figure 3).
Figure 3.
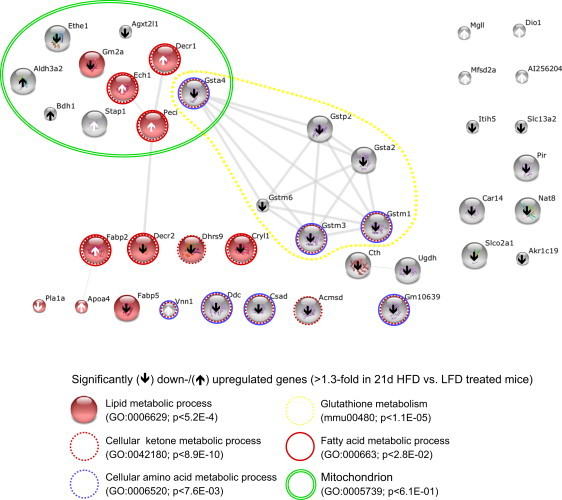
Functional characterization of genes regulated independent from the genetic background in early HFD-induced hepatosteatosis. The network construction was based on the overlapping, significantly down- and upregulated (arrow down/up) transcripts (40 out of 44 matched in STRING database) identified in livers of all four mouse strains after 21 days of HFD exposure in comparison to the respective LFD-fed controls. Major overrepresented terms in KEGG pathways, GO term categories (cellular component, biological function), and the respective p-values (Bonferroni correction) are indicated. All analyses were performed using the STRING database. AI256204 is represented by 1810055G02Rik.
3.7. Analysis of significantly over-represented transcription factor binding sites in liver in HFD-induced hepatosteatosis
Transcription factors regulate the expression of genes and are vital for a variety of important cellular processes. Therefore, we analyzed motifs of significantly over-represented transcription factor binding sites in liver. Results predict the activation of e.g. GATA3, and GFI in 129 mice, ESR1, CTCF, and HAND1 in B6J mice, RORA2 in B6N mice, as well as TBP, GATA1, GATA2, several fork head transcription factors, IRF2, and CBPA in C3H mice in moderate hepatosteatosis (Table 2). Findings also corroborate strain-independent adaptations in the development of fatty liver that potentially involve activation of the transcription factors HNF4A, PPARG, NR2F1 and NR2E1 in liver (Table 2).
Table 2.
Overrepresented transcription factor binding sites identified in livers of all strains or in specific strains after 21 days of HFD intervention compared to LFD-fed controls of each strain.
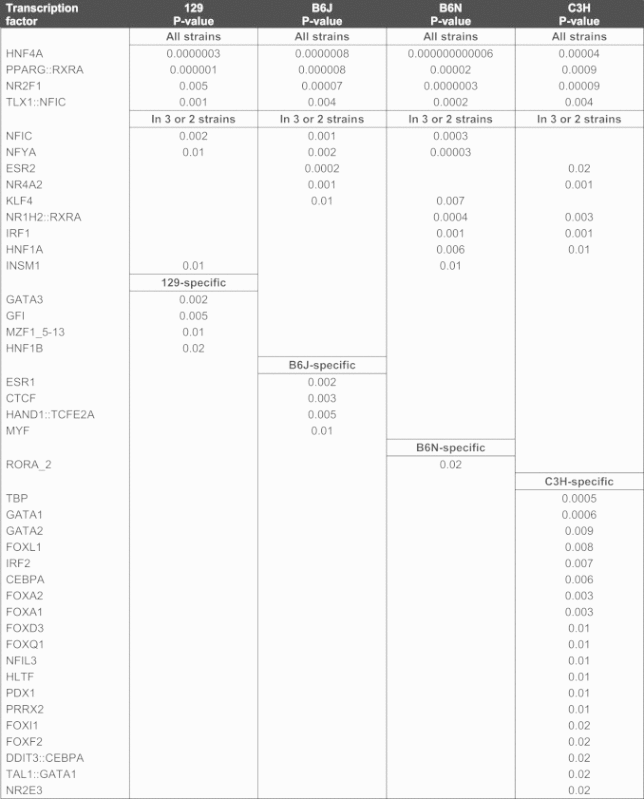 |
4. Discussion
Genetic and environmental factors contribute to the development of NAFLD but their individual shares in disease pathophysiology are not clearly understood. To assess physiological and molecular hepatic adaptations during induction of hepatosteatosis caused by a HFD-intervention, we extensively examined four widely used inbred mouse strains. We performed these studies in collaboration with the German Mouse Clinic II, as its expertise is to mimic major environmental risk factors for human health in mice applying various challenges (diet, air, stress, infection and physical activity [22]). Comparison of phenotypic alterations in four, commonly used, inbred mouse strains exposed to an environmental challenge, a HFD, outlined distinct genotype-dependent differences regarding their susceptibility to store fat in extrahepatic or hepatic tissue as well as associated transcript patterns in mild hepatosteatosis.
