

Abstract
Solid state NMR spectroscopy has evolved rapidly in recent years into an excellent tool for the characterization of membrane proteins and their complexes. In the past few years it has also become clear that the structure of membrane proteins, especially helical membrane proteins is determined, in part, by the membrane environment. Therefore, the modeling of this environment by a liquid crystalline lipid bilayer for solid state NMR has generated a unique tool for the characterization of native conformational states, local and global dynamics, and high resolution structure for these proteins. Protein-protein interactions can also benefit from this solid state NMR capability to characterize membrane proteins in a native-like environment. These complexes take the form of oligomeric structures and hetero-protein interactions both with water soluble proteins and other membrane proteins.
Introduction
Most membrane proteins interact with other membrane and/or water soluble proteins, thereby generating important biochemistry for the cell, as well as complexity for researchers interested in understanding how the biochemistry is achieved through membrane protein structure, dynamics, function and regulation. Furthermore, it is well known that membrane protein structure is, in part, dictated by the protein’s interactions with its native environment through the unique biophysical properties of the cellular membranes.[••1] Fortunately, solid state NMR (ssNMR) spectroscopy can be used to characterize membrane proteins in liquid crystalline lipid bilayers,[2,3, ••4, •5, •6] a much more native-like membrane mimetic environment than those that support isotropic motions for solution NMR spectroscopy or crystallization conditions for X-ray diffraction. Indeed, a few studies have shown that ssNMR spectroscopy can be used to characterize membrane proteins in cellular membranes where they have been inserted by the cellular machinery and have not been purified with detergents and reconstituted into a synthetic sample environment.[•7,8,9, •10] Furthermore, NMR is well known as a technique that can characterize protein-protein interactions, even when complexes have only modest stability.
Helical membrane protein structural characterization has lagged far behind the characterization of water soluble proteins, because membrane proteins are difficult to purify and to stabilize in non-native environments. In part, this is due to their heterogeneous environment and in part, due to weak interactions that stabilize the transmembrane (TM) domain. Even in the interior of the TM domain the amino acid composition is very hydrophobic and consequently the tertiary and quaternary stability arises primarily from weak van der Waals interactions and other weak electrostatic interactions.[•11] As a result, the tertiary and quaternary structure is easily disrupted or distorted. The influence of the membrane environment on protein structure and function is complex involving gradients in water concentration, dielectric and order parameters, as well as the degree of membrane curvature frustration, the lateral pressure profile, and the membrane phase behavior.[••1,12,13] This is not to say that detergent based crystals and micelles, despite the lack of many of these physical properties of the membrane environment, cannot stabilize native-like membrane conformations. Most of the structural biology of membrane proteins that we know today has come from X-ray diffraction and solution NMR, but there exists many distorted membrane protein structures in the Protein Data Bank from these techniques.[••1] Synthetic lipid bilayers, however, appear to be effective membrane mimetics displaying many of the native membrane properties that stabilize native structure as shown by the functional assays of numerous membrane proteins.
Protein-Protein Interactions
Membrane proteins form a variety of protein-protein interactions within native membranes. Oligomeric proteins, such as the glycerol transporter and aquaporins form homotetramers where each monomer is a functional domain.[14] Bowie in his review in 2005 pointed out the interrupted TM helix on the surface of this glycerol transporter facing the fatty acyl environment, as well as a proline kinked helix also exposed to the membrane environment.[12] Indeed, this suggests the exposure of multiple hydrophilic sites to the fatty acyl environment. Even in the tetramer these features face the lipids, but multiple aromatic residues shield the hydrophilic backbone sites from the low-dielectric environment.[••1] On the opposite side of the protein in the tetramer interface many hydrophilic atoms are exposed to the monomer-monomer interfaces. The result is that the protein-protein interface is significantly more hydrophilic than the fatty acyl protein interface. Such oligomeric structures generate a high concentration of functional activity in a membrane local. Another type of oligomeric structure is exemplified by the well-studied K+ channel, KcsA, and the H+ channel, M2 from Influenza A.[11,15] Both of these structures are homotetramers forming a single functional entity that save on the amount of DNA needed to code for the functional protein. For viruses this conservation of the genetic material is particularly important, while in some bacteria, such as E. coli, a K+ transporter, KdpA has all four ‘monomers’ coded in a single protein. [16] While KcsA and M2 have various functional states as a tetramer, other proteins are active or inactive depending on their oligomerization state. In bacterial chemotaxis oligomerization has long been recognized as a mechanism for forming a functional state, yet the extent and structure of the various oligomeric active states is still a subject of considerable debate.[17]
Hetero-protein complexes in membranes are also very common. TM proteins form complexes with water soluble proteins, such as the complex of the TM protein DsbB with the water soluble protein DsbA.[18] Transient hetero-protein complexes between membrane proteins can also be formed such as the binding of the regulatory TM protein, KdpF to the KdpABC complex.[19] Other complexes are not transient such as KdpABC and the 13 polypeptides that form the functional Cytochrome c Oxidase structure.[20]
The tertiary and quaternary stability of many helical TM proteins is rather fragile. Some are significantly stabilized by cofactors that interact often through electrostatic interactions, as in the electron transport complexes and photosynthetic receptors. Many helical TM proteins have a remarkably high content of glycine and proline residues, known from water soluble proteins to be helix breakers. However, in TM helices 2° structure has enhanced stability due to the low dielectric environment, and the presence of glycine motifs (e.g. GxxxG) are known to facilitate helix-helix packing.[21,22] Consequently, glycine residues in TM helices sacrifice some 2° structure stability for enhanced 3° stability.[•23] Furthermore, it is known that both Pro and Gly residues can induce helix kinks.[24,25] Absolutely uniform and rigid helices would have minimal interaction surfaces with adjacent helices, especially if the helix tilt angles with respect to the bilayer normal are large. However, if the helices were somewhat distorted by Gly and Pro residues they may have greater interaction surfaces. Moreover, it is known that many membrane proteins have a large array of functional conformations. Relatively weak tertiary and quaternary stability facilitates the interconversion between these functional states and Gly residues have been shown to facilitate this through helix kinks.
