

Abstract
Zebrafish embryos are an exceptional system for studying vertebrate development. Historically, studies using zebrafish to uncover key players in developmentally regulated gene expression have entailed detailed analysis of transcription factors. It is now apparent that epigenetic modifications of both DNA and histone tails are equally important in the regulation of gene expression during development. As such, blocking the function of key epigenetic modifiers impairs development, albeit with surprising tissue specificity. For instance, DNA methylation is an important epigenetic mark that is depleted in embryos lacking dnmt1 and uhrf1. These embryos display developmental defects in the eye, liver, pancreas, and larval lethality. Interestingly, human tumors derived from these same organs have aberrant changes in DNA methylation and altered expression of genes that are thought to contribute to formation of these cancers. These observations have provided a mechanistic basis for treating cancer with drugs that block the enzymes that facilitate DNA and histone modifications. Thus, it is important to understand the consequences of targeting these factors in a whole animal. We review the use of zebrafish for probing the genetic, cellular, and physiological response to alterations in the epigenome and highlight exciting data illustrating that epigenetic studies using zebrafish can inform and impact cancer biology.
Keywords: DNA methylation, histone acetylation, histone methylation, development, DNMT1, UHRF1
INTRODUCTION
Embryonic development is largely driven by changes in gene expression, and patterns of developmentally regulated genes are frequently recapitulated in cancer cells. As such, there are numerous analogies between organogenesis and tumorigenesis. In rare but celebrated cases, a single gene, such as a potent oncogene, can drive cells to completely change their fate, proliferation status, or migratory behavior. However, more typically, these changes are driven by the collective effect of a constellation of genes. Thus, in both development and in cancer, the cumulative effects of gene activation and silencing impact cell identity, proliferative capacity, survival, and fate.
Alterations in the epigenetic landscape and chromatin structure are major, yet incompletely understood, mechanisms that can simultaneously activate and repress the expression of multiple genes. Histone modifications and DNA methylation alter nucleosome positioning, chromatin compaction, and transcription factor access to DNA. It is now clear that epigenetic marks collaborate with the underlying genetic code to dictate whether a gene is active, silenced, or inactive, but poised for activation when the appropriate conditions fire the transcriptional machinery.
The complex molecular and biochemical interactions between epigenetic players have been elucidated using genetic and biochemical tools that are extremely useful in tissue culture cells. However, as drugs that target enzymes that write the epigenetic code, such as DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), are now used in patients, it is important to study how systemic inhibition of select epigenetic modifiers impacts the epigenome in a whole animal. Moreover, the demonstrated teratogenic potential of these drugs (Branch et al., 1996; Rosen and Chernoff, 2002; Gurvich et al., 2005; Menegola et al., 2006) underscores the importance of understanding the effects of these drugs not only on the diseases they are designed to treat but also on embryogenesis.
Zebrafish are an excellent vertebrate system to study embryonic development and, as such, have proven useful for probing the requirement of individual epigenetic modifiers in embryogenesis. Imprinted genes aside, studies from zebrafish and other organisms unambiguously provide data demonstrating that altering the epigenome disrupts development. Epigenetic marks are sprinkled throughout the genome, and as such, mice lacking the key mediators of many of these marks die very early in development, many before implantation. The hefty maternal contribution of many of these factors in zebrafish embryos enables their prolonged survival despite bearing mutations in embryonic essential genes. This attribute of zebrafish, combined with the use of pharmacological inhibitors and the many advantages of this system for studying development, allows investigation of the role for epigenetic mediators in developmental processes occurring both early and later in embryogenesis. Interestingly, few of these processes, such as the rapid cell division that occurs during outgrowth and morphogenesis of the liver, are reminiscent of events that occur during neoplastic transformation. As more zebrafish mutants are evaluated and as increasingly sophisticated tools for identifying epigenetic marks are developed and applied to zebrafish, it is becoming evident that although all tissues have loci which are regulated by DNA methylation and histone modifications, some cells are more sensitive than others to losing key epigenetic regulators.