In B6N males a decrease in hepatic TAG concentrations between d14 and 21 of HFD intervention was associated with a relative increase in visceral adipose tissue mass (epididymal white adipose tissue mass 13%, 13%, 15% of terminal body fat mass at HFD d7, 14, 21) that was not observed in B6J males (epididymal white adipose tissue mass 10%, 12%, 12% of terminal body fat mass at HFD d7, 14, 21). In contrast to observations in the other three strains, HFD-challenged B6J males did neither increase plasma glucose nor insulin concentrations under random fed conditions (Figure 4). In our experimental setting B6J are susceptible to develop HFD-induced glucose intolerance, however they display a more modest degree when compared to age-matched B6N males (unpublished data). Throughout the study time-course B6J mice also showed the weakest HFD-mediated gain in whole body and intra-abdominal fat mass among the four strains. As lipogenesis is considered an insulin-dependent process the observed low plasma insulin concentrations in HFD-fed B6J mice might have co-contributed to their phenotype. Impaired glucose-stimulated insulin secretion in B6J has been related to a nicotine amide-nucleotide transhydrogenase (Nnt) loss-of-function mutation, its gene product implicated in mitochondrial energy metabolism [23,24]. Changes in mitochondrial physiology are also discussed to play a role in the pathogenesis of NAFLD [25]. It remains an open question to whether the Nnt-loss of function mutation and regulation of the Nt5e, Nt5m, and Pnp1 genes – the latter implicated in nicotinamide metabolism – in steatotic B6J livers are related to the extent of hepatocellular lipid accumulation. To gain insights into molecular mechanisms it might prove useful to compare genetically closely related and well characterized C57BL substrains.
Figure 4.
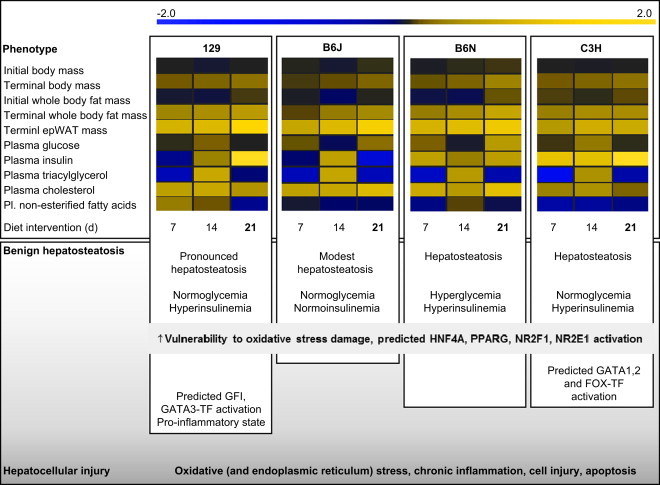
Comparison of hepatosteatosis mouse models. Heat maps visualize HFD-induced changes in physiological parameters in each strain compared to LFD-fed littermates between day 7 and 21 of HFD intervention down- (blue) and up-regulated (yellow) parameters in HFD-fed mice during the survey are compared to their corresponding LFD-fed littermates. Furthermore strain-specific and genotype-independent predicted alterations in the activity of transcription factors after 21 days of HFD exposure in liver are indicated. However, mice – especially when hepatosteatosis is induced via a HFD – appear more protected to develop fully blown NASH compared to humans. Even if in this survey 129 mice appear more susceptible to HFD-induced hepatic disease progression compared to the other three strains, it is unclear if prolonged HFD exposure induces hepatic fibrosis and cirrhosis in this mouse model. Abbreviations: HFD, high-fat diet; LFD, low-fat diet; epWAT, epididymal white adipose tissue; Pl, plasma; HNF4A, hepatocyte nuclear factor 4 alpha; PPARG, peroxisome proliferator-activated receptor gamma; NRF2F1, nuclear receptor subfamily 2 group F member 1; NR2E1, nuclear receptor subfamily 2 group E member 1; GFI, growth factor independent 1 transcription repressor; GATA, GATA-binding factor.