While Gly residues in the interior of the TM domain are highly conserved, Gly residues on the fatty acyl facing surface appear not to be conserved and consequently not available for forming inter-protein helix-helix interactions.[23] Presumably this is because of the exposure of hydrophilic backbone atoms to the low dielectric environment. Ala and Ser residues are also well recognized for their helix-helix packing motifs and these residues are often observed on the fatty acyl facing surface of TM proteins, providing binding sites for other proteins without excessive exposure of hydrophilic atoms to the fatty acyl environs when the protein partners are not bound. Hence these Ala/Ser motifs are important for forming a significant van der Waals surface and interaction energy for protein-protein interactions.
Membrane Protein ssNMR
More than three decades ago pioneering studies of proteins[26], protein complexes[27], membranes[28,29] and membrane proteins[30,31] identified the potential of ssNMR in this realm of membrane biophysics. Two important approaches for utilizing ssNMR were also identified in the same era; that of using magic angle sample spinning coupled with cross polarization[32] to obtained a solution NMR-like spectra with isotropic resonances[27,31] and that of studying uniformly aligned samples[33–35]. The years between these pioneering studies and the present has increased the field strengths by a factor of four, the magic angle spinning rate by more than an order of magnitude, the orientational uniformity of oriented samples for membrane proteins by a factor of five, and dramatically enhanced the sophistication of the experimental design, the dimensionality of the experiments and the instrumentation to perform the experiments. The result is that structures of membrane proteins can be characterized by ssNMR in lipid environments and that membrane protein-ligand and protein-protein interfaces can be characterized. To accomplish this the sophistication of sample preparation had to evolve from lyophilized powders to preparations of membrane proteins in liquid crystalline bilayers while avoiding aggregation and denaturation of the proteins. Furthermore, sample heating due to high power RF absorption during ssNMR experiments by waters in the bilayer interface had to be minimized[36,37]
The characterization of membrane proteins in native-like environments remains a very challenging task. The low dielectric environment in the membrane interstices coupled with the very hydrophobic amino acid composition in the TM domains[•11] leads to a relatively uniform helical secondary structure for most membrane proteins despite the presence of Gly and Pro residues. The result is very little isotropic chemical shift dispersion for the resonances of the side chains for each amino acid type. Coupled with this lack of dispersion is the breadth of the chemical shift resonances, which makes it very difficult to make sequence specific assignments for the common amino acids in the TM domains. In other words the Val, Leu, Ile, Ala and Phe residues which account for 70% of the residues on the protein surface facing the fatty acyl domain of the lipid bilayer and more than 50% of the amino acid residues in the TM domain interior[•11] are very difficult to uniquely assign in the ssNMR spectra. While, the structural strategy for water soluble proteins that has been so successful in solution and solid state NMR by obtaining large numbers of distance and torsional restraints between uniquely assigned backbone and side chain resonances is not readily applicable to membrane proteins. Instead, a different strategy has been developed in which, a combination of orientational and sparse distance restraints are used for each structural characterization.[38,39] Orientational restraints are derived from uniformly aligned samples of a membrane protein, in which each protein molecule has the same orientation with respect to the bilayer normal and the magnetic field axis of the spectrometer. These restraints are known as absolute restraints since the errors in anisotropic tensor orientations do not accumulate as one progresses across the protein structure from one restraint to another. For the characterization of helix tilt over the length of a helix such restraints are very useful. The rotational orientation of the helices can also be defined by these restraints. The result is that relatively few distances between each helix-helix interface is needed to characterize the tertiary and/or quaternary structure of the protein or protein-protein complex.
Solid-State NMR of Protein-Protein Interactions
To date the majority of protein-protein complexes studied by ssNMR have been homo-oligomeric, such as the K+ channel, KcsA, the M2 from Influenza A, and the light-harvesting complexes. Initial hetero-protein studies, such as the spectroscopy on the DsbB-DsbA complex are showing how such complex systems can be approached by solid state NMR. These solid state NMR contributions aim to characterize the structure, dynamics and function and/or chemical properties of these protein-protein complexes. Examples of these studies are presented below illustrating the diversity of the science, the depths of information that can be achieved and the unique ability of ssNMR to characterize the native biochemical and biophysical properties of this important and still poorly understood class of proteins and their complexes. For crystallography data collection is typically restricted to cryogenic temperatures and consequently insights into protein dynamics are limited; conversely, the dynamics of these proteins are enhanced in detergent micelles compared to lipid bilayers. Solid stateNMR spectra of liquid crystalline lipid bilayer samples provides an opportunity to characterize native-like membrane protein dynamics. These latter experiments can be tailored to characterize a model for molecular motions from anisotropic averaging of spin interaction tensors or to characterize frequencies from a variety of experiments including relaxation parameters.
Early ssNMR Studies of Protein-Protein Interactions
The first membrane bound structure characterized by ssNMR was the dimeric peptide, gramicidin A in which all of the side chains, as well as the backbone was determined using orientational restraints and inter-monomer REDOR distances.[40,41] Most of the other structures that have utilized orientational restraints are primarily backbone structures with a few if any experimentally restrained side chain conformations. In this approach, the tilt and rotational orientation of each helix is defined, thereby severely restricting the range of potential helix packing geometry. So, even though the side chains are not characterized and few if any inter-helical distances are observed the critical tertiary and quaternary structure can be approximated. The addition of sparse distances between the helices can result in a high resolution, high order structure with modeled side chain conformations from molecular dynamics. Such structures are far more informative for the biochemist than structures with completely characterized structures but disordered helices.