We speculate that some common epigenetic themes contribute to embryonic development and to cancer, and here we highlight relevant studies using zebrafish. We emphasize how findings in zebrafish embryos can provide insight into the teratogenic potential of epigenetic modifiers and the mechanism of action of chemotherapeutics that inhibit epigenetic modifiers and impact cancer biology.
ZEBRAFISH AS A TOOL TO UNDERSTAND HUMAN DISEASE: ADVANTAGES AND CHALLENGES
Zebrafish embryos develop ex vivo and thus can be manipulated, injected, and imaged as they develop. The use of fluorescent transgenes enables analysis of developmental processes with spectacular detail. Forward and reverse genetics are powerful tools that are widely used and growing in sophistication. Embryos can be easily bathed in compounds, providing an easy and even automatable means of identifying the toxicity, teratogenic, and therapeutic potential of water soluble drugs. Thus, the attributes of zebrafish and the tools available to manipulate the factors that modulate the epigenome during development are unparalleled.
Zebrafish development is extremely rapid. Important developmental milestones can be observed in the course of a day using a simple microscope: gastrulation and axis formation are complete within a few hours post-fertilization (hpf) and the brain, eyes, heart, trunk muscle, fins, germ cells, and many other structures have formed by 24 hpf. Patterning, differentiation, growth, and morphogenesis are complete for most organs and the brain by 5 days postfertilization (dpf). Although there are a few cases, such as adipose tissue (Flynn et al., 2009; Imrie and Sadler, 2010), where entire organs form during the postembryonic period, tissue remodeling and expansion on existing structures typically occurs in larval and juvenile zebrafish (Parichy et al., 2009).
The many advantages of using zebrafish as a tool for understanding human disease include (i) the use of genetic and pharmacological agents, (ii) the high genetic homology between zebrafish and humans, and (iii) the similarity in tissue architecture and organ development, function, and cellular composition. However, there are also some drawbacks. Developing models of chronic diseases is difficult because the main advantage of zebrafish is based on the use of embryos and larvae. There are also obvious physiological differences between aquatic and terrestrial species. Although fish are afflicted with many of the same diseases found in humans, including developmental defects and cancer, it remains to be determined whether the pathophysiological processes are the same in both species. Clearly, in many cases, they are: human and zebrafish embryos exposed to alcohol develop cyclopia (Blader and Strahle, 1998), and oncogenes such as MYC (Yang et al., 2004; Langenau et al., 2005) and RAS (Davison et al., 2008) drive cancer in both species. Moreover, the gene expression profiles of liver cancers are similar (Lam et al., 2006; Mirbahai et al., 2011), as is the histopathology of several liver diseases (Sadler et al., 2005; Passeri et al., 2009). Thus, there are several aspects of zebrafish that can inform our understanding of human disease.
Genetic Tools
The technologies to manipulate gene expression in zebrafish are evolving. Many approaches to study epigenetics have been used successfully in zebrafish. Knockout/knockdown approaches used in other animals, such as homologous recombination and RNA interference, are not yet developed in zebrafish. Instead, emerging technologies for gene targeting including Targeting Induced Local Lesions in Genomes (TILLING), gene traps, and zinc-finger nucleases (Moens et al., 2008; Foley et al., 2009; Urasaki and Kawakami, 2009), combined with conventional mutagenesis screening, have generated a vast collection of mutants. While most of these have not been characterized, a few thousand mutants have been cataloged at the Zebrafish Model Organism Database (ZFIN; http://zfin.org) and the stocks are maintained in the Zebrafish International Resource Center (ZIRC; http://zebrafish.org/zirc/home/guide.php). In many of these, the disrupted gene has been identified, revealing the existence of zebrafish mutants in several chromatin modifying genes (Table 1).
TABLE 1.