During excess dietary lipid intake 129 males exhibited the highest preference among the four strains to expand whole body fat mass. This was accompanied by a progressive increase in visceral fat deposits (epididymal white adipose tissue mass 11%, 12%, 13% of terminal body fat mass at HFD d7, 14, 21) and liver TAG content, the latter in humans suggested to contribute to the risk of developing metabolic diseases [8]. It has been reported, that a large proportion of 129P2/OlaHsd mice (129P2/OlaHsd formerly known as 129/OlaHsd) expresses major shunts between the hepatic portal system and the Vena cava [26]. Such an anatomical abnormality might impact on liver physiology and metabolism by causing a decrease in hepatic blood pressure and alterations in the supply of hormones and metabolites to and from the liver. In our survey, the transcription factors zinc finger protein GFI-1 (GFI-1) and GATA binding protein 3 (GATA3) were predicted activated in liver biopsies obtained from HFD-treated 129 males. Both transcription factors modulate immune system competence, inflammatory processes, and stress responses [27,28]. 129-specific transcript signatures and enriched cell surface genes in liver biopsies likely do not indicate local liver cell type-specific alterations but rather systemic effects in neutrophil, lymphoid, leukocyte, granulocyte and macrophage linages, in the classical and alternative complement pathways (e.g. concerted transcriptional induction of C1qa, C1qb, C1qc, C4a, Cfp), and apoptosis-related events (transcriptional induction of Casp8). In addition, in male 129 mice a higher than normal concentration of serum ceruloplasmin has been reported [29]. Ceruloplasmin is an acute-phase reactant and seems to have a function in copper and ferric ion transport. Taken together, the 129 strain appears valuable for studying the implication of inflammation-associated processes in the development of hepatosteatosis (Figure 4).
Despite a continuous increase in whole body fat mass gain during high energy intake, C3H males displayed an early peak in hepatic TAG concentrations that thereafter progressively declined suggesting early adaptive responses in the liver such as a decrease in lipogenesis, increase in lipolysis and/or TAG export, or decrease in lipid clearance. In this strain preferential storage of lipids in subcutaneous fat depots is assumed as the relative proportion of visceral fat (epididymal white adipose tissue mass 12%, 13%, 12% of terminal body fat mass at HFD d7, 14, 21) remained relatively stable throughout HFD exposure. Hepatoma incidence (72–91% in males at age 14 months) is high in C3H [30] and has been associated with six chromosomal regions [31]. In this survey, induction of hepatosteatosis was associated with a C3H genotype-dependent activation of several forkhead and GATA transcription factors in liver. GATA transcription factors are involved in the coordinated maturation and cell cycle withdrawal in terminally differentiating cells and induce cellular differentiation [32]. Also FOXD3 and FOXF2 are involved in cell differentiation and proliferation [33]. As alterations in FOXO and GATA activity might contribute to cancer development, C3H might serve as a suitable mouse model for studying the implication of genetic predisposition to hepatocarcinogenesis in NAFLD (Figure 4).
It was suggested, that oxidative stress damage and increased lipid peroxidation in liver co-contribute to the transition of ‘benign’ hepatosteatosis to steatohepatitis [25]. This might originate from an increase in lipid peroxidation products and free radicals as a consequence of impaired mitochondrial physiology, an imbalance between glutathione and oxidized equivalents, and alterations in fatty acid oxidation [34]. Noteworthy, the transcripts of Cth, Gsta2, Gsta4, Gstm1, Gstm3, Gstm6, Gstp2, and Vnn1 were concertedly repressed in fatty livers of all four mouse strains and independent from phenotypic differences during HFD-inducted hepatosteatosis. Family members of the glutathione-S-transferase family detoxify lipid-peroxidation products and are considered to protect cells from reactive oxygen species [35]. According to previous studies cystathionine gamma-lyase (Cth)-deficient mice are prone to systemic oxidative injury, Cth-deficient hepatocytes display a greater sensitivity to oxidative stress, and CTH mutations in humans occur in association with liver disease [36]. Vanin1 (VNN1) has been demonstrated to possess pantetheinase activity [37], and therefore may play a role in oxidative-stress response and possibly lymphocyte migration. Taken together, a decrease in the liver's defense capacity against oxidative stress damage during steatosis might promote progression towards a chronic pro-inflammatory state, cell injury, and apoptosis (Figure 4).