The pentameric structure of the M2δ peptide from the nicotinic acetylcholine receptor,[42] the tetrameric structure of the Influenza A M2 proton channel TM domain,[39] and the tetrameric or pentameric HIV-1 channel forming VpU structure obtained from glass slide aligned samples[43] and magnetically aligned bicelles[44] were additional early structures. The drug bound state of the M2 proton channel was also characterized[45] and later refined with distances between the drug and protein.[46] A larger and more functionally relevant construct of this protein with an amphipathic helix was also characterized in 2010 and a set of sparse inter-helical distances were observed through 13C-13C correlation spectra.[3,47]
Present ssNMR studies
Recognizing the importance, but also the challenge for obtaining inter-helical distances from solid state NMR several additional strategies have been used to provide this data. For the pentameric structure of phospholamban orientational restraints from ssNMR experiments were coupled with solution NMR NOEs obtained from detergent micelle preparations of the protein to gain the inter-helical restraints.[••48] Veglia and coworkers validated these solution NMR restraints with ssNMR 13C-13C correlation spectra for a mix of proteins 50% labeled with 13C Leu and 50% labeled with 13C Ile. The observed Leu-Ile crosspeaks even though they were not sequence specifically assigned, provided validation for the NOE restraints. Another approach is to use X-ray diffraction restraints. Reinstra and coworkers have been studying the DsbA/DsbB complex responsible for disulfide bond formation in E. coli.[•49] DsbB is a helical TM protein that has been structurally characterized by X-ray diffraction of detergent based crystals and by solution NMR in detergent micelles. These structures display numerous charged and hydrophilic sidechains in what would be the hydrophobic interstices of the lipid bilayer similar to that of some other structures that have been characterized in detergent based environments.[••1] With the resonance assignments from many sites in the protein generating many torsional restraints and a couple of long range distance restraints, Reinstra and coworkers co-refined the DsbB/DsbA complex using crystallographic restraints.[•49] The co-refinement led to a substantial improvement upon the resolution of the crystal structure. In addition, Glu26 was shown to form a hydrogen bond with Tyr153 in the middle of the TM domain. Previously, this hydrogen bond that protects the hydrophilic atoms from the fatty acyl environment of a lipid bilayer had not been observed. While the co-refinement represents an interesting, if not important approach, caution must be exercised if the sample environments are significantly different, as this can imply that the structures may also be different. Recently, Rienstra and coworkers published another study of the DsbB/DsbA complex with the DsbA active site Cys33 mutated to Ser trapping the complex in a stable intermediate. Using 15N-13C TEDOR correlation spectroscopy specific changes in the Pro31 chemical shifts and dynamics in this region of the protein’s active site were characterized.[•50]
Multiple members of the Rhodopsin family of proteins are known to be oligomeric. Now Ladizhansky and coworkers have shown through a combination of ssNMR derived PREs in lipid bilayers, visible CD, non-denaturing SDS gels, and cross-linking experiments that Anabena Sensory Rhodopsin is trimeric.[••51] While the trimeric structure appears to be more stable than that of bacteriorhodopsin, where it is easily disrupted by detergents, the interaction interface in sensory rhodopsin is not the same as that observed in crystal lattices of the sensory rhodopsin characterized by x-ray diffraction. Progress has been made with this protein toward a ssNMR structural characterization in lipid bilayers with a secondary structural analysis and H/D exchange data.[•52] Naito and coworkers have pursued an understanding of Pharaonis sensory rhodopsin II (ppR) photo-signaling through experiments of ppR bound to its transducer, pHtrII.[53] Asp75 of ppR has been proposed to be the counter ion for the Schiff base and once protonated a salt bridge between helices C and G breaks and hence the mutant D75N of ppR can be used as a quasi activated receptor. Through labeling the Ala methyls and Val carbonyls with 13C they have been able to assign some of the resonances. Changes in the dynamics monitored at these labeled sites using cross-polarization and direct detection MAS experiments for both the WT and mutants resulted in a proposed model for the molecular mechanism of photo-induced signaling.
Extensive studies of KcsA in lipid bilayers illustrate the challenges associated with describing the functional mechanism and regulation of this bacterial channel even though it is not voltage regulated. KcsA is a symmetric tetramer and the K+ selectivity filter is on the tetramer axis. The conductance is pH sensitive, as well as sensitive to the internal and external K+ concentrations. There is an activation gate controlled by the inner TM helix that kinks in the vicinity of Gly99 and an inactivation gate associated with the selectivity filter. While it is not possible to separately modulate the internal and external [K+] in the NMR samples, it is possible to titrate pH and regulate overall [K+]. Chemical shift changes in sequence specifically assigned resonances in and near the selectivity filter have been used to demonstrate a Kd for K+ on the order of a few μM obtained from full length KcsA in bilayers[4] (Figure 1) and from a KcsA chimera with Kv1.3.[54,55] This chimera is blocked in a voltage independent fashion by a porphyrin derivative and in a voltage dependent process by kaliotoxin. The porphyrin derivative was found to bind deeply in the selectivity filter inducing the voltage dependence, while the kaliotoxin bound at the channel entrance lacks voltage dependence.[55] These pore blockers were further used to trap the K+ channel in distinct states confirming that the two gates function in a coordinated fashion.[54] Conformational changes were monitored by chemical shift changes in the selectivity filter and in the hinge region. C-type inactivation of the selectivity filter has also been shown at low K+ concentrations through chemical shift changes and that the E71A inactivation resistant mutant does not display these changes.[•56] The shift changes in the wild type were observed at both 1 and 10 μM and the fact that they were not broadened meant that the exchange rate was slower than 500 s−1 in the bilayer environment. Moreover, this later manuscript reported on the chemical shift anisotropy of the E71 and D80 showing that while D80 is deprotonated at pH 7.5, E71 is protonated and hence has a highly shifted pKa. Previously, the isotropic chemical shifts for the carboxyl carbon, which are also sensitive to protonation, had been reported for E71 as a function of pH by Baldus and coworkers to indicate that this pKa was highly shifted.[57] Recently, Baldus and coworkers have investigated lipid binding to KcsA and to the KcsA-Kv1.3 chimera.
Figure 1.

Chemical shift differences induced by two different K+ conditions in the full length UL 13C and 15N KcsA channel reconstituted into DOPE/DOPS (9:1 weight ratio) proteoliposomes. Protein/lipid ratio was mixed in a 1:1 weight ratio. The 13C-13C and 13C-15N correlation spectroscopy was performed 750 MHz for 1H and at an estimated sample temperature of 280K. Multiple sites show significant shifts between a [K+] of 1 μM and 50mM.