Zebrafish orthologs and mutants lines of key epigenetic modifiers
| Human gene | Zebrafish ortholog | Zebrafish mutant |
|---|---|---|
| DNA methyltransferases | ||
| DNMT1 | dnmt1 | dandylion |
| DNMT3A | dnmt8, dnmt6 | |
| DNMT3B | dnmt3, 4, 5, 7 | |
| Histone acetyltransferases | ||
| HAT1 | hat1 | |
| CREBP | crebbp a, crebbp b | |
| MYST 1–4 | myst 1–4 | moz |
| KAT2A | kat2a | |
| KAT5/TIP60 | zgc:92510 | |
| Histone deacetylases | ||
| HDAC1 and 2 | hdac1 | hdac1 |
| HDAC3 | hdac3 | |
| Others | ||
| UHRF1 | uhrf1 | uhrf1 |
| BRPF1 | brpf1 | brpf1 |
Many mutations that cause early embryonic lethality in mammals are not as severe in zebrafish because wild-type mRNA or protein deposited into the egg by the mother can support early development of the embryo. Zygotic transcription begins during the mid-blastula transition around 3.5 hpf, and thus the mutant phenotype is only revealed after the maternal supply is depleted. Therefore, while early embryonic lethality occurs in mammals with null mutations in the genes required for DNA methylation, such as DNMT1, DNMT3A, DNMT3B, and UHRF1 (Li et al., 1992; Okano et al., 1999; Muto et al., 2002), zebrafish which lack these same genes survive to later stages of development (Sadler et al., 2007; Anderson et al., 2009; Tittle et al., 2011). This unique aspect of zebrafish development allows investigation of the requirement for DNA methylation in later developmental events, such as organogenesis. Depleting both maternal and zygotic gene products using morpholinos is the standard zebrafish approach to carrying out reverse-genetics experiments. Not surprisingly, morpholinos targeting dnmt1 (Rai et al., 2006) and uhrf1 (J. Chu and K.C. Sadler, unpublished data) display earlier and more severe phenotypes than their respective mutants. Finally, transgenics are widely used to express specific genes at a given time and cell type during development. This has been particularly useful for creating a zebrafish line that serves as a reporter for defects in DNA methylation (Goll et al., 2009; Feng et al., 2010a; Akitake et al., 2011).
Genetic Conservation
The zebrafish genome has been assembled at high resolution (http://www.sanger.ac.uk/Projects/D_rerio/ and http://www.ensembl.org/Danio_rerio/Info/Index). Most zebrafish genes have mammalian orthologs, and most epigenetic regulators are highly conserved: there is 75 and 92% identity between zebrafish and human DNMT1 and HDAC1, respectively. However, genome duplication occurred during evolution of the ray-fin phylogeny, and over 20% of these genes are retained in zebrafish (Postlethwait et al., 2000). This presents a difficulty in using in silico analysis to identify some of the zebrafish orthologs of human genes. For instance, there are two human DNMTs involved in de novo DNA methylation, whereas there are six corresponding genes in zebrafish (Table 1). Further experimentation is required to decipher which are orthologs and which are paralogs.
Despite the improved quality of the zebrafish genome, some issues remain a challenge for comparative genomics. Bioinformatics analysis of promoters is widely used to determine the transcriptional potential of a gene of interest. For instance, clusters of cytosines 5′ to guanosines (i.e., CpG islands) in a gene promoter are conserved from human to zebrafish and algorithms to detect these CpG islands can indicate potential genes that may be repressed by DNA methylation. However, while coding sequences are typically highly conserved from zebrafish to human, the noncoding regions of genes are not. Therefore, although the quality of the zebrafish genome has greatly improved, some issues remain a challenge. The lack of complete genomes from fish with a close evolutionary proximity to zebrafish impairs our ability to identify all but the most highly conserved DNA sequences.
It is clear that many genes, pathways and, especially, histone modifications are conserved from humans to zebrafish. As such, many of the antibody reagents to identify these modifications work equally well in fish and mammals (Fig. 2B; Anelli et al., 2009; Lindeman et al., 2010a; Vastenhouw et al., 2010). Thus, zebrafish provide an opportunity to use biochemical and genetic approaches to study the requirement for epigenetic modifications during embryonic development, with the hope that insight from such studies may inform work to use epigenetic drugs to treat cancer.
Figure 2.