Major facilitator superfamily domain-containing protein 2 (Mfsd2a) was identified as a fasting-inducible gene regulated by both, PPARA and glucagon signaling in liver. Genotype-independent induction of the Mfsd2a gene in steatotic livers in the current survey is consistent with findings in gene-targeted Mfsd2a-deficient mice that are smaller, leaner, and display decreased liver TAG concentrations compared to wild-type gene carriers [38]. Thus, data suggest a potentially important role for Mfsd2a in the regulation of hepatic lipid metabolism.
Ectopic lipid accumulation leads to cell dysfunction and apoptosis, a phenomenon known as lipotoxicity. In hepatocytes nutritional stimuli such as fatty acids appear capable of initiating a compensatory endoplasmic reticulum stress response (ERSR) and unfolded protein response (UPR) that include transcriptional regulation of genes whose protein products increase the capacity of the ER for protein degradation, protein folding, and apoptosis [39]. However, in steatotic livers in the four mouse strains no significantly differentially expressed transcripts directly involved in ERSR or UPR were detected (e.g. Grp78, Edem, Chop/Gadd153, Casp3). This might be related to the relatively early hepatosteatosis stage and the high content of polyunsaturated alpha-linoleic acid in the HFD. There is evidence that specifically long-chain saturated fatty acids induce ER stress in liver [40] and experiments in liver cells indicate that ER stress is mediated by saturated fatty acids but augmented by conjugated linoleic acid [41].
NAFLD presents a risk factor for T2D development which in turn aggravates hepatosteatosis [2,5,34]. We identified an array of genes and transcription factors in steatotic livers that were regulated independent from the genetic background and pathophenotype during HFD-intervention (Figure 4). Some of these candidates might play a molecular role in the development of insulin resistance, T2D, or cancer. Non-esterified fatty acids are endogenous ligands of several transcription factors controlling hepatic metabolism. Previously, the n-6 fatty acid linoleic acid was identified to serve as an endogenous ligand of hepatocyte nuclear factor 4-alpha (HNF4A) and seems to permanently bind to the transcription factor [42]. HNF4A controls the expression of a large array of genes in liver involved in regulation of glucose metabolism, gluconeogenesis, hepatic lipogenesis, lipid transport, and liver morphogenesis [43,44]. In the current study genotype-independent HNF4A activation could be related to the high abundance of linoleic acid (~78% of dietary fatty acids) in the safflower oil-containing HFD. In addition in all mouse strains HFD-induced hepatosteatosis was associated with a predicted activation of peroxisome proliferator-activated receptor gamma (PPARG). Long-chain saturated and unsaturated fatty acids or their derivatives stimulate ligand-dependent PPARG activation [45,46]. Due to its transcriptional control of many enzymes responsible for the oxidation and synthesis of fatty acids, PPARG activation contributes to lipid accumulation and therefore has been implicated in the pathology of diabetes, obesity, and cancer. Despite a decrease in PPARG receptor activity, humans carrying the Pro12Ala mutation express enhanced insulin sensitivity [47] and heterozygous PPARG-null mice are protected from the development of HFD-mediated insulin resistance compared to wild-type allele carriers [48]. In contrast, PPARG loss-of-function mutations identified in humans have been reported to also cause insulin resistance [49].
Currently, the NAFLD-cancer link attracts attention as epidemiologic evidence recently supported an association between NAFLD and an increased risk of hepatocellular carcinoma [50]. Underlying molecular mechanisms are unclear but might involve enoyl CoA hydratase 1, peroxisomal (Ech1) or pirin (Pir), both genes significantly regulated in steatotic livers of all mouse strains. Ech1 and Pir play a role in proliferation, cell adhesion and migration capacities, DNA regulation, the control of cellular senescence and melanoma [51,52]. Furthermore, individual expression levels of the Ech1 gene in livers of all HFD-fed mice in this study tended negatively correlated with individual plasma glucose concentrations on day 21 of HFD intervention (Supplemental Table 5A).
By inducing the expression of several nuclear encoded OXPHOS genes [53] or direct regulation of mitochondrial DNA transcription [54] thyroid hormone regulates mitochondrial biogenesis and respiratory function [55,56]. Deiodinase (Dio1), its expression induced in a genotype-independent manner in steatotic livers, encodes a protein that activates thyroid hormone by conversion of the prohormone thyroxine (T4) to bioactive 3,3′,5-triiodothyronine (T3). DIO1 also degrades both, T3 and T4 by inner ring deiodination. Hyper- as well as hypothyroidism have been associated with insulin resistance likely related to differential effects of thyroid hormones at the liver and peripheral tissues [57]. Targeted disruption of the Dio1 gene results in elevated serum T4 and reverse 3,3′,5′-triiodothyronine (rT3) concentrations but no changes in those of thyroid-stimulating hormone (TSH) and T3 [58]. Genotype-independent induction of Dio1 in fatty liver in this survey was paralleled by a trend towards decreased plasma T4 concentrations (Supplemental Table 6B). Furthermore, the thyroxine-binding globulin (Serpina7) gene was statistically significant induced in steatotic livers of three strains and its expression tended increased in the 129 strain (Supplemental Table 6A). SERPINA7 is primarily synthetized in liver and compared to albumin and transthyretin, expresses the highest affinity for T4 and T3, and carries the majority of T4 in blood. Therefore, data from the current study suggest a relationship between hepatosteatosis and hepatic lipid metabolism and alterations in thyroid hormone metabolism.
Besides differences in HFD composition and exposure duration, substantial genetic variations among mouse strains explain “discrepancies” in metabolic phenotypes observed in the current survey and published studies. Biddinger et al. compared responses of 129S6/SvEvTac and C57Bl/6J mice to long-term HFD exposure [59]. In contrast to data from our survey, 18 weeks HFD intervention produced a modestly higher body mass gain in C57Bl/6J (~2.0 fold) than in 129S6/SvEvTac mice (~1.6 fold). However, when Paigen compared 43 mouse strains following 8 weeks HFD (15% fat content) challenge, C57BL/6J males (~0.98 g) were no more prone to body mass gain than 19S1/SvImJ males (~1.17 g) (http://phenome.jax.org/db/qp?rtn=views/measplot&brieflook=2933&projhint=Paigen; accessed 24 July 2013). The Jackson Laboratory identified a large degree of genetic diversity among the numerous 129 substrains [60,61]. The latter either derive from a congenic strain by outcrossing to introduce the steel mutation (designated by the letter S), from the original parent strain (designated by the letter P), or from a 129 congenic originally carrying the teratoma mutation (designated by the letter T). 129 substrains are widely used for gene targeting experiments. Due to the genetic variability among mouse (sub)strains heterogeneous phenotypes can arise that are not fully explained by differences in targeting constructs, “leakiness” of induced mutations, or environmental factors. Thus correct identification and designation of the strain is of great importance.
In conclusion, systemic phenotypic characterization of four globally used mouse strains outlined distinct genotype-dependent and -independent differences in the development of HFD-induced hepatosteatosis (Figure 4). The 129 strain seems suitable for studying alterations in HFD-induced, early-onset, mild inflammation-associated NAFLD and mechanisms implicated in the progression towards hepatocellular injury. Hepatosteatosis in B6N males was associated with hyperglycemia, in C3H males with hyperinsulinemia, whereas B6J males maintained normoglycemia and normoinsulinemia. Further functional studies in these mouse models could advance our understanding of the relationship between hepatocellular lipid content, obesity, IR, and the role of mitochondrial physiology in the development of NAFLD. Analyses also revealed novel genetic background independent phenotypic and transcriptional adaptations accompanying hepatocellular lipid accumulation. These insights can aid the search for molecular mechanisms linking human NAFLD with insulin resistance, cancer, or thyroid metabolism.
Conflict of interests
The participating authors declared they do not have anything to disclose regarding conflict of interests with respect to this manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank E. Holupirek for outstanding technical assistance, the animal caretakers for their support, G.K.H. Przemeck, P. Huypens, and K. Schwarzenbacher for excellent scientific advice, and B. Naton for editing.
This work was funded in part by a grant from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.) and to the German Mouse Clinic (NGFN-Plus: 01GS0850, 01GS0851; Infrafrontier: 01KX1012) and by BMBF Grant no. 0315494A (SysMBo).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2013.07.009.
Appendix A. Supplementary materials
Supplementary material
Supplementary material
Supplementary material
References
- 1.Milic S., Stimac D. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Digestive Diseases. 2012;30:158–162. doi: 10.1159/000336669. [DOI] [PubMed] [Google Scholar]
- 2.Roden M. Mechanisms of disease: hepatic steatosis in type 2 diabetes-pathogenesis and clinical relevance. Nature Clinical Practice Endocrinology & Metabolism. 2006;2:335–348. doi: 10.1038/ncpendmet0190. [DOI] [PubMed] [Google Scholar]
- 3.Stefan N., Kantartzis K., Haring H.U. Causes and metabolic consequences of fatty liver. Endocrine Reviews. 2008;29:939–960. doi: 10.1210/er.2008-0009. [DOI] [PubMed] [Google Scholar]
- 4.Day C.P. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Samuel V.T., Liu Z.X., Qu X., Elder B.D., Bilz S., Befroy D. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. Journal of Biological Chemistry. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 6.Day C.P. The potential role of genes in nonalcoholic fatty liver disease. Clinics in Liver Disease. 2004;8:673–691. doi: 10.1016/j.cld.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Dowman J.K., Tomlinson J.W., Newsome P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM: Monthly Journal of the Association of Physicians. 2010;103:71–83. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browning J.D., Szczepaniak L.S., Dobbins R., Nuremberg P., Horton J.D., Cohen J.C. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 9.Guerrero R., Vega G.L., Grundy S.M., Browning J.D. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology. 2009;49:791–801. doi: 10.1002/hep.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rainer J., Sanchez-Cabo F., Stocker G., Sturn A., Trajanoski Z. CARMAweb: comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Research. 2006;34:W498–503. doi: 10.1093/nar/gkl038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveros, J.C., 2007. VENNY. An interactive tool for comparing lists with Venn Diagrams. Available from: 〈http://bioinfogp.cnb.csic.es/tools/venny/index.html〉.
- 13.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zambelli F., Pesole G., Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Research. 2009;37:W247–252. doi: 10.1093/nar/gkp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gailus-Durner V., Fuchs H., Becker L., Bolle I., Brielmeier M., Calzada-Wack J. Introducing the German Mouse Clinic: open access platform for standardized phenotyping. Nature Methods. 2005;2:403–404. doi: 10.1038/nmeth0605-403. [DOI] [PubMed] [Google Scholar]
- 16.Bradley A., Anastassiadis K., Ayadi A., Battey J.F., Bell C., Birling M.C. The mammalian gene function resource: the international knockout mouse consortium. Mammalian Genome. 2012;23:580–586. doi: 10.1007/s00335-012-9422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soewarto D., Fella C., Teubner A., Rathkolb B., Pargent W., Heffner S. The large-scale Munich ENU-mouse-mutagenesis screen. Mammalian Genome. 2000;11:507–510. doi: 10.1007/s003350010097. [DOI] [PubMed] [Google Scholar]
- 18.Nolan P.M., Peters J., Strivens M., Rogers D., Hagan J., Spurr N. A systematic, genome-wide, phenotype-driven mutagenesis program for gene function studies in the mouse. Nature Genetics. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 19.Ayadi A., Birling M.C., Bottomley J., Bussell J., Fuchs H., Fray M. Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mammalian Genome: Official Journal of the International Mammalian Genome Society. 2012;23:600–610. doi: 10.1007/s00335-012-9418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown S.D., Moore M.W. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mammalian Genome. 2012;23:632–640. doi: 10.1007/s00335-012-9427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neschen S., Morino K., Dong J., Wang-Fischer Y., Cline G.W., Romanelli A.J. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes. 2007;56:1034–1041. doi: 10.2337/db06-1206. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs H., Gailus-Durner V., Neschen S., Adler T., Afonso L.C., Aguilar-Pimentel J.A. Innovations in phenotyping of mouse models in the German Mouse Clinic. Mammalian Genome. 2012;23:611–622. doi: 10.1007/s00335-012-9415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman H.C., Hugill A., Dear N.T., Ashcroft F.M., Cox R.D. Deletion of nicotinamide nucleotide transhydrogenase. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson A., Reifsnyder P.C., Malcolm R.D., Lucas C.A., MacGregor G.R., Zhang W. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity. 2010;10:1902–1905. doi: 10.1038/oby.2009.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Begriche, K., Massart, J., Robin, M.A., Bonnet, F., and Fromenty, B., 2013. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology (Epub ahead of print). 10.1002/hep.26226. [DOI] [PubMed]
- 26.Coulson P.S., Wilson R.A. Portal shunting and resistance to Schistosoma mansoni in 129 strain mice. Parasitology. 1989;3:383–389. doi: 10.1017/s0031182000059102. [DOI] [PubMed] [Google Scholar]
- 27.Li H., Ji M., Klarmann K.D., Keller J.R. Repression of Id2 expression by Gfi-1 is required for B-cell and myeloid development. Blood. 2010;116:1060–1069. doi: 10.1182/blood-2009-11-255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng W.P. All things considered: transcriptional regulation of Th2 differentiation from precursor to effector activation. Immunology. 2013;140:31–38. doi: 10.1111/imm.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier H., MacPike A.D. Levels and heritability of serum ceruloplasmin activity in inbred strains of mice. Proceedings of the Society for Experimental Biology and Medicine. 1968:1185–1190. [Google Scholar]
- 30.Liebelt A.G., Liebelt R.A., Dmochowski L. Cytoplasmic inclusion bodies in primary and transplanted hepatomas of mice of different strains. Journal of the National Cancer Institute. 1971;47:413–427. [PubMed] [Google Scholar]
- 31.Dragani T.A., Manenti G., Gariboldi M., De Gregorio L., Pierotti M.A. Genetics of liver tumor susceptibility in mice. Toxicology Letters. 1995;82–83:613–619. doi: 10.1016/0378-4274(95)03505-2. [DOI] [PubMed] [Google Scholar]
- 32.Zheng R., Blobel G.A. GATA transcription factors and cancer. Genes & Cancer. 2010;1:1178–1188. doi: 10.1177/1947601911404223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuteja G., Kaestner K.H. SnapShot: forkhead transcription factors I. Cell. 2007;130:1160. doi: 10.1016/j.cell.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Krawczyk M., Bonfrate L., Portincasa P. Nonalcoholic fatty liver disease. Best Practice & Research Clinical Gastroenterology. 2010;24:695–708. doi: 10.1016/j.bpg.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Rohrdanz E., Nguyen T., Pickett C.B. Isolation and characterization of the human glutathione S-transferase A2 subunit gene. Archives of Biochemistry and Biophysics. 1992;298:747–752. doi: 10.1016/0003-9861(92)90475-c. [DOI] [PubMed] [Google Scholar]
- 36.Ishii I., Akahoshi N., Yamada H., Nakano S., Izumi T., Suematsu M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. Journal of Biological Chemistry. 2010;285:26358–26368. doi: 10.1074/jbc.M110.147439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maras B., Barra D., Dupre S., Pitari G. Is pantetheinase the actual identity of mouse and human vanin-1 proteins? FEBS Letters. 1999;461:149–152. doi: 10.1016/s0014-5793(99)01439-8. [DOI] [PubMed] [Google Scholar]
- 38.Berger J.H., Charron M.J., Silver D.L. Major facilitator superfamily domain-containing protein 2a (MFSD2A) has roles in body growth, motor function, and lipid metabolism. PloS One. 2012;7:e50629. doi: 10.1371/journal.pone.0050629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schönthal A.H. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. 2012 Scientifica. 2012:26. doi: 10.6064/2012/857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D., Wie Y., Pagliassotti M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 41.Wie Y., Wang D., Pagliassotti M.J. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Molecular and Cellular Biochemistry. 2007;303:105–113. doi: 10.1007/s11010-007-9461-2. [DOI] [PubMed] [Google Scholar]
- 42.Yuan X., Ta T.C., Lin M., Evans J.R., Dong Y., Bolotin E. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PloS One. 2009;4:e5609. doi: 10.1371/journal.pone.0005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang-Verslues W.W., Sladek F.M. HNF4alpha-role in drug metabolism and potential drug target? Current Opinion in Pharmacology. 2010;10:698–705. doi: 10.1016/j.coph.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin L., Ma H., Ge X., Edwards P.A., Zhang Y. Hepatic hepatocyte nuclear factor 4alpha is essential for maintaining triglyceride and cholesterol homeostasis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:328–336. doi: 10.1161/ATVBAHA.110.217828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forman B.M., Tontonoz P., Chen J., Brun R.P., Spiegelman B.M., Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 46.Jump D.B., Tripathy S., Depner C.M. Fatty acid-regulated transcription factors in the liver. Annual Review of Nutrition. 2013;33:249–269. doi: 10.1146/annurev-nutr-071812-161139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deeb S.S., Fajas L., Nemoto M., Pihlajamaki J., Mykkanen L., Kuusisto J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nature Genetics. 1998;20:284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 48.Miles P.D., Barak Y., He W., Evans R.M., Olefsky J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. Journal of Clinical Investigation. 2000;105:287–292. doi: 10.1172/JCI8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barroso I., Gurnell M., Crowley V.E., Agostini M., Schwabe J.W., Soos M.A. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 50.White D.L., Kanwal F., El-Serag H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clinical Gastroenterology and Hepatology: the Official Clinical Practice Journal of the American Gastroenterological Association. 2012;10(1342–1359):e1342. doi: 10.1016/j.cgh.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Licciulli S., Luise C., Scafetta G., Capra M., Giardina G., Nuciforo P. Pirin inhibits cellular senescence in melanocytic cells. American Journal of Pathology. 2011;178:2397–2406. doi: 10.1016/j.ajpath.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J., Song M., Wang J., Sun M., Wang B., Li R. Enoyl coenzyme A hydratase 1 is an important factor in the lymphatic metastasis of tumors. Biomedicine & Pharmacotherapy=Biomedicine & Pharmacotherapie. 2011;65:157–162. doi: 10.1016/j.biopha.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Li R., Luciakova K., Zaid A., Betina S., Fridell E., Nelson B.D. Thyroid hormone activates transcription from the promoter regions of some human nuclear-encoded genes of the oxidative phosphorylation system. Molecular and Cellular Endocrinology. 1997;128:69–75. doi: 10.1016/s0303-7207(97)04023-9. [DOI] [PubMed] [Google Scholar]
- 54.Enriquez J.A., Fernandez-Silva P., Garrido-Perez N., Lopez-Perez M.J., Perez-Martos A., Montoya J. Direct regulation of mitochondrial RNA synthesis by thyroid hormone. Molecular and Cellular Biology. 1999;19:657–670. doi: 10.1128/mcb.19.1.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pillar T.M., Seitz H.J. Thyroid hormone and gene expression in the regulation of mitochondrial respiratory function. European Journal of Endocrinology/European Federation of Endocrine Societies. 1997;136:231–239. doi: 10.1530/eje.0.1360231. [DOI] [PubMed] [Google Scholar]
- 56.Wrutniak-Cabello C., Casas F., Cabello G. Thyroid hormone action in mitochondria. Journal of Molecular Endocrinology. 2001;26:67–77. doi: 10.1677/jme.0.0260067. [DOI] [PubMed] [Google Scholar]
- 57.Brenta G. Why can insulin resistance be a natural consequence of thyroid dysfunction? Journal of Thyroid Research. 2011;2011:152850. doi: 10.4061/2011/152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider M.J., Fiering S.N., Thai B., Wu S.Y., St, Germain E., Parlow A.F. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147:580–589. doi: 10.1210/en.2005-0739. [DOI] [PubMed] [Google Scholar]
- 59.Biddinger S.B., Almind K., Miyazaki M., Kokkotou E., Ntambi J.M., Kahn C.R. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes. 2005;54:1314–1323. doi: 10.2337/diabetes.54.5.1314. [DOI] [PubMed] [Google Scholar]
- 60.Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E., Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nature Genetics. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 61.Threadgill D.W., Yee D., Matin A., Nadeau J.H., Magnuson T. Genealogy of the 129 inbred strains: 129/SvJ is a contaminated inbred strain. Mammalian Genome: Official Journal of the International Mammalian Genome Society. 1997;8:390–393. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material
Supplementary material