While KcsA appears to function primarily as a symmetric tetramer, the fundamental mechanism of the tetrameric M2 proton channel from influenza A requires that the tetrameric symmetry be broken as protons are shuttled through the His37 selectivity filter in the middle of the lipid bilayer. The non-conducting state of the channel is the +2 charge state for the His37 tetrad that forms a dimer of dimer state in which a pair of His37 side chains share a proton.[45,58] The His37 pH titration showed two pKas at 8.2 indicating cooperative H+ binding in forming the imidazole-imidazolium dimers. The pH activated state is formed around pH 6.3 when the third proton is bound to the histidine tetrad. This breaks one of the imidazole-imidazolium dimers forming a pair of adjacent charged histidine residues that then transfers the proton to a water wire leading to the interior of the viral particle when the Trp41 gate opens.[•11,47,59] The structure of the conductance domain (residues 22–62 – has the same conductance properties as the full length protein) of this channel has been characterized by ssNMR using a combination of orientational and distance restraints.[47] Differences between this structure and those that have been characterized from detergent based crystals and detergent micelles have been the subject of considerable debate.[•11] Recently, spectra of the full length protein (55 kDa tetramer including His tag) in liposomes have been shown to be superimposable on those of the conductance domain, in particular the chemical shifts for the His37 residues and multiple distance restraints are consistent with the ssNMR structure of the conductance domain (Figure 2).[3, •7, •60] Moreover, excellent dynamic nuclear polarization experiments have provided multiple restraints for rimantadine bound in the conductance domain.[••61] The location of the rimantadine is similar to the location previously characterized for amantadine binding.[46]
Figure 2.
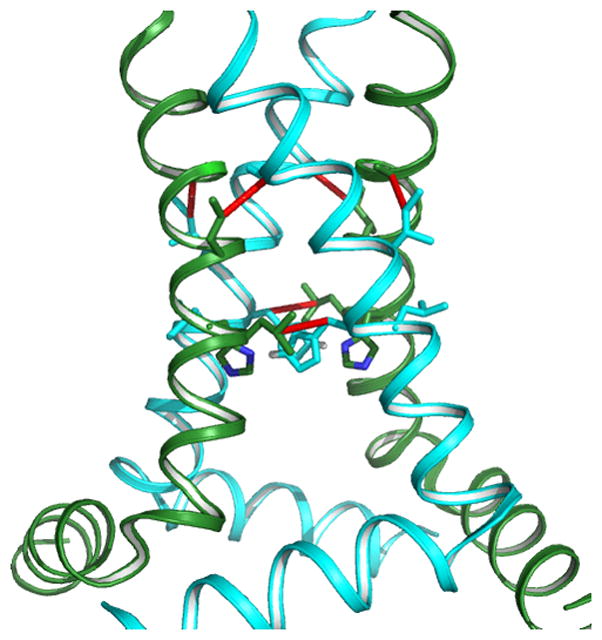
In the conductance domain of the M2 proton channel the high resolution monomeric backbone structure is characterized by orientational restraints. Sparse distance restraints and four fold symmetry (observed in the PISEMA spectra) were used to assemble the tetrameric structure.[47] Shown here is the conductance domain structure (PDB 2L0J) obtained from liquid crystalline DOPC/DOPE bilayer (4:1 molar ratio) samples for the orientational restraints and superimposed on the structure are observed distance restraints from the full length M2 protein also in DOPC/DOPE bilayers.[60]
Phospholamban (PLN) is a 52 residue protein that regulates the sarcoplasmic reticulum Ca2+ ATPase (SERCA) in cardiac muscle. While PLN binds to SERCA as a monomer it frequently forms a pentameric structure in various membrane mimetic environments and is thought to be a storage form of this regulatory protein and possibly a Ca2+ or Cl− channel in the sarcoplasmic reticulum. Early orientational restraints from OS ssNMR described a C-terminal TM helix with a tilt of 13 ± 6° in POPC as well as POPC/POPE bilayers,[62] and ~15° in DOPC/DOPE bilayers,[63] while REDOR distance measurements between 5-15N-Gln29 and 5-13C-Gln29 on adjacent proteins defined the rotational orientation of the TM helix in the oligomeric state.[64] Although considerable debate has existed over the years as to the orientation of the N-terminus and its interactions with the bulk aqueous environment or the membrane interface, the latter has been confirmed by characterizing the significant amphipathic helical segment bound to the surface of bilayers OS ssNMR.[62,63] In between these two helical segments is a dynamic loop. More recently a complete structure of phospholamban has been characterized using a combination of solution and ssNMR.[••48] Primary distance restraints were obtained from solution NMR NOESY experiments followed by selective 13C-13C MAS ssNMR correlation experiments validating the restraints in a lipid environment.
Monomeric PLN has a regulatory role through binding to both Protein Kinase A (PKA) and to SERCA.[65,66] While the conformation of the N-terminus influences the binding of PLN to its targets, there has been as much effort focused on the dynamics as on the structural aspects of the transition from the oligomeric state to the SERCA bound phosphorylated (Ser16 & Ser17) monomer of PLN as reviewed by Veglia.[65] The PLN structure can be roughly characterized as a helix-loop-helix motif with four segments having differing dynamics that appear to be functionally important. Not only the peptide structure and dynamics are important, but the lipid composition may modulate the interactions of PLN with SERCA.[67] Studies of the monomer have been facilitated by a fully functional mutant that does not oligomerize (C36A, C41F, C46A) abbreviated AFA-PLN. Baldus and coworkers have made multiple sequence specific assignments for AFA-PLN bound to SERCA.[•68] The chemical shift data support the two AFA-PLN helices bound to SERCA spanning a significantly different amino acid residue surface than modeled previously based on cross linking studies. This relaxed or ‘R’ state has enhanced regulatory activity for PKA, whereas a more structured (relatively long amphipathic helix) form or ‘T’ state inhibits SERCA. The conformational equilibrium appears to be influenced by the membrane mimetic environment with negatively charged lipids stabilizing the T state.[••69]
The light harvesting complexes were some of the earliest crystal structures of membrane protein complexes. de Groot and coworkers have published an extensive literature on the antenna complexes and reaction centers from a variety of sources.[70] The protein-protein interactions and the interactions with and between the chlorophylls generate the unique chemistry that can be dissected with NMR and DFT calculations to further analyze the chemical bonding. Light harvesting complexes of plants and some algae switch between a light harvesting state and an energy-dissipating state. MAS experiments on the LHCII complex from Chlamydomonas reinhardtii have been characterized structural changes between these states.[71] Because of small changes in the electronic structure of the chlorophylls, the isotropic chemical shifts of multiple 13C sites in the chlorophylls change. Figure 3 shows such changes in the aromatic region of the 13C-13C correlation spectra. Small changes in the chemical shifts for C4, C5 and C6 are hypothesized to reflect the perturbations in the chlorophylls in the terminal emitter domain supporting mechanisms that suggest subtle modification to chlorophyll-lutein interactions associated with the dissipative state.[••71]
Figure 3.
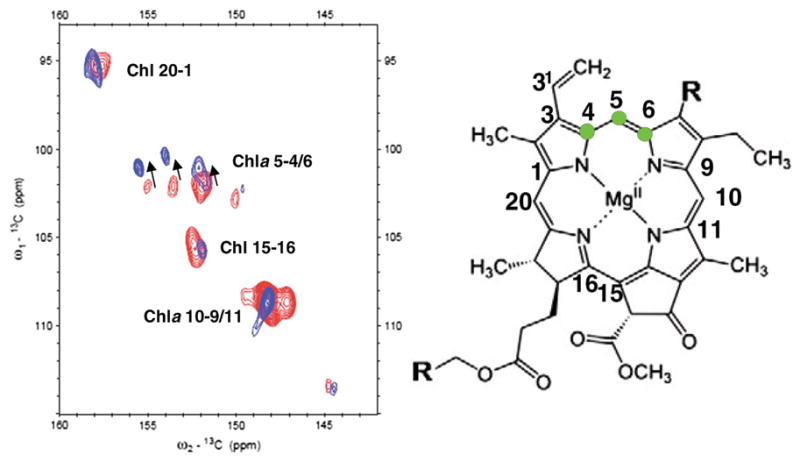
13C-13C correlation spectra for the light harvesting state (blue) and the energy dissipative state (red). Significant shifts are observed, particularly with the carbons 4,5 and 6 as indicated on the chlorophyll ring.
Receptor tyrosine kinase receptors are a particularly important class of proteins where the phosphorylation in the cytoplasm is induced by extracellular ligand binding and receptor dimerization. A mutation in the highly hydrophobic TM sequence (V664E) results in dimerization through inter-helical hydrogen bonding and full activation of the kinase. Placement of the Glu residues in a different rotational position around the helix fails to achieve activation. Smith and coworkers confirmed the helix-helix interacting surface through MAS 2H spectra of deuterated Leu668 and Leu670.[••72] Only the residue in the i to i+4 position showed reduced dynamics upon dimerization. Following the TM helix is a juxta-membrane (JM) domain that has many positively charged residues that can interact with the negatively charged membrane. By comparing direct polarization spectra of 13C labeled residues in the JM domain with INEPT spectra of the same residues they showed dramatically increased dynamics in the JM domain upon dimerization.[••72] In other words the binding of ligand in the native protein induces dimerization, which releases the JM domain from the membrane leading to kinase activation.
The bacterial chemotaxis receptor is a large complex formed by receptor dimers, and two additional proteins CheA and CheW forms a complex that in turn trimerizes forming a hexameric state of the receptor under some conditions. While such large complexes are beyond the scope of present day ssNMR for analyzing uniformly labeled samples, targeted distance measurements with REDOR experiments can be performed on fully functional complexes, even in structures exceeding 100kDa, to evaluate and or validate structural models. For the serine receptor Thompson and coworkers have measured an intra-dimer distance of 8.8Å between a unique Cys residue and a 5F-Phe residue.[••73] For models of the kinase-active signaling complex the predicted inter-dimer distances were much shorter, however the REDOR measurements indicated the same distance as that measured previously for the intra-dimer subunit of the complex suggesting that the high protein density of the crystal lattice may be influencing the structure of this functional state.
We have focused on helical membrane proteins, but outer membrane proteins are typically β-barrel structures in which the extensive hydrogen bonding between strands (and for oligomeric structures, between proteins) results in a much more rigid tertiary structure. This generates numerous advantages for the ssNMR spectroscopist. The sensitivity to environment is much reduced because of the high energy intra-protein (and inter-protein) interactions and consequently the stability of the native structure in various media is high and spectral linewidths could also be anticipated to be narrower. Indeed, the microcrystalline spectra of YadA, a trimeric autotransporter adhesion obtained by van Rossum and colleagues are excellent generating nearly 1200 non-redundant distance restraints where 219 of these were long range distances.[••74] The structural calculation was complicated by the fact that many of the restraints reflect both intra- and inter-monomer contacts. The excellent structure was calculated using inferential structure determination, an approach in which ambiguous restraints can be used,[75] similar to that used by Reinstra in the DsbA/DsbB structure calculation.[•49]
Conclusions
Here, we have discussed just the ssNMR spectroscopy of protein-protein interactions in membrane environments leaving aside numerous monomeric structures that have been studied from a structural, dynamic and functional perspective with this technology. Solid state NMR is ideally suited for the study of this very important class of membrane proteins and in particular for the study of membrane proteins involved in weak protein-protein interactions. Indeed, membrane proteins represent more than 50% of pharmaceutical targets and most membrane proteins function at least part time as a protein complex. Since their structural, dynamic and functional properties are all sensitive to their environment in is significant that ssNMR can characterize these proteins and their complexes in liquid crystalline lipid bilayers. The strategies are rapidly developing for characterizing these proteins. Combining ssNMR restraints with those from other structural techniques, when justified, will speed the process towards biochemical and biophysical conclusions. Enhanced computational methods from Rosetta[76] to inferrential structure determination[75] and from restrained molecular dynamics[47] to ab initio calculations[59] will enhance the structural, dynamic and functional understanding of these proteins and their complexes, one of the great frontiers in structural and functional biology.
Highlights.
Multiple examples of homo- or hetero-oligomeric protein-protein complexes discussed
Membrane environments influence membrane protein-protein interactions
Solid state NMR characterizes protein complexes in native-like membrane environs
Absolute and relative restraints provide unique membrane protein structural tools
Structural, dynamic, chemical and functional characterizations by solid state NMR
Acknowledgments
This review was, in part, supported by NIH grants AI 074805, AI 073891 and AI 023007 and the National Science Foundation through Cooperative Agreement 0654118 between the Division of Materials Research and the State of Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ••1.Zhou HX, Cross TA. Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. A review of membrane protein structures identifying native and non-native appearances of the structures resulting from non native-like environments used for the structural characterization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renault M, Cukkemane A, Baldus M. Solid-state NMR spectroscopy on complex biomolecules. Angew Chem Int Ed Engl. 2010;49:8346–8357. doi: 10.1002/anie.201002823. [DOI] [PubMed] [Google Scholar]
- 3.Can TV, Sharma M, Hung I, Gor’kov PL, Brey WW, Cross TA. Magic angle spinning and oriented sample solid-state NMR structural restraints combine for influenza A M2 protein functional insights. J Am Chem Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••4.Bhate MP, Wylie BJ, Tian L, McDermott AE. Conformational dynamics in the selectivity filter of KcsA in response to potassium ion concentration. J Mol Biol. 2010;401:155–166. doi: 10.1016/j.jmb.2010.06.031. Mechanistic insights into the KcsA channel were achieved through isotropic chemical shift changes as a function [K+] and by the observation that these K+ dependent changes did not occur with the E71A inactivation resistant mutant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •5.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. The first structure of a G-Protein coupled receptor observed in its wild-type full length sequence and in a liquid crystalline lipid bilayer environment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •6.Judge PJ, Watts A. Recent contributions from solid-state NMR to the understanding of membrane protein structure and function. Curr Opin Chem Biol. 2011;15:690–695. doi: 10.1016/j.cbpa.2011.07.021. Recent review of solid state NMR spectroscopic strategies for characterizing membrane protein structure and function in lipid bilayers. [DOI] [PubMed] [Google Scholar]
- •7.Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew Chem Int Ed Engl. 2012;51:8383–8386. doi: 10.1002/anie.201204666. The first ssNMR spectra of the full length tetrameric M2 protein and the observation of this protein in E. coli membranes as it was inserted by the celular apparatus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc Natl Acad Sci U S A. 2012;109:4863–4868. doi: 10.1073/pnas.1116478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinsey RA, Kintanar A, Oldfield E. Dynamics of amino acid side chains in membrane proteins by high field solid state deuterium nuclear magnetic resonance spectroscopy. Phenylalanine, tyrosine, and tryptophan. J Biol Chem. 1981;256:9028–9036. [PubMed] [Google Scholar]
- •10.Fu R, Wang X, Li C, Miranda AN, Pielak GJ, Tian F. In Situ Detection of a Recombinant Human Protein in Native E. coli Membranes by Solid-State NMR. J Am Chem Soc. 2011;133:12370–12373. doi: 10.1021/ja204062v. The first stucural study of a membrane protein, LR11, in an E. coli membrane. LR11 is a critical regulator of amyloid precursor protein transport and prcessing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •11.Zhou HX, Cross TA. Modeling the membrane environment has implications for membrane protein structure and function: Influenza A M2 protein. Protein Sci. 2013;22:381–394. doi: 10.1002/pro.2232. A detailed comparison of M2 Proteon channel structures from X-ray crystallography, solution and solid state NMR highlighting the influence of the protein environment used in the structural characterization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 13.Botelho AV, Huber T, Sakmar TP, Brown MF. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu D, Libson A, Miercke LJ, Weitzman C, Nollert P, Krucinski J, Stroud RM. Structure of a glycerol-conducting channel and the basis for its selectivity. Science. 2000;290:481–486. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- 15.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 16.Durell SR, Bakker EP, Guy HR. Does the KdpA subunit from the high affinity K+ translocating P-type Kdp-ATPase have a structure similar to that of K+ channels? Biophys J. 2000;78:188–199. doi: 10.1016/S0006-3495(00)76584-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sferdean FC, Weis RM, Thompson LK. Ligand affinity and kinase activity are independent of bacterial chemotaxis receptor concentration: insight into signaling mechanisms. Biochemistry. 2012;51:6920–6931. doi: 10.1021/bi3007466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 19.Gassel M, Siebers A, Epstein W, Altendorf K. Assembly of the Kdp complex, the multi-subunit K+-transport ATPase of Escherichia coli. Biochim Biophys Acta. 1998;1415:77–84. doi: 10.1016/s0005-2736(98)00179-5. [DOI] [PubMed] [Google Scholar]
- 20.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 21.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 22.Javadpour MM, Eilers M, Groesbeek M, Smith SO. Helix packing in polytopic membrane proteins: Role of glycine in transmembrane helix association. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •23.Dong H, Sharma M, Zhou HX, Cross TA. Glycines: Role in alpha-helical membrane protein structures and a potential indicator of native conformation. Biochemistry. 2012;51:4779–4789. doi: 10.1021/bi300090x. While it has been known that glycine residues participate in motifs that facilitate helix packing here we show that conserved glycine residues are rarely exposed to the fatty acyl environment and that while they stabilize 3° structure they destabilize 2° structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y, Lee A, Chen J, Chadene M, Chait B, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;4176:523–552. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 25.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Torchia DA, VanderHart DL. 13C Magnetic resonance evidence for anisotropic molecular motion in collagen fibrils. J Mol Biol. 1976;104:315–321. doi: 10.1016/0022-2836(76)90018-8. [DOI] [PubMed] [Google Scholar]
- 27.Opella SJ, Frey MH, Cross TA. Detection of Individual Carbon Resonances in Solid Proteins. J Am Chem Soc. 1979;101:5856–5857. [Google Scholar]
- 28.Oldfield E, Chapman D. Effects of cholesterol and cholesterol derivatives on hydrocarbon chain mobility in lipids. Biochem Biophys Res Commun. 1971;43:610–616. doi: 10.1016/0006-291x(71)90658-9. [DOI] [PubMed] [Google Scholar]
- 29.Seelig J, Niederberger W. Two Pictures of a Lipid Bilayer. A Comparison Between Deuterium Label and Spin-Label Experiments. Biochemistry. 1974;13:1585–1588. doi: 10.1021/bi00705a005. [DOI] [PubMed] [Google Scholar]
- 30.Kinsey RA, Kintanar A, Tsai MD, Smith RL, Janes N, Oldfield E. First observation of amino acid side chain dynamics in membrane proteins using high field deuterium nuclear magnetic resonance spectroscopy. J Biol Chem. 1981;256:4146–4149. [PubMed] [Google Scholar]
- 31.Harbison GS, Herzfeld J, Griffin RG. Solid-state nitrogen-15 nuclear magnetic resonance study of the Schiff base in bacteriorhodopsin. Biochemistry. 1983;22:1–4. doi: 10.1021/bi00270a600. [DOI] [PubMed] [Google Scholar]
- 32.Schaefer J, Stejskal EO. Carbon-13 Nuclear Magnetic Resonance of Polymers Spinning at the Magic Angle. J Am Chem Soc. 1976;98:1031–1032. [Google Scholar]
- 33.Griffin RG, Powers L, Pershan PS. Head-Group Conformation in Phospholipids: A Phosphorus-31 Nuclear Magnetic Resonance Study of Oriented Monodomain Dipalmitoylphosphatidylcholine Bilayers. Biochemistry. 1978;17:2718–2722. doi: 10.1021/bi00607a004. [DOI] [PubMed] [Google Scholar]
- 34.Rothgeb TM, Oldfield E. Nitrogen-14 nuclear magnetic resonance spectroscopy as a probe of lipid bilayer headgroup structure. J Biol Chem. 1981;256:6004–6009. [PubMed] [Google Scholar]
- 35.Cross TA, Opella SJ. Protein Structure by Solid State NMR. J Am Chem Soc. 1983;105:306–308. [Google Scholar]
- 36.Li C, Mo Y, Hu J, Chekmenev E, Tian C, Gao FP, Fu R, Gor’kov P, Brey W, Cross TA. Analysis of RF heating and sample stability in aligned static solid-state NMR spectroscopy. J Magn Reson. 2006;180:51–57. doi: 10.1016/j.jmr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Gor’kov PL, Chekmenev EY, Li C, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J Magn Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Murray DT, Das N, Cross TA. Solid State NMR Strategy for Characterizing Native Membrane Protein Structures. Acc Chem Res. 2013 doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimura K, Kim S, Zhang L, Cross TA. The Closed State of a H+ Channel Helical Bundle: Combining Precise orientational and Distance Restraints from Solid State NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 40.Ketchem RR, Roux B, Cross TA. High-Resolution Polypeptide Structure in a Lamellar Phase Lipid Environment from Solid-State NMR Derived Orientational Constraints. Structure. 1997;5:1655–1669. doi: 10.1016/s0969-2126(97)00312-2. [DOI] [PubMed] [Google Scholar]
- 41.Fu R, Cotten M, Cross TA. Inter- and intramolecular distance measurements by solid-state MAS NMR: determination of gramicidin A channel dimer structure in hydrated phospholipid bilayers. J Biomol NMR. 2000;16:261–268. doi: 10.1023/a:1008372508024. [DOI] [PubMed] [Google Scholar]
- 42.Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M. Structures of the M2 Channel-Lining Segments from Nicotinic Acetylcholine and NMDA Receptors by NMR Spectroscopy. Nat Struct Biol. 1999;6:374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333:409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 44.Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys J. 2006;91:3032–3042. doi: 10.1529/biophysj.106.087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••48.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci U S A. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. The structure of the pentameric phospholamban in lipid bilayers. A ombination of orientational restraints and solution NMR NOEs were combned for the structure. The distances were validated through 13C-13C correlation spectra of bilayer preparations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •49.Tang M, Sperling LJ, Berthold DA, Schwieters CD, Nesbitt AE, Nieuwkoop AJ, Gennis RB, Rienstra CM. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR. 2011;51:227–233. doi: 10.1007/s10858-011-9565-6. A refinement of the crystal structure of DsbB in the presence of DsbA. The sample included lipids, but was not a liquid crystalline lipid bilayer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •50.Sperling LJ, Tang M, Berthold DA, Nesbitt AE, Gennis RB, Rienstra CM. Solid-state NMR study of a 41 kDa membrane protein complex DsbA/DsbB. J Phys Chem B. 2013;117:6052–6060. doi: 10.1021/jp400795d. Changes in chemical shift are observed in a conformational state of the complex induced by the DsbA C33S mutation. [DOI] [PubMed] [Google Scholar]
- ••51.Wang S, Munro RA, Kim SY, Jung KH, Brown LS, Ladizhansky V. Paramagnetic relaxation enhancement reveals oligomerization interface of a membrane protein. J Am Chem Soc. 2012;134:16995–16998. doi: 10.1021/ja308310z. Monomer-monomer interactions are characterized used paramagnetic relaxation enhancements through cysteine mutagenesis and spin labeling. [DOI] [PubMed] [Google Scholar]
- •52.Shi L, Kawamura I, Jung KH, Brown LS, Ladizhansky V. Conformation of a seven-helical transmembrane photosensor in the lipid environment. Angew Chem Int Ed Engl. 2011;50:1302–1305. doi: 10.1002/anie.201004422. Assignment of most bb and sidechain resonance in the TM domain of ASR leading to secondary structural analysis throughout the TM domain. [DOI] [PubMed] [Google Scholar]
- 53.Kawamura I, Yoshida H, Ikeda Y, Yamaguchi S, Tuzi S, Saito H, Kamo N, Naito A. Dynamics change of phoborhodopsin and transducer by activation: study using D75N mutant of the receptor by site-directed solid-state 13C NMR. Photochem Photobiol. 2008;84:921–930. doi: 10.1111/j.1751-1097.2008.00326.x. [DOI] [PubMed] [Google Scholar]
- 54.Ader C, Schneider R, Hornig S, Velisetty P, Vardanyan V, Giller K, Ohmert I, Becker S, Pongs O, Baldus M. Coupling of activation and inactivation gate in a K+-channel: potassium and ligand sensitivity. EMBO J. 2009;28:2825–2834. doi: 10.1038/emboj.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ader C, Schneider R, Hornig S, Velisetty P, Wilson EM, Lange A, Giller K, Ohmert I, Martin-Eauclaire MF, Trauner D, et al. A structural link between inactivation and block of a K+ channel. Nat Struct Mol Biol. 2008;15:605–612. doi: 10.1038/nsmb.1430. [DOI] [PubMed] [Google Scholar]
- •56.Bhate MP, McDermott AE. Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc Natl Acad Sci U S A. 2012;109:15265–15270. doi: 10.1073/pnas.1211900109. A mechanism for C-type inactivation of the selectivity filter is further supported by data for the elevated pKa of E71 and lack of the selectivity collapse for E71A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, Baldus M. Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy. J Am Chem Soc. 2009;131:170–176. doi: 10.1021/ja806306e. [DOI] [PubMed] [Google Scholar]
- 58.Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A. J Am Chem Soc. 2012;134:7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong H, Yi M, Cross TA, Zhou HX. Ab Initio calculations and validation of the pH-dependent structures of the His37-Trp41 quartet, the heart of acid activation and proton conductance in the M2 protein of Influenza A virus. Chemical Science. 2013 doi: 10.1039/C3SC50293G. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •60.Miao Y, Cross TA, Fu R. Identifying inter-residue resonances in crowded 2D C- C chemical shift correlation spectra of membrane proteins by solid-state MAS NMR difference spectroscopy. J Biomol NMR. 2013 doi: 10.1007/s10858-013-9745-7. Difference spectra between two different mixing times in 13C-13C correlation spectra are used to enhance resolution, facilitate resonance assignments and to identify distance restraints. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••61.Andreas LB, Barnes AB, Corzilius B, Chou JJ, Miller EA, Caporini M, Rosay M, Griffin RG. Dynamic nuclear polarization study of inhibitor binding to the m218-60 proton transporter from influenza a. Biochemistry. 2013;52:2774–2782. doi: 10.1021/bi400150x. Dynamic Nuclear Polization spectra of the M2 conductance domain with rimantadine bound provides multiple distance restraints between the protein and bound drug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abu-Baker S, Lu JX, Chu S, Shetty KK, Gor’kov PL, Lorigan GA. The structural topology of wild-type phospholamban in oriented lipid bilayers using 15N solid-state NMR spectroscopy. Protein Sci. 2007;16:2345–2349. doi: 10.1110/ps.072977707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Traaseth NJ, Verardi R, Torgersen KD, Karim CB, Thomas DD, Veglia G. Spectroscopic validation of the pentameric structure of phospholamban. Proc Natl Acad Sci U S A. 2007;104:14676–14681. doi: 10.1073/pnas.0701016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu W, Fei JZ, Kawakami T, Smith SO. Structural constraints on the transmembrane and juxtamembrane regions of the phospholamban pentamer in membrane bilayers: Gln29 and Leu52. Biochim Biophys Acta. 2007;1768:2971–2978. doi: 10.1016/j.bbamem.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, Veglia G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of Ca(2+)-ATPase. Biochemistry. 2008;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masterson LR, Yu T, Shi L, Wang Y, Gustavsson M, Mueller MM, Veglia G. cAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. J Mol Biol. 2011;412:155–164. doi: 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hughes E, Clayton JC, Middleton DA. Cytoplasmic residues of phospholamban interact with membrane surfaces in the presence of SERCA: a new role for phospholipids in the regulation of cardiac calcium cycling? Biochim Biophys Acta. 2009;1788:559–566. doi: 10.1016/j.bbamem.2008.10.029. [DOI] [PubMed] [Google Scholar]
- •68.Seidel K, Andronesi OC, Krebs J, Griesinger C, Young HS, Becker S, Baldus M. Structural characterization of Ca(2+)-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry. 2008;47:4369–4376. doi: 10.1021/bi7024194. Chemical shift restraints were used to characterize the bound state of phospholamban to SERCA. [DOI] [PubMed] [Google Scholar]
- ••69.Gustavsson M, Traaseth NJ, Veglia G. Probing ground and excited states of phospholamban in model and native lipid membranes by magic angle spinning NMR spectroscopy. Biochim Biophys Acta. 2012;1818:146–153. doi: 10.1016/j.bbamem.2011.07.040. Characterizing the sensitivity of Phospholamban in different lipid environments and the influence of various functional states. Conformational kinetcs were found to be different between micelles and lipid bilayers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alia A, Buda F, de Groot HJ, Matysik J. Solid-state NMR of nanomachines involved in photosynthetic energy conversion. Annu Rev Biophys. 2013;42:675–699. doi: 10.1146/annurev-biophys-083012-130415. [DOI] [PubMed] [Google Scholar]
- ••71.Pandit A, Reus M, Morosinotto T, Bassi R, Holzwarth AR, de Groot HJ. An NMR comparison of the light-harvesting complex II (LHCII) in active and photoprotective states reveals subtle changes in the chlorophyll a ground-state electronic structures. Biochim Biophys Acta. 2013;1827:738–744. doi: 10.1016/j.bbabio.2013.02.015. The critically important switch between a light havesting versus energy dissipative state is documented through chemical shift changes to occur in a chlorophyll ring presumed to be in the vicinity of the interactions associated with modifications necessitated for the dissipative state. [DOI] [PubMed] [Google Scholar]
- ••72.Matsushita C, Tamagaki H, Miyazawa Y, Aimoto S, Smith SO, Sato T. Transmembrane helix orientation influences membrane binding of the intracellular juxtamembrane domain in Neu receptor peptides. Proc Natl Acad Sci U S A. 2013;110:1646–1651. doi: 10.1073/pnas.1215207110. The binding interface for the helices of the Neu receptor are defined through changes in the sidechain dynamics characterized by 2H ssNMR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••73.Fowler DJ, Weis RM, Thompson LK. Kinase-active signaling complexes of bacterial chemoreceptors do not contain proposed receptor-receptor contacts observed in crystal structures. Biochemistry. 2010;49:1425–1434. doi: 10.1021/bi901565k. A 19F-13C REDOR measurement between chemorecptor monomers elliminates possible oligomeric structures for the sugnaling complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••74.Shahid SA, Bardiaux B, Franks WT, Krabben L, Habeck M, van Rossum BJ, Linke D. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods. 2012;9:1212–1217. doi: 10.1038/nmeth.2248. The structural characterization of a trimeric outer membrane protein, YadA with 1200 non-redundant distance restraces. [DOI] [PubMed] [Google Scholar]
- 75.Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309:303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 76.Tian Y, Opella SJ, Marassi FM. Improved chemical shift prediction by Rosetta conformational sampling. J Biomol NMR. 2012;54:237–243. doi: 10.1007/s10858-012-9677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]