

Abstract
Cytokine signaling has been connected to regulation of metabolism and energy balance. Numerous cytokine gene expression changes are stimulated by accumulation of bile acids in livers of young Foxa2 liver-conditional null mice. We hypothesized that bile acid-induced inflammation in young Foxa2 mutants, once chronic, affects metabolic homeostasis. We found that loss of Foxa2 in the liver results in a premature aging phenotype, including significant weight gain, reduced food intake, and decreased energy expenditure. We show that Foxa2 antagonizes the mammalian target of rapamycin (mTOR) pathway, resulting in increased hepatic lipogenesis and adiposity. While much prior work has focused on adipose tissue in obesity, we discovered a novel age-onset obesity phenotype in a model where gene deletion occurs only in the liver, underscoring the importance of the role hepatic lipogenesis plays in the development of obesity.
Keywords: Bile acids, Aging, Winged helix transcription factors, mTOR pathway, Hepatic lip genesis, Foxa proteins
1. Introduction
Bile acids, amphipathic derivatives of cholesterol, facilitate absorption of dietary lipids. Bile salts also regulate glucose homeostasis [1], liver regeneration [2], and energy balance [3]. Accumulation of hepatic bile acids activates inflammatory signaling [4] and leads to cholestatic liver disease. We have shown that hepatocyte-specific ablation of a liver-enriched transcription factor Foxa2, previously known as HNF-3β, results in decreased expression of genes encoding bile acid transporters, leading to intrahepatic cholestasis [5].
Numerous studies have connected cytokine signaling to regulation of metabolic homeostasis and energy balance [6,7]. Obesity, caused either by excessive food intake or genetic predisposition, is associated with low-grade chronic inflammation characterized by secretion of pro-inflammatory cytokines (adipokines), small immunomodulatory molecules, by adipose tissue and activation of inflammatory signaling pathways [8]. Cytokine signaling, propagated by the receptors on the plasma membrane, converges on phosphorylation and nuclear translocation of signal transducer and activator of transcription (Stat) DNA-binding proteins and activation of their target genes. Important in this context are defects in Stat5b regulation, which, like Foxa2, regulates targets involved in synthesis, transport, and detoxification of bile acids [9]. Stat5b plays an anti-inflammatory role and activates expression of hepatoprotective genes in a cholestatic setting [10]. Mutations in Stat5b, which also mediates growth hormone-dependent sexually dimorphic gene expression in the male liver [11], have been associated with spontaneous age-onset obesity in males [12–14].
Activation of the inflammatory response regulated by NF-κB is involved in accelerated aging [15]. Aging is associated with increased prevalence of metabolic disease [16]. Inflammatory cytokines also activate mammalian “Target of rapamycin” or mTOR, an evolutionarily conserved serine/threonine protein kinase that integrates nutrient and growth factor signals to regulate metabolism and cellular growth [17,18]. Reducing mTOR pathway activity is beneficial to slow aging and the onset of age-related diseases [19]. Several observations indicate that bile acids are likely to be important in regulating metabolic changes during aging. In C. elegans, bile acid-like compounds, named dafachronic acids (DA), are endogenous ligands of DAF-12, a nuclear receptor homologous to the farnesoid X (FXR) and liver X receptors (LXR), that mediates responses to environmental conditions during development and aging [20]. In addition, expression analysis in long-lived Little mice revealed upregulation of bile acid-induced FXR-dependent xenobiotic detoxification targets [21], indicating that FXR plays a protective role in increased stress resistance, beneficial for longevity.
We hypothesized that bile acid-induced inflammation in young Foxa2 mutants, once chronic, affects global metabolic homeostasis, and found that loss of Foxa2 in the liver of aged mice results in a premature aging phenotype, as Foxa2loxP/loxPAlfp.Cre mice exhibit significant weight gain, reduced food and drink intake, and decreased energy expenditure. We show that ablation of Foxa2 in the liver leads to activation of the mTOR pathway, resulting in increased hepatic lipogenesis and overall adiposity. These findings underscore the importance of hepatic lipogenesis in the development of obesity.
2. Materials and methods
2.1. Animals
The derivation of the Foxa2loxP/loxP;Alfp.Cre mouse model has been reported previously [22]. Male mice of different age groups as indicated in the text were used for metabolic studies. Mice were genotyped by PCR of tail DNA as described [22].
2.2. Serum chemistry
Serum acute phase proteins and adipokines were measured using Luminex kits (Millipore). HDL cholesterol levels were determined by AniLytics, Inc. Growth hormone levels were measured using an ELISA kit (ALPCO Diagnostics).
2.3. Metabolic measurements
Energy expenditure, food and drink intake, oxygen consumption and carbon dioxide production, as well as locomotor activity were assessed with a Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) [23]. Body composition was measured in conscious mice by nuclear magnetic resonance (NMR) (Echo Medical Systems, Houston, TX).
2.4. De novo lipogenesis
Rapamycin treatment was performed as reported previously [24]. Briefly, mice were fasted overnight (~16 h), re-fed with a high carbohydrate diet, injected with either vehicle or vehicle containing rapamycin (20 mg/kg) an hour into re-feeding, and sacrificed after 6 h of refeeding. The mice were also injected with deuterated water at the onset of re-feeding (20 µl/g body weight). Lipids were extracted from the liver and fatty acid composition was analyzed using gas chromatography–mass spectrometry as described [25].
2.5. RNA isolation and expression analysis
Liver RNA was isolated from Foxa2loxP/loxP;Alfp.Cre and control littermates, and quantitative reverse transcription-PCR performed as described [22]. Hybridization to Agilent 4×44 k Whole Mouse Genome Oligo Microarray and microarray analysis were completed as reported previously [26]. Four individual samples for each genotype were analyzed. The microarray data from this study can be accessed at ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession nos. E-MEXP-2106 (young Foxa2 mutants) and E-MTAB-1825 (old Foxa2 mutants).
2.6. Western blot analysis
Whole cell lysates preparation and western blot analysis was as completed as described [27]. The primary antibodies used were against Akt (Cell Signaling, #9272), phospho-Akt (Ser 473)(Cell Signaling, #9272), Foxo1 (Cell Signaling, #9454), phospho- Foxo1 (Ser316) (Cell Signaling, #2486), p70 S6-kinase (Refs. [42,28]), phospho-p70 S6 Kinase (Thr389) (Cell Signaling, #9206), 4E-BP1 (Cell Signaling, #9644), phospho-4E-BP1 (Thr37/46) (Cell Signaling, #2855), and β-actin (Abcam, ab8227).
2.7. Chromatin immunoprecipitation
ChIP and the real-time PCR reactions that followed were performed as previously described [5]. Snap-frozen mouse liver (100 mg) from wild type mice was used to prepare chromatin. We performed immunoprecipitation with a rabbit antiserum specific to Foxa2 [29].
2.8. Data analysis
The DAVID 6.7 suite of software and Ingenuity Pathway Analysis (IPA, Ingenuity Systems, http://www.ingenuity.com) were used to determine enriched biological functional categories in the set of differentially expressed genes as reported previously [26,30]. Ingenuity Pathways Analysis was utilized to determine transcription factor networks enriched in Foxa2 targets. IPA calculates a statistical overlap between the gene set known to be regulated by each transcription factor (manually curated by Ingenuity) and the subset of those genes found in the given list of differentially expressed genes. The heatmap was generated using GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E/index.html).
2.9. Statistical analyses
We compared quantitative PCR data by unpaired Student's t-test and metabolic measurements by Mann–Whitney U test or ANCOVA. A p-value below 0.05 was considered significant for all analyses. All values are represented as means±s.e.m.
3. Results
3.1. Accumulation of bile acids in livers of young Foxa2-deficient mice induces inflammatory gene expression
Deletion of the winged helix transcription factor Foxa2 in the liver leads to mild cholestasis in young mice (Foxa2loxP/loxPAlfp.Cre, 2–3 months of age), without causing liver injury [5]. Similarly, treatment of isolated hepatocytes with bile acids in vitro activates expression of numerous inflammatory markers without causing cell toxicity [4]. Based on this finding, we re-examined the gene expression profile of young Foxa2-deficient mice. Functional analysis revealed that ‘Inflammatory Response’, ‘Acute Phase Signaling’, and ‘Hepatic Stellate Cell Activation’ pathways were highly overrepresented among these targets. Hepatic stellate cells are activated by proinflammatory cytokines during liver inflammation [31].
We employed computational network analysis to investigate which transcription factors mediated the gene expression changes observed in Foxa2loxP/loxPAlfp.Cre mice. The Ingenuity Pathway Analysis (IPA) tool calculates a statistical overlap between the gene set known to be regulated by each transcription factor (as curated by Ingenuity) and the subset found in a list of differentially expressed genes. Targets of Stat3 (p-value 4.21e-10), a transcription factor that transduces cytokine signaling, and Irf3 (p-value 4.76e-4) and NF-κB (p-value 3.90e-4), which modulate inflammatory signaling, were activated in Foxa2 mutants (Figure 1A). The regulatory network featuring inflammatory signaling activated by Stat3, Irf3, and NF-κB is comprised of fifty-eight genes that are differentially expressed in livers of Foxa2-deficient mice (53 upregulated in red, 5 downregulated in green, Figure 1B, Supplementary Table 1). The transcription factors Irf5, Egr1, and Stat3, and its targets, Stat1 and Socs3, are among genes induced in young Foxa2 mutants. Stat3 regulates the majority of the genes shown, including the cytokines, chemokines, and adhesion molecules that are also induced by bile acids [4], and expression of many targets is controlled by two of the transcription factors in the network.
Figure 1.
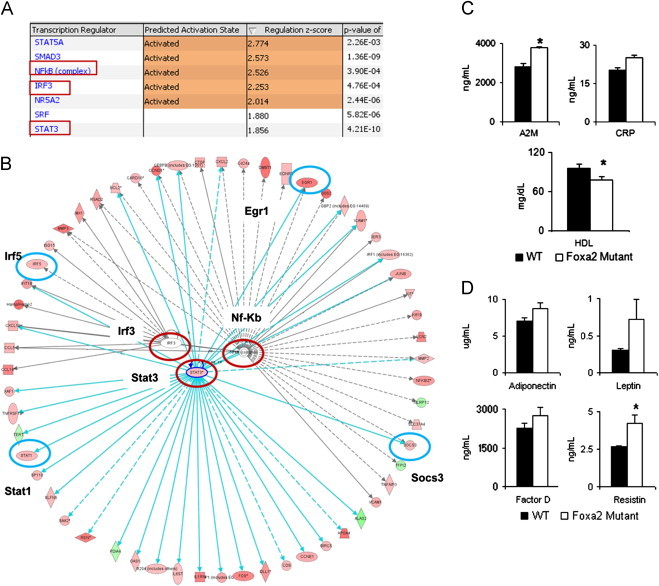
Inflammatory signaling is induced in young Foxa2 mutants. (A) A list of transcription factors shown to be activated and responsible for gene expression changes observed in livers of Foxa2 mutants as determined by Ingenuity Pathway Analysis. These include Stat3, which transduces cytokine signaling, and Irf3 and NF-κB, which modulate inflammatory signaling. The statistical significance of the overlap between the gene set known to be regulated by each transcription factor (curated by Ingenuity) and the subset of that set in differentially expressed genes regulated by Foxa2 is indicated as the p-value in the last column. (B) The regulatory network featuring inflammatory signaling activated by Stat3, Irf3, and NF-κB is comprised of fifty-eight genes differentially expressed in livers of Foxa2-deficient mice (53 upregulated in red, 5 downregulated in green). Transcription factors Irf5, Egr1, as well as Stat3, and its targets, Stat1 and Socs3, are among the genes that are induced in young Foxa2 mutants. Stat3 regulates the majority of the genes shown, and expression of many targets is controlled by two of the transcription factors in the network. Color intensity indicates the magnitude of change in gene expression in the absence of Foxa2. Genes in the network are listed in Supplementary Table 1. (C) Serum levels of acute phase reactants alpha-2-Macroglobulin (A2M) and C-reactive protein (CRP) are increased in Foxa2loxP/loxPAlfp.Cre mice (p<0.0002 for A2M, p<0.07 for CRP, Figure 1C). High-density lipoprotein (HDL) cholesterol levels, reduced during acute phase response, are significantly decreased in young Foxa2-deficient mice. (D) Circulating levels of adipokines increase in Foxa2 mutants (adiponectin, p<0.08, complement factor D, p<0.15, leptin, p<0.09, resistin, p<0.03).
The acute phase response (APR) is the organism's defense reaction to inflammation, involving synthesis and secretion of acute phase proteins by the liver into circulation. We analyzed acute phase reactants in serum of young Foxa2 mutants and found that levels of alpha-2-Macroglobulin (A2M) and C-reactive protein (CRP), proteins synthesized by the liver and induced by APR, were increased in Foxa2loxP/loxPAlfp.Cre mice (p<0.0002 for A2M, p<0.07 for CRP, Figure 1C). High-density lipoprotein (HDL) cholesterol levels, reduced during APR [32], were also significantly decreased in young Foxa2-deficient mice (Figure 1C). In addition, mRNA levels of Apoa1, a lipoprotein associated with HDL cholesterol, were reduced in Foxa2loxP/loxPAlfp.Cre mice. Together, these data suggest that inflammatory gene expression activated by bile acids leads to activation of the APR in the liver.
In order to ascertain whether hepatic inflammation and induction of the APR affected Foxa2 mutants systemically, we also measured serum concentration of adipokines, inflammatory mediators and hormones secreted by the adipose tissue. Serum levels of adiponectin showed a trend towards an increase (p<0.08, Figure 1D). Serum adiponectin is elevated in cholestatic patients and correlates with hepatic inflammation [33,34]. In addition, serum levels of resistin were induced two-fold (p<0.03, Figure 1D) in young Foxa2-deficient mice. Resistin is a pro-inflammatory protein whose levels have been positively correlated with markers of liver injury [35]. Circulating levels of leptin and complement factor D, both elevated in obese individuals, showed a trend towards an increase (p-value <0.09 for leptin, p-value<0.15 for factor D, Figure 1D). Hence, changes in inflammatory gene expression in the liver induced by bile acids lead to changes in adipokine secretion in young Foxa2 mutants prior to onset of obesity.
3.2. Growth hormone-dependent Stat5b signaling is reduced in Foxa2loxP/loxPAlfp.Cre mice
Similar to Foxa2, Stat5b is important in bile acid homeostasis, regulating targets involved in synthesis, transport and detoxification of these lipophilic compounds [9]. Stat5b plays an anti-inflammatory role and activates expression of hepatoprotective genes, especially in the cholestatic setting [10]. Since Stat3 signaling was upregulated in young Foxa2 mutants and members of the Stat transcription factor family compensate for each other [36], we next explored whether signaling by Stat5b was dysregulated in Foxa2loxP/loxPAlfp.Cre mice. Stat5b is a mediator of circulating growth hormone (GH) pulses on sexually dimorphic hepatic gene expression [37] and plays an important role in the male liver [11]. Indeed, out of 229 transcripts differentially expressed in livers of young Stat5b knockout mice and Foxa2 mutants, 192 changed in the same direction (128 upregulated in red, 64 downregulated in blue, Figure 2A, Supplementary Table 2). In addition, overlap analysis of binding sites revealed that Foxa2 and Stat5 [38] share 3276 common regulatory elements in the liver (Figure 2B). Hence, Foxa2 and Stat5b co-regulate several hundred genes in the male liver, since deletion of either Foxa2 or Stat5b has the same effect on expression of these genes.
Figure 2.
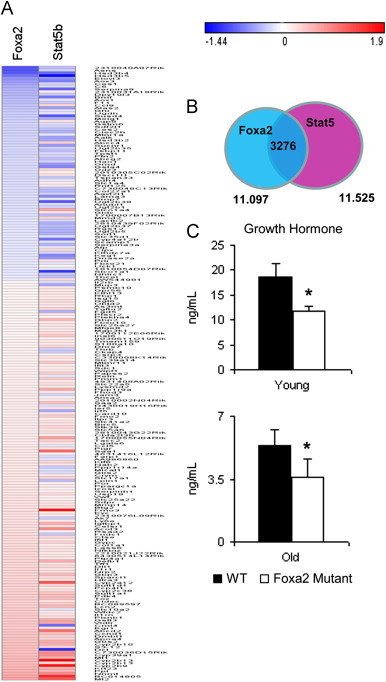
Growth hormone-dependent Stat5b signaling is reduced in Foxa2loxP/loxPAlfp.Cre mice. (A) Heat map generated for genes that are differentially expressed in both Foxa2 (2–3 months old males) and Stat5b (8–9 week old males) liver-specific mutants (|FC|>=1.3, FDR<10%). Out of 229 genes, 192 are changed in the same direction (128 upregulated and 64 downregulated). Scale of the heatmap is log based. Genes and fold changes in both Foxa2 and Stat5b mutants are listed in Supplementary Table 2. (B) Venn diagram showing the overlap of genome-wide location analysis for Foxa2 (11,097 sites) and Stat5 (11,525 sites peaks identified in Stat5 high-activity livers)[38] in the adult liver (2–3 months old). The two transcription factors share 3276 sites in common (30% of total Foxa2 sites and 28% of total Stat5 sites). (C) Serum growth hormone (GH) levels are significantly decreased in Foxa2-deficient mice of all ages.
Next, we measured circulating growth hormone (GH) levels in young and old Foxa2 mutants. GH pulses decline in magnitude and GH-dependent gene expression is reduced in older males [39]. Serum growth hormone (GH) levels were significantly decreased in young Foxa2-deficient mice are reduced even further in older mutants (Figure 2C). Together, changes in circulating GH levels and expression of hundreds of Stat5b targets in Foxa2loxP/loxPAlfp.Cre mice suggest that gene expression in young Foxa2 mutants exhibits features of old males.
3.3. Foxa2 mutants develop age-onset obesity
Stat5b regulates body composition and is associated with maturity-onset obesity in humans and rodents [13,14]. Therefore, we investigated how metabolic homeostasis of Foxa2-deficient mice was affected with aging. While young Foxa2loxP/loxPAlfp.Cre mice (2–3 months of age) weighed the same as their control littermates, 8-month and 11-month old mutants were 10% and 20% heavier than controls, respectively (Figure 3A). The 8-month old Foxa2 mutants (intermediate phenotype) already exhibited much larger triglyceride depots than those of control mice (Figure 3A, right panels). In addition, NMR composition analysis of these animals revealed that weight gain was mediated by increases in both fat and lean mass (Figure 3A, lower left panel).
Figure 3.
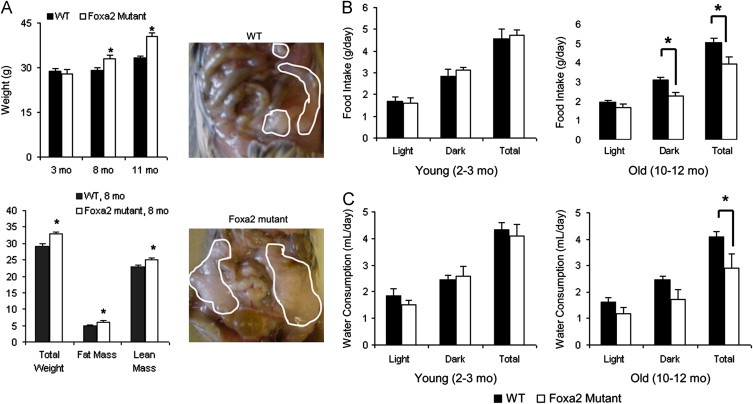
Foxa2 mutants exhibit features of a premature aging phenotype. (A) Young mice deficient for Foxa2 in the liver (Foxa2loxP/loxPAlfp.Cre, 2–3 months of age, n=8 per group) have the same weight as their control littermates, while cohorts of older mutants (8–9 months, n=7 or 9 per group and 10–12 months, n=4 or 6 per group) are 10% and 20% heavier than controls, respectively (upper left panel). The middle group (8–9 months old Foxa2 mutants, intermediate phenotype) already exhibits fat depots (outlined by white circles) that are much larger than those of control mice of the same age (right panels). NMR composition analysis of 8–9 months old Foxa2 mutants animals shows that weight gain is mediated by increases in both fat and lean mass (lower left panel). (B and C) Young Foxa2-deficient animals (2–3 months old, n=8 per group) maintain the same food and drink intake as their control littermates, while aged Foxa2 mutants (10–12 months old, n=4 or 6 per group) eat and drink less than controls. Values are represented as means plus standard error. p-Values were determined by Mann–Whitney U test. *p-value <0.05.
In order to ascertain why Foxa2 mutants gained more weight, we utilized the Comprehensive Laboratory Animal Monitoring System (CLAMS) to measure a variety of physiological and behavioral parameters. While young Foxa2-deficient animals maintained the same food and drink intake as their control littermates, paradoxically, aged Foxa2 mutants ate and drank less than controls (Figure 3B and C). We utilized ANCOVA or generalized linear modeling, the current consensus method, to normalize the effects of body weight on the parameters of energy expenditure, including VO2 consumption, CO2 production, and the amount of heat generated [40]. Old Foxa2loxP/loxPAlfp.Cre mice consumed less oxygen (Figure 4A) and exhaled less carbon dioxide during the observation period (Figure 4B). Analysis of covariance to test the dependence of VO2 consumption and CO2 production on genotype adjusted for body weight was highly significant for reduced energy expenditure (ANCOVA p<0.001 for both VO2 and CO2, Supplementary Figure 1). Heat production was similar for young Foxa2 mutants and their control littermates as well as older Foxa2-deficient mice (ANCOVA p<0.15, Figure 4C). Physical activity, measured as a sum of infrared beam breaks per day, although lower in all older animals, also did not differ significantly between mutant and control mice (Figure 4C). A recent report demonstrated that deletion of a cytokine (cardiotrophin-1, a member of the gp130 family) also results in mature-onset obesity despite reduced food intake [7]. However, while we see multiple changes in circulating cytokine levels in young Foxa2 mutants, leptin levels rise only with the onset of the obesity phenotype in cardiotrophin-1-deficient animals.
Figure 4.
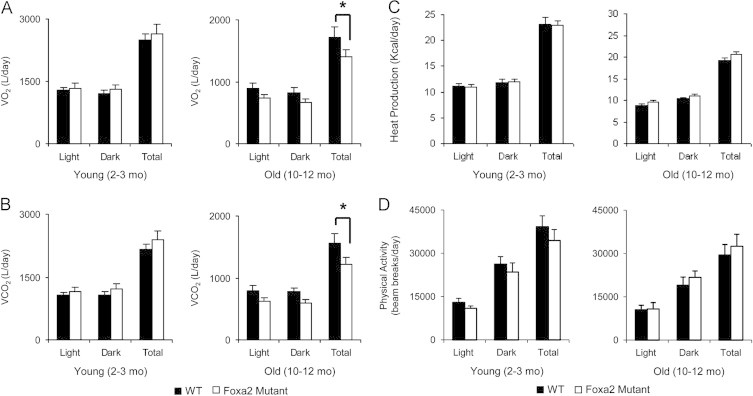
Respiration, energy expenditure, and physical activity in Foxa2-deficient mice. (A) While young Foxa2loxP/loxPAlfp.Cre mice (2–3 months, n=8 per group) consume the same amount of oxygen as controls, aged Foxa2 mutants (10–12 months, n=4 or 6 per group) utilize less oxygen compared to control littermates (ANCOVA, p<0.001). (B) Older Foxa2-deficient mice (10–12 months, n=4 or 6 per group) exhale less carbon dioxide (ANCOVA, p<0.001). (C) Heat production is similar for young Foxa2 mutants and their control littermates (2–3 months, n=8 per group) and old Foxa2-deficient animals (10–12 months, n=4 or 6 per group) (ANCOVA, p<0.15). (D) Physical activity, measured as a sum of infrared beam breaks per day, although lower in all older animals, did not differ significantly between mutant and control mice. Values are represented as means plus standard error. p-Values were determined by ANCOVA. *p-value<0.05.
In summary, aged Foxa2loxP/loxPAlfp.Cre mice display significant weight gain, reduced food and drink intake, and decreased energy expenditure. Fat accumulation, anorexia, and reduced energy expenditure are associated with aging [41,42], and by these parameters Foxa2 mutants exhibit a premature aging phenotype. Changes in cytokine signaling modulated by Stat3, NF-κB, Irf3, and Stat5b, and in circulating levels of cytokines preceded the manifestation of the metabolic phenotype. While defects in Stat5b signaling have been associated with spontaneous age-onset obesity, no clear mechanism to the cause of the phenotype has been elucidated previously.
3.4. Foxa2 represses the mTOR pathway
Inflammatory cytokines have been shown to activate mTOR [17,18]. In addition, a collection of targets regulated by mTOR signaling was upregulated in livers of aged Foxa2 mutant mice (Supplementary Table 3). Therefore, we hypothesized that mTOR signaling might be dysregulated in the Foxa2 mutants. mTOR forms two structurally and functionally distinct complexes, mTORC1 and mTORC2 [43]. We analyzed expression of multiple pathway components in liver extracts of old Foxa2loxP/loxPAlfp.Cre mice and their control littermates in both basal fasted condition and following intraperitoneal (IP) injection of insulin [44]. Phosphorylation of S6 kinase (S6K), the primary substrate of mTORC1, was upregulated in fasted Foxa2 mutants compared to littermate controls and comparable to the levels in Foxa2 deficient animals injected with insulin (Figure 5A). Phosphorylation of eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), another substrate of mTORC1, was also elevated in aged Foxa2loxP/loxPAlfp.Cre mice (Figure 5A). Next, we performed western blot analysis for Akt and its target Foxo1, regulated by the mTORC2 complex, and saw no differences in phosphorylation of either protein in livers of Foxa2-deficient mice in both control and insulin-injected conditions (Figure 5A). Thus, mTORC1, but not mTORC2, is activated in the liver in the absence of Foxa2.
Figure 5.
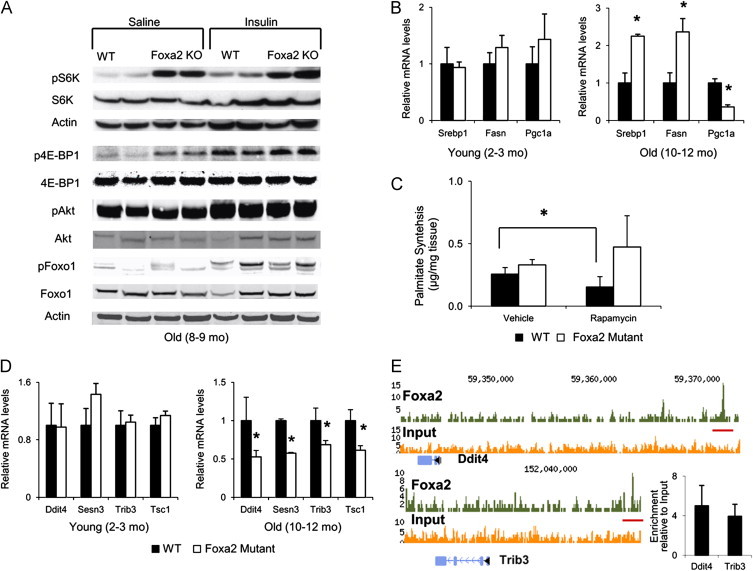
Foxa2 represses the mTOR pathway. (A) Western blot analysis of protein whole cell liver extracts for expression of p70 S6 kinase and eukaryotic translation initiation factor 4E binding protein 1, primary substrates of mTORC1, and Akt and Foxo1, substrates of mTORC2. Phosphorylation of S6 kinase, the primary substrate of mTORC1, is already upregulated in fasted Foxa2 mutants (10–12 months old) compared to littermate controls, and comparable to the levels in Foxa2-deficient animals (10–12 months old) injected with insulin. Phosphorylation of 4E-BP1 is increased in old Foxa2 mutants in both conditions. Western blot analysis for Akt and its target Foxo1 shows no difference in phosphorylation of either in livers of aged Foxa2-deficient mice in both conditions. (B) Expression of Srebp1, Fasn, and Pgc1α in liver RNA of wild-type and Foxa2loxP/loxPAlfp.Cre mice shows a significant increase for Srebp1 and Fasn, and a significant decrease for Pgc1α in old (10–12 months old) Foxa2 mutants as compared to controls. Values are represented as means plus standard error. p-Values were determined by Student's t-test. *p-value<0.05. (C) The hepatic de novo lipogenesis rate was measured in vivo by tracing newly synthesized palmitate from deuterated water, in presence and absence of the mTOR inhibitor rapamycin. While rapamycin treatment decreased the rate in old wild type mice (12 months old), no significant changes were observed in Foxa2 mutants (n=4 per group). Values are represented as means plus standard error. p-Values were determined by Mann–Whitney U test. *p-value<0.05. (D) Quantitative RT-PCR analysis for mRNA of Ddit4, Sesn3, Trib3, and Tsc1, repressors of mTOR. mRNA levels of these genes, while not changed in young Foxa2 mutants (2–3 months old, n=4 per group), are significantly decreased in older Foxa2-deficient mice (10–12 months old, n=4 or 6 per group) compared to controls. Values are represented as means plus standard error. p-Values were determined by Student's t-test. *p-value<0.05. (E) ChIP-Seq analysis of promoter occupancy of Ddit4 and Trib3 in liver chromatin from wild-type or Foxa2loxP/loxPAlfp.Cre mice. Upstream Foxa2 binding sites are indicated by the orange bars. Enrichment of promoter amplicons is confirmed by qPCR (n=4).
Considering that aged Foxa2-deficient animals accumulate fat, we hypothesized that de novo lipogenesis was also increased in the livers of Foxa2-deficient mice. Hwahng and colleagues showed that S6K activates LXRα-dependent gene transcription [45], inducing insulin-dependent transcription of Srebp1-c, a transcription factor mediating fatty acid synthesis in the liver [46,47], and suppressing expression of Pgc1α, a co-activator important for gluconeogenesis [48]. While expression of these genes was not changed in the livers of 3-month old Foxa2loxP/loxPAlfp.Cre mice compared to controls, mRNA levels of Srebp1-c and its target Fasn were significantly increased, and expression of Pgc1α was significantly decreased in the livers of aged Foxa2 mutant mice, confirming the results of the gene expression profile (Figure 5B, Supplementary Table 4). A recent study by Mueller and colleagues reported that Stat5 regulates de novo lipogenesis in the liver, as deficiency of Stat5 results in upregulation of Srebp1-c and its transcriptional targets [49]. Hence, reduced Stat5b signaling could also contribute to changes in lipid metabolism observed in Foxa2-deficient mice.
Although mTORC1 and mTORC2 are functionally distinct, both complexes can regulate de novo lipogenesis [43]. In order to ascertain whether activation of mTORC1 signaling was responsible for changes observed in Foxa2-deficient animals, we measured the hepatic de novo lipogenesis rate in vivo, by tracing newly synthesized palmitate from deuterated water, in the presence or absence of the mTOR inhibitor rapamycin. While a recent study has reported that chronic rapamycin treatment can disrupt mTORC2 in addition to mTORC1 signaling [50], we administered only one acute rapamycin injection (details in Materials and Methods), which suppresses only the activity of mTORC1. In this experiment, we employed physiological conditions to maximally stimulate lipogenesis by analyzing mice first fasted and then re-fed a carbohydrate-rich diet. The de novo lipogenesis rate of wild type mice decreased as expected when treated with rapamycin, but not in Foxa2 mutants (Figure 5C), confirming that the mTORC1 pathway is constitutively active in aged Foxa2loxP/loxPAlfp.Cre mice.
Next, we pursued the connection between loss of Foxa2 and increased mTORC1 signaling. Since Foxa2 is a transcriptional activator [51], we hypothesized that a repressor of mTOR is regulated by Foxa2. While expression of four such repressors, Ddit4, Sesn3, Trib3, and Tsc1 [52–55], did not differ between young Foxa2-deficient animals and their control littermates, mRNA levels of these genes were significantly reduced in livers of older Foxa2 mutants (Figure 5D). We identified Foxa2 binding sites upstream of the transcription start sites of Ddit4 and Trib3 by ChIP-Seq [56] and confirmed that Foxa2 indeed occupies these cis-regulatory regions by quantitative PCR, indicating that at least two of these mTOR repressors are direct Foxa2 targets (Figure 5E).
In summary, aged Foxa2 mutant mice exhibit a premature aging phenotype, with reduced feeding behavior, decreased energy expenditure, and increased triglyceride accumulation. Other features of the phenotype include: (1) inflammatory gene expression modulated by NF-κB, which is hyperactivated in mouse models of premature aging [57], (2) hepatic gene expression in young Foxa2-deficient mice resembling that of older males due to reduced growth hormone levels and Stat5b signaling, and (3) decreased expression of FXR-dependent detoxification genes [26] that leads to reduced stress resistance in long-lived mice [21] (Figure 6A and B). The metabolic changes are preceded by improper regulation of cytokine signaling in young Foxa2loxP/loxPAlfp.Cre mice. Foxa2 and its targets antagonize the mammalian target of rapamycin (mTOR) pathway, and absence of this inhibition, in addition to reduced Stat5b signaling in liver-specific Foxa2 mutants, contributes to induction of Srebp1-c, increased hepatic lipogenesis, and overall adiposity in aged mice (Figure 6С).
Figure 6.
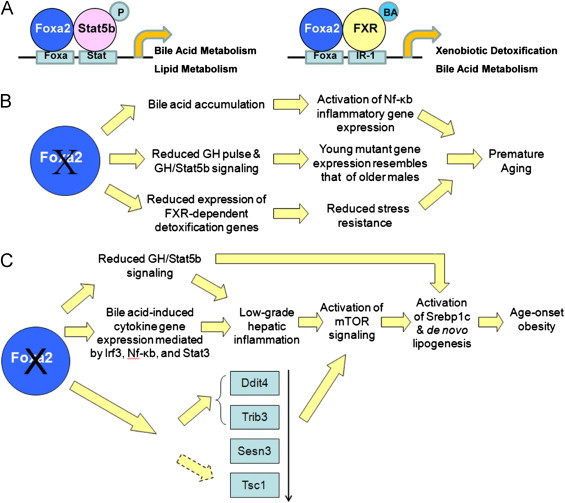
A model of Foxa2 deletion leading to premature aging and age-onset obesity. (A) Cis-regulatory modules regulated by Foxa2. Once Stat5b is activated, phosphorylated (P), and translocates to the nucleus, together with Foxa2 the factor turns on genes important in synthesis, transport, and detoxification of bile acids (left panel). A bile acid (BA) ligand bind to FXR, activating the nuclear receptor, and FXR in synergy with Foxa2 induces targets essential for xenobiotic detoxification (right panel). (B) Deletion of Foxa2 leads to a premature aging phenotype. Accumulation of bile acids in Foxa2 mutants leads to activation of inflammatory gene expression modulated by Nf-κb, which is hyperactivated in mouse models of premature aging. Hepatic gene expression in young Foxa2-deficient mice resembles that of older males due to reduced growth hormone levels and Stat5b signaling. Decreased expression of FXR-dependent detoxification genes leads to reduced stress resistance in a long-lived mouse model. These features of Foxa2 mutants lead to premature aging. (C) Deletion of Foxa2 in the liver leads to age-onset obesity. Young Foxa2 mutants accumulate bile acids that induce cytokine signaling and low-grade inflammation mediated by activated Irf3, Stat3, and NF-κB. In addition, Foxa2 co-regulates many genes with Stat5b, a hepatoprotective and anti-inflammatory factor. Together, activation of Stat3 and reduced Stat5b signaling lead to low-grade hepatic inflammation in young Foxa2 mutants. Foxa2 and its targets, two of which are direct, normally antagonize the mammalian target of rapamycin (mTOR) pathway. Reduced expression of repressors of mTOR and presence of inflammatory cytokines contribute to activation of mTOR in livers of older Foxa2-deficient mice. Increased mTOR activity and reduced Stat5b signaling lead to induction of Srebp1-c and de novo lipogenesis in the liver, resulting in age-onset obesity.
4. Discussion
Although bile acids have been linked to energy homeostasis, bile-acid induced changes in Foxa2 mutants and their contribution to premature aging are unique to our model. Bile acids are known to reduce triglyceride levels via repression of Srebp-1c [58] and increase energy expenditure by activating thyroid hormone in brown adipose tissue [3]. However, young Foxa2-deficient mice with mild cholestasis do not exhibit changes in expression of Srebp1-c. We also observed no changes in energy expenditure in young Foxa2loxP/loxPAlfp.Cre mice, as their circulating bile acid levels do not differ from those in control littermates and increase only when the mice are fed a cholic acid-enriched diet [5]. However, bile-acid induced inflammatory signaling leads to activation of NF-κB that is involved in accelerated aging [15], and induction of mTOR, resulting in metabolic changes in older animals. In C. elegans, bile acid-like compounds, named dafachronic acids (DA), are endogenous ligands of DAF-12, a nuclear receptor homologous to farnesoid X (FXR) and liver X receptor (LXR), that mediates responses to environmental conditions during development and aging [20]. It is likely that bile acids are also important in regulating metabolic changes during mammalian aging, as suggested by our model. In addition, microarray expression analysis in long-lived Little mice revealed upregulation of bile acid-induced FXR-dependent xenobiotic detoxification targets [21], indicating that FXR plays a protective role in increased stress resistance. We have shown previously that activation of FXR by bile acids is Foxa2-dependent [26], suggesting that Foxa2 is also important in modulating adaptive changes in these mice.
While obesity-induced inflammation leads to insulin resistance, hepatic inflammatory signaling in our mice leads to changes downstream of the insulin receptor at the level of mTORC1, and connects Foxa2 to mTOR signaling. Similarly, in C. elegans, pha-4, a forkhead box transcription factor orthologous to the vertebrate Foxa transcription factor family, and TOR antagonize each other [59]. We have observed that expression of FOXA2 is severely downregulated in patients with cholestatic liver disease [5], a set of conditions associated with inflammation and injury, while lipopolysaccharide administration greatly reduces Foxa2 protein levels in the mouse liver [60]. These observations suggest that Foxa2 is repressed by cytokine signaling and could be downregulated in inflammatory conditions in order to induce the mTOR pathway. In fact, Tnfα signaling has recently been shown to suppress Foxa2 [61], akin to repressing Tsc1, to activate the mTOR pathway [17].
We have also demonstrated that Foxa2 co-regulates expression of numerous genes with Stat5b in the liver of male mice. Stat5b regulates a number of targets involved in synthesis, transport and detoxification bile acids [9]. A recent study reported that Stat5 regulates de novo lipogenesis in the liver, as deficiency of Stat5 resulted in upregulation of Srebp1-c and its transcriptional targets. Stat5loxP/loxP; Alfp.Cre mutants also exhibit elevated circulating resistin levels [49]. Hence, Foxa2 and Stat5b play overlapping roles in hepatic gene regulation as similar changes are observed in Foxa2 and Stat5b liver-specific mutants. While defects in Stat5b signaling have been associated with spontaneous age-onset obesity in males, no clear mechanism to the cause of the phenotype had been elucidated thus far. We propose that Foxa2 is the essential link between coordinating proper gene expression with Stat5b in the male liver by repressing the mTOR pathway in inflammatory conditions, preventing lipogenesis and increased adiposity. While most investigators have focused on the role of adipose tissue in obesity [62–64], we describe a novel age-onset obesity phenotype in a mouse model where gene deletion occurs only in the liver, underscoring the importance of the role hepatic lipogenesis plays in the development of obesity. Novel approaches to target lipogenesis in the liver through activation of Foxa2 or by other means might benefit in combating obesity and the metabolic syndrome.
Conflict of interest
The authors have declared that no conflict of interest exists.
Author contributions
I.M.B performed experiments and data analysis. S.S. performed experiments. I.M.B and K.H.K wrote the manuscript. K.H.K directed the study.
Acknowledgments
We thank M. Wan, K. Leavens, and M. Birnbaum for advice on insulin injections and critical reading of the manuscript; J.A. Whittsett (Cincinnati Children's Hospital Medical Center and The University of Cincinnati College of Medicine) for providing rabbit polyclonal antibodies to Foxa2; V. Kameswaran, R. Dhir, J. Millar, and H. Collins for contributions to this project. This work was supported by NIH Grant DK-P01-049210 to K.H.K. We thank the Mouse Phenotyping, Physiology and Metabolism Core and Functional Genomics Core of the Penn Diabetes Research Center (P30-DK19525), for experimental assistance.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2013.08.005.
Appendix A. Supplementary materials
Supplementary Material
References
- 1.Ma K., Saha P.K., Chan L., Moore D.D. Farnesoid X receptor is essential for normal glucose homeostasis. Journal of Clinical Investigation. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang W., Ma K., Zhang J., Qatanani M., Cuvillier J., Liu J. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe M., Houten S.M., Mataki C., Christoffolete M.A., Kim B.W., Sato H. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 4.Allen K., Jaeschke H., Copple B.L. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. American Journal of Pathology. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bochkis I.M., Rubins N.E., White P., Furth E.E., Friedman J.R., Kaestner K.H. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nature Medicine. 2008;14:828–836. doi: 10.1038/nm.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Febbraio M.A. gp130 receptor ligands as potential therapeutic targets for obesity. Journal of Clinical Investigation. 2007;117:841–849. doi: 10.1172/JCI30453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno-Aliaga M.J., Perez-Echarri N., Marcos-Gomez B., Larequi E., Gil-Bea F.J., Viollet B. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metabolism. 2011;14:242–253. doi: 10.1016/j.cmet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 9.Baik M., Yu J.H., Hennighausen L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Annals of the New York Academy of Sciences. 2011;1229:29–37. doi: 10.1111/j.1749-6632.2011.06100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaas L., Kornfeld J.W., Schramek D., Musteanu M., Zollner G., Gumhold J. Disruption of the growth hormone--signal transducer and activator of transcription 5--insulinlike growth factor 1 axis severely aggravates liver fibrosis in a mouse model of cholestasis. Hepatology. 2010;51:1319–1326. doi: 10.1002/hep.23469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clodfelter K.H., Holloway M.G., Hodor P., Park S.H., Ray W.J., Waxman D.J. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Molecular Endocrinology. 2006;20:1333–1351. doi: 10.1210/me.2005-0489. [DOI] [PubMed] [Google Scholar]
- 12.Lichanska A.M., Waters M.J. How growth hormone controls growth, obesity and sexual dimorphism. Trends in Genetics. 2008;24:41–47. doi: 10.1016/j.tig.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Udy G.B., Towers R.P., Snell R.G., Wilkins R.J., Park S.H., Ram P.A. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vidarsdottir S., Walenkamp M.J., Pereira A.M., Karperien M., van Doorn J., van Duyvenvoorde H.A. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. Journal of Clinical Endocrinology and Metabolism. 2006;91:3482–3485. doi: 10.1210/jc.2006-0368. [DOI] [PubMed] [Google Scholar]
- 15.Osorio F.G., Barcena C., Soria-Valles C., Ramsay A.J., de Carlos F., Cobo J. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes and Development. 2012;26:2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez A., Muller D.C., Metter E.J., Maggio M., Harman S.M., Blackman M.R. Aging, androgens, and the metabolic syndrome in a longitudinal study of aging. Journal of Clinical Endocrinology and Metabolism. 2007;92:3568–3572. doi: 10.1210/jc.2006-2764. [DOI] [PubMed] [Google Scholar]
- 17.Lee D.F., Kuo H.P., Chen C.T., Hsu J.M., Chou C.K., Wei Y. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 18.Chen C., Liu Y., Liu Y., Zheng P. Mammalian target of rapamycin activation underlies HSC defects in autoimmune disease and inflammation in mice. Journal of Clinical Investigation. 2010;120:4091–4101. doi: 10.1172/JCI43873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapahi P., Chen D., Rogers A.N., Katewa S.D., Li P.W., Thomas E.L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metabolism. 2010;11:453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S.S., Schroeder F.C. Steroids as central regulators of organismal development and lifespan. PLoS Biology. 2012;10:e1001307. doi: 10.1371/journal.pbio.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amador-Noguez D., Dean A., Huang W., Setchell K., Moore D., Darlington G. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. 2007;6:453–470. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L., Rubins N.E., Ahima R.S., Greenbaum L.E., Kaestner K.H. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metabolism. 2005;2:141–148. doi: 10.1016/j.cmet.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Varela G.M., Antwi D.A., Dhir R., Yin X., Singhal N.S., Graham M.J. Inhibition of ADRP prevents diet-induced insulin resistance. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2008;295:G621–G628. doi: 10.1152/ajpgi.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S., Brown M.S., Goldstein J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Z., Miller R.A., Patel R.T., Chen J., Dhir R., Wang H. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nature Medicine. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochkis I.M., Schug J., Rubins N.E., Chopra A.R., O’Malley B.W., Kaestner K.H. Foxa2-dependent hepatic gene regulatory networks depend on physiological state. Physiological Genomics. 2009;38:186–195. doi: 10.1152/physiolgenomics.90376.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Lay J., Tuteja G., White P., Dhir R., Ahima R., Kaestner K.H. CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metabolism. 2009;10:55–62. doi: 10.1016/j.cmet.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers S.A., Whiteman E.L., Cho H., Lipfert L., Birnbaum M.J. Differentiation-dependent suppression of platelet-derived growth factor signaling in cultured adipocytes. Journal of Biological Chemistry. 1999;274:23858–23867. doi: 10.1074/jbc.274.34.23858. [DOI] [PubMed] [Google Scholar]
- 29.Besnard V., Wert S.E., Hull W.M., Whitsett J.A. Immunohistochemical localization of Foxa1 and Foxa2 in mouse embryos and adult tissues. Gene Expression Patterns. 2004;5:193–208. doi: 10.1016/j.modgep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 30.White P., May C.L., Lamounier R.N., Brestelli J.E., Kaestner K.H. Defining pancreatic endocrine precursors and their descendants. Diabetes. 2008;57:654–668. doi: 10.2337/db07-1362. [DOI] [PubMed] [Google Scholar]
- 31.Hui A.Y., Friedman S.L. Molecular basis of hepatic fibrosis. Expert Reviews in Molecular Medicine. 2003;5:1–23. doi: 10.1017/S1462399403005684. [DOI] [PubMed] [Google Scholar]
- 32.Cabana V.G., Lukens J.R., Rice K.S., Hawkins T.J., Getz G.S. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. Journal of Lipid Research. 1996;37:2662–2674. [PubMed] [Google Scholar]
- 33.Salman T.A., Allam N., Azab G.I., Shaarawy A.A., Hassouna M.M., El-Haddad O.M. Study of adiponectin in chronic liver disease and cholestasis. Hepatology International. 2010;4:767–774. doi: 10.1007/s12072-010-9216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honsawek S., Chayanupatkul M., Chongsrisawat V., Theamboonlers A., Praianantathavorn K., Udomsinprasert W. Serum adiponectin and transient elastography as non-invasive markers for postoperative biliary atresia. BMC Gastroenterology. 2011;11:16. doi: 10.1186/1471-230X-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durazzo M., Niro G., Premoli A., Morello E., Rizzotto E.R., Gambino R. Type 1 autoimmune hepatitis and adipokines: new markers for activity and disease progression? Journal of Gastroenterology. 2009;44:476–482. doi: 10.1007/s00535-009-0023-0. [DOI] [PubMed] [Google Scholar]
- 36.Hosui A., Hennighausen L. Genomic dissection of the cytokine-controlled STAT5 signaling network in liver. Physiological Genomics. 2008;34:135–143. doi: 10.1152/physiolgenomics.00048.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gebert C.A., Park S.H., Waxman D.J. Termination of growth hormone pulse-induced STAT5b signaling. Molecular Endocrinology. 1999;13:38–56. doi: 10.1210/mend.13.1.0235. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y., Laz E.V., Waxman D.J. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Molecular and Cellular Biology. 2012;32:880–896. doi: 10.1128/MCB.06312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhir R.N., Shapiro B.H. Interpulse growth hormone secretion in the episodic plasma profile causes the sex reversal of cytochrome P450s in senescent male rats. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15224–15228. doi: 10.1073/pnas.2434273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tschop M.H., Speakman J.R., Arch J.R., Auwerx J., Bruning J.C., Chan L. A guide to analysis of mouse energy metabolism. Nature Methods. 2011;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hays N.P., Roberts S.B. The anorexia of aging in humans. Physiology and Behavior. 2006;88:257–266. doi: 10.1016/j.physbeh.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 42.Manini T.M. Energy expenditure and aging. Ageing Research Reviews. 2010;9:1–11. doi: 10.1016/j.arr.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cornu M., Albert V., Hall M.N., 2013. mTORin aging, metabolism, and cancer. Current Opinion in Genetics and Development 23:53–62. [DOI] [PubMed]
- 44.Leavens K.F., Easton R.M., Shulman G.I., Previs S.F., Birnbaum M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metabolism. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwahng S.H., Ki S.H., Bae E.J., Kim H.E., Kim S.G. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology. 2009;49:1913–1925. doi: 10.1002/hep.22887. [DOI] [PubMed] [Google Scholar]
- 46.Chen G., Liang G., Ou J., Goldstein J.L., Brown M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Repa J.J., Liang G., Ou J., Bashmakov Y., Lobaccaro J.M., Shimomura I. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes and Development. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laffitte B.A., Chao L.C., Li J., Walczak R., Hummasti S., Joseph S.B. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5419–5424. doi: 10.1073/pnas.0830671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mueller K.M., Themanns M., Friedbichler K., Kornfeld J.W., Esterbauer H., Tuckermann JP. Hepatic growth hormone and glucocorticoid receptor signaling in body growth, steatosis and metabolic liver cancer development. Molecular and Cellular Endocrinology. 2012;361:1–11. doi: 10.1016/j.mce.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamming D.W., Ye L., Katajisto P., Goncalves M.D., Saitoh M., Stevens D.M. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lai E., Prezioso V.R., Tao W.F., Chen W.S., Darnell J.E., Jr. Hepatocyte nuclear factor 3 alpha belongs to a gene family in mammals that is homologous to the Drosophila homeotic gene fork head. Genes and Development. 1991;5:416–427. doi: 10.1101/gad.5.3.416. [DOI] [PubMed] [Google Scholar]
- 52.Corradetti M.N., Inoki K., Guan K.L. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. Journal of Biological Chemistry. 2005;280:9769–9772. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 53.Zhang H., Cicchetti G., Onda H., Koon H.B., Asrican K., Bajraszewski N. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. Journal of Clinical Investigation. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salazar M., Carracedo A., Salanueva I.J., Hernandez-Tiedra S., Lorente M., Egia A. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. Journal of Clinical Investigation. 2009;119:1359–1372. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen C.C., Jeon S.M., Bhaskar P.T., Nogueira V., Sundararajan D., Tonic I. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Developmental Cell. 2010;18:592–604. doi: 10.1016/j.devcel.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuteja G., White P., Schug J., Kaestner K.H. Extracting transcription factor targets from ChIP-Seq data. Nucleic Acids Research. 2009;37:e113. doi: 10.1093/nar/gkp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Osorio F.G., Lopez-Otin C., Freije J.M. NF-kB in premature aging. Aging (Albany NY) 2012;4:726–727. doi: 10.18632/aging.100502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe M., Houten S.M., Wang L., Moschetta A., Mangelsdorf D.J., Heyman R.A. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheaffer K.L., Updike D.L., Mango S.E. The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Current Biology. 2008;18:1355–1364. doi: 10.1016/j.cub.2008.07.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zollner G., Wagner M., Fickert P., Geier A., Fuchsbichler A., Silbert D. Role of nuclear receptors and hepatocyte-enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2005;289:G798–G805. doi: 10.1152/ajpgi.00319.2004. [DOI] [PubMed] [Google Scholar]
- 61.Liu M., Lee D.F., Chen C.T., Yen C.J., Li L.Y., Lee H.J., et al., 2012. IKKalpha Activation of NOTCH Links Tumorigenesis via FOXA2 Suppression. Molecular Cell 45:171–184. [DOI] [PMC free article] [PubMed]
- 62.Rosen E.D., Spiegelman B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng Y., Scherer P.E. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Annals of the New York Academy of Sciences. 2010;1212:E1–E19. doi: 10.1111/j.1749-6632.2010.05875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. Journal of Clinical Investigation. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material