DNMT and HDAC inhibitors disrupt zebrafish embryogenesis and liver development. (A) Zebrafish embryos treated with VPA or 5-Aza develop small livers. VPA treatment starting at 6 hpf delayed the appearance of the liver on day 3 and resulted in 100% of the affected embryos displaying a small, ball-shaped liver. 5-Aza treatment at 1.5 hours resulted in 50% mortality (not shown). The majority of survivors (84%) display a smaller, misshapen liver. (B) Treatment of zebrafish embryos at 6 hpf with 20 μM VPA results in increased acetylated histone H3 (Ac-H3) at 5 dpf. (C) Treatment of zebrafish embryos with 5-Aza at 1.5 hpf results in a global loss of DNA methylation. The methylation sensitive (HpaII) and methylation insensitive (MspI) enzymes were used to assess changes in methylation levels in untreated and 5-Aza treated embryos. The smear in the HpaII lane of the 5-Aza treated embryos indicates increased digestion of the DNA, reflecting decreased methylation.
EPIGENETICS OVERVIEW
DNA or histone modifications, that is, epigenetic marks, are complex, interdependent, and not fully understood. It is now appreciated that epigenetic mechanisms of gene regulation control embryogenesis, cell fate, and carcinogenesis. As these marks are not coded for in the DNA sequence, vary among cell types, and are mediated in large part by enzymes, they are attractive targets for therapies. Epigenetic marks can be roughly divided between those that occur on the DNA—namely, DNA methylation—and those that occur on histone tails (Fig. 1). We briefly review these here and direct the reader to recent reviews which cover these topics in more depth (Bonasio et al., 2010; Chi et al., 2010; Feng et al., 2010b).
Figure 1.
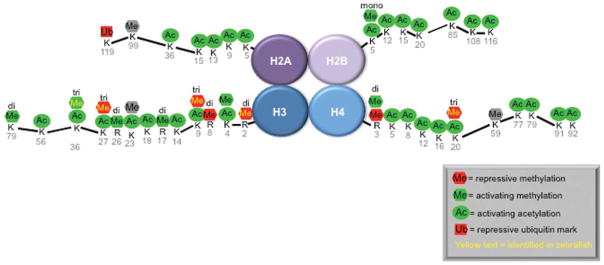
Modifications of the tails of histones play an important role in regulating gene expression. This figure demonstrates some of the better studied histone modifications on the tails of H2A, H2B, H3, and H4. Those in green are activating marks, in red are repressive marks, and in gray are those that are unknown.
DNA methylation is the best studied epigenetic mark. DNMT3A and DNMT3B carry out de novo methylation of DNA, whereas DNMT1 carries out maintenance of the DNA methylation pattern from mother to daughter cell. De novo DNA methylation is essential for X-inactivation and genomic imprinting, whereas maintenance DNA methylation is required for cell fate decisions, differentiation, and potency. In maintenance methylation, DNMT1 and UHRF1 cooperate to create the stable attachment of a methyl group to CpG sites (Bostick et al., 2007; Sharif et al., 2007; Arita et al., 2008; Avvakumov et al., 2008; Hashimoto et al., 2008; Qian et al., 2008). The CpG islands found in the promoters of approximately 60% of genes tend to be unmethylated in normal cells but upon methylation are silenced. DNA methylation frequently occurs in tandem with other repressive epigenetic marks. The two mechanisms postulated for how DNA methylation represses gene transcription are that methyl groups hinder the binding of transcriptional activators or that methyl groups signal recruitment of transcriptional repressor complexes to methylated CpGs (Jones et al., 1998).
Posttranslational modifications of histone tails also serve to regulate chromatin structure and gene expression (Fig. 1). Histones are among the most highly conserved proteins in eukaryotes. The four core histones (H2A, H2B, H3, and H4) form an octomer around which 147 base pairs of DNA is wrapped. Multiple residues on histone tails can be modified by acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, and ADP-ribosylation. However, the most well investigated histone modifications include histone acetylation and methylation. Histone acetylation results in opening of chromatin structure, whereas histone deacetylation causes chromatin compaction resulting in transcriptional repression. In contrast, histone methylation marks can be repressive or activating, depending on which lysine residues are methylated and to what degree (Fig. 1). For instance, addition of three methyl groups to the lysine at position 9 in the tail of histone H3 (i.e., H3K9me3) is a common repressive mark mediated primarily by the methyltransferases Suv39h1 and G9a (Peters et al., 2001; Tachibana et al., 2001), whereas a dimethylation of lysine at position 4 (H3K4Me2) is a mark associated with gene activation. These same marks can occur at a single locus, indicating that the function of these modifications in gene activation or repression are context dependent.
Epigenetic Changes in Cancer
Dynamic changes in epigenetic modifiers during development help to regulate tissue specific gene expression and cell fate decisions, but in cancer, aberrant establishment of these same epigenetic marks drive tumorigenesis. Multiple changes in most epigenetic marks have been documented in human cancers (Berdasco and Esteller, 2010). The complex crosstalk between histone modifications has made it difficult to draw general conclusions about these marks and cancer development or progression. However, DNA methylation changes in tumors are better characterized and understood. Tumors tend to exhibit genome wide hypomethylation, which can result in chromosomal instability (Eden et al., 2003), loss of genomic imprinting (Jirtle, 2004), and activation of oncogenes (Cheah et al., 1984). Thus, DNA hypomethylation in cancer cells may reflect both their undifferentiated state and also contribute to transformation. Additionally, regional hypermethylation at CpG islands results in tumor suppressor silencing (Jones and Baylin, 2002).
For instance, in human hepatocellular carcinoma (HCC), promoter methylation of the cell cycle regulatory gene CDKN2A (also called p16INK4a) is a common occurrence and hypermethylation is the primary mechanism underlying the transcriptional repression of CDKN2A, whereas deletions and mutations of the gene are rarely seen (Ko et al., 2008; Tischoff and Tannapfe, 2008). Other frequently hypermethylated tumor suppressor genes in HCC include the key apoptotic genes, Caspase 8 and TMS1 (Tischoff and Tannapfe, 2008), the cell adhesion molecule, E-cadherin (Lee et al., 2003), and a regulator of the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway, Suppressor of Cytokine Signaling 1 (SOCS-1) (Yoshikawa et al., 2001). Interestingly, in zebrafish HCC samples, changes in the expression levels of many genes are correlated with DNA methylation changes at their promoters, and many of these genes are also similarly altered in human HCC (Mirbahai et al., 2011). This suggests that DNA methylation changes in HCC might be similar in both humans and zebrafish and supports the use of zebrafish to study epigenetic alterations in cancer.
Given that epigenetic modifications are both heritable and reversible makes them attractive targets for cancer therapy. Treating zebrafish embryos with the DNMT inhibitor, 5-Azacytidine (5-Aza), results in some lethality and abnormal development (Fig. 2A; Martin et al., 1999). 5-Aza and its deoxycitidine analog, Decitabine, have thus been used in the clinic to treat myelodysplastic syndromes (Oki et al., 2007; Vigna et al., 2011) and are also being investigated as chemotherapy for solid tumors (Aparicio and Weber, 2002). However, genome hypomethylation resulting in nonspecific gene activation can be a serious side effect of using these drugs to treat cancer patients. Recently, two HDAC inhibitors have also received approval from the Food and Drug Administration (FDA) for use in treating T-cell lymphoma (Shabason et al., 2010), and many more HDAC inhibitors are being evaluated as anticancer agents. However, our understanding of histone modification aberrations in cancer is incomplete. The effects of such inhibitors on the acetylation status of nonhistone proteins and on animal development and physiology must also be taken into account.
EPIGENETICS AND ZEBRAFISH
There is such variety in the changes in chromatin structure and epigenetic marks across loci in a single cell, that on first glance, the feasibility of identifying important epigenetic marks in the four dimensions of embryonic development appears intractable. Moreover, crosstalk between epigenetic marks, the duplicitous roles of some marks as both activating and silencing and the dynamic changes in gene expression during development all present challenges. Nevertheless, advances in our understanding of how epigenetics controls development have been made using zebrafish. Additionally, a few recent reports have described epigenetic changes in zebrafish cancers (Anelli et al., 2009; Mirbahai et al., 2011). Thus, zebrafish may prove useful for studying the epigenetic factors that control both development and cancer. Here, we describe the phenotypes resulting from changes in the epigenetic marks that are best studied in zebrafish: DNA methylation, histone acetylation, and some histone methylation marks (Fig. 1).
Genome wide studies have been used to describe the epigenetic landscape in zebrafish embryos (Wardle et al., 2006; Lindeman et al., 2010a, 2010b; Vastenhouw et al., 2010) and many approaches are being developed in zebrafish to study epigenetics. For instance, several epigenetic modifiers have been mutated (Table 1) or targeted by morpholinos or inhibitors. However, only a subset of their phenotypes has been described, which is partially attributed to the nature of forward genetic screening. For instance, we identified uhrf1 in our screen for mutants that have a defect in hepatic outgrowth (Sadler et al., 2007). Although these mutants clearly have other abnormalities, we focused only on the hepatic phenotype. At the same time, another group identified this same line in a screen for defects in eye development (Gross et al., 2005; Tittle et al., 2011), but have not investigated the phenotypes in the liver or other organs. Whether the same mechanism cause these two phenotypes has yet to be established.
DNA Methylation
DNA methylation is typically correlated with gene repression. There is a striking similarity in the methylation pattern of genomic DNA isolated from whole mouse and zebrafish embryos, with a distinct dip in methylation at the beginning of genes and an enrichment in the body of protein coding genes and in repeated sequences (Feng et al., 2010a). This indicates that zebrafish are an excellent vertebrate model to study the role of DNA methylation in development.
In early embryogenesis, the methylation pattern of the genome is erased and entirely reset by the process of de novo DNA methylation. This results in the silencing of imprinted genes, such as no tail, which is suppressed by dnmt7 (a homolog of mammalian DNMT3; Table 1; Shimoda et al., 2005). The mechanisms of DNA demethylation have recently been uncovered through elegant experiments using zebrafish (Rai et al., 2008, 2010b) and have been expertly reviewed elsewhere (Fritz and Papavasiliou, 2010; Goll and Bestor, 2005). Here, we focus on studies using zebrafish that provide insight into the function of maintenance DNA methylation, as this is likely to be the predominant DNA methylation pathway that is altered in cancer. We predict that findings from studies in zebrafish embryos will provide a unique perspective to our understanding of the regulation and function of the factors that control DNA methylation.
Maintenance DNA methylation requires two factors: DNMT1, which catalyzes the methyltransferase reaction on the unmethylated cytosine of the CpG pair, and UHRF1, which recruits DNMT1 to the DNA (Bostick et al., 2007; Sharif et al., 2007) and flips the methylated cytosine of the parental strand out of the way so that DNMT1 can access the unmethylated cytosine on the opposite strand (Arita et al., 2008; Avvakumov et al., 2008; Hashimoto et al., 2008; Qian et al., 2008). Zebrafish dnmt1 and uhrf1 mutants are strikingly similar. Embryos depleted of Dnmt1, either by morpholino injection (Rai et al., 2006) or mutation (Anderson et al., 2009), have developmental defects in the eye (Rai et al., 2006; Tittle et al., 2011) and endodermal organs (Rai et al., 2006; Anderson et al., 2009). The early stages of development of these tissues are normal, but failure in their growth and morphogenesis is accompanied by massive apoptosis (Rai et al., 2006; Sadler et al., 2007; Anderson et al., 2009; Tittle et al., 2011). This is strikingly similar to the phenotype in uhrf1 mutants (Sadler et al., 2007; Tittle et al., 2011) but different from the phenotype induced by morpholino knockdown of dnmt3 (Rai et al., 2010a). Although there are many similarities between uhrf1 and dnmt1 loss in zebrafish, some embryos with simultaneous mutation of both genes display a more severe phenotype than either mutant does alone (Tittle et al., 2011). Therefore, while it is likely that Dnmt1 serves a single, primary function in maintenance DNA methylation, Uhrf1 has been implicated in mediating other epigenetic marks, and therefore could have multiple, Dnmt1-independent roles in the embryo.
It is difficult to envision how studies using whole embryos, which are rapidly undergoing development, may be used to probe biochemical interactions between different chromatin modifying enzymes. However, epistatic interaction analyses in zebrafish embryos demonstrate that the phenotype caused by loss of Dnmt1, but not Dnmt3, can be reversed by providing excess of the histone methyltransferase, Suv39h1 (Rai et al., 2006, 2010a), which catalyzes the repressive H3K9 methylation mark in heterochromatic regions (Peters et al., 2001). Conversely, a different histone methyltransferase G9a, which also trimethylates H3K9, but selectively in euchromatic regions (Tachibana et al., 2001), acts downstream of Dnmt3 and rescues the brain and retinal defects induced by dnmt3 knockdown (Rai et al., 2010a). This suggests that the phenotypic consequences of wiping out DNA methylation may not be directly due to the methylation defect, per se, but instead could be a secondary consequence of altered histone methylation.
Transgenesis in zebrafish is a widely used and excellent means of marking cells of interest by expressing a fluorescent protein, for labeling subcellular structures with fluorescent proteins and for expressing proteins that may alter a developmental process. Transgenic reporters that are silenced by DNA methylation are being used to examine mutants which fail to methylate DNA (Goll et al., 2009; Feng et al., 2010a; Akitake et al., 2011), including dnmt1 (Anderson et al., 2009) and uhrf1 (Feng et al., 2010a). Such tools will make it possible to carry out screens for other modifiers of DNA methylation.
Histone Acetylation and Deacetylation
Histone deacetylation is carried out by HDACs and is typically associated with gene silencing. Histone acetyltransferases (HAT) act to neutralize the positively charged lysines on the tails of core histones (Fig. 1) and reduce the interaction between histones and DNA. This forces the chromatin into an open configuration that allows transcription factors to more easily bind their target sequences. There are four different HDAC classes. HDAC1 and HDAC3 are nuclear proteins that belong to class I. Their ability to remove acetyl groups from the lysines of histone tails is well defined but it must be noted that they can also deacetylate other nonhistone proteins.
Newly synthesized histones are acted on by HATs just after synthesis in the cytoplasm and are then deacetylated when they are incorporated into the nucleosome. Mutation of the HAT, myst3, in zebrafish causes patterning defects in the facial skeleton (Miller et al., 2004; Crump et al., 2006). The myst3 phenotype is thought to result from the inappropriate acetylation and silencing of hox genes. Interestingly, expression of these genes and the skeletal defects are partially rescued by incubating myst3 mutants in an HDAC inhibitor (HDACi) (Miller et al., 2004). Although non-histone acetylated proteins could contribute to this rescue of the myst3 phenotype, these data underscore the importance of proper histone acetylation in regulating tissue development.
Mutants, morphants, and inhibitors have been used to investigate the role of histone deacetylation in zebrafish development. An increase in global histone acetylation is observed in all of these cases (Fig. 2A; Noel et al., 2008). Many hdac1 mutant alleles have been identified in screens for embryonic essential genes (Amsterdam et al., 2004), hematopoietic stem cells (Burns et al., 2009), development of the liver, pancreas (Noel et al., 2008), retina (Stadler et al., 2005), neural crest (Nambiar et al., 2007; Harrison et al., 2011), and oligodendrocytes (Cunliffe and Casaccia-Bonnefil, 2006). Among other phenotypes, hdac1 mutants have a small liver and expanded foregut (Noel et al., 2008), raising the intriguing possibility that alterations in chromatin structure as a result of persistent histone acetylation directs cell fate decisions.
hdac1 morphants phenocopy most mutant alleles, although in several cases morphants have a more severe phenotype (Cunliffe, 2004; Pillai et al., 2004; Stadler et al., 2005; Yamaguchi et al., 2005; Harrison et al., 2011). This demonstrates that both maternally and zygotically provided Hdac1 contributes to embryonic development. The phenotypes of hdac1 mutants and morphants reflect the first developmental events that require Hdac1 activity. On the other hand, HDACis provide the flexibility of blocking Hdac activity at any stage of development and reveal stage-specific functions. For instance, adding the HDACi valproic acid (VPA) to early embryos at 6 hpf results in a small liver phenotype (Fig. 2A) but adding it later at 72 hpf does not (Farooq et al., 2008). This indicates that the VPA target is required for an early stage of hepatic development. As RNA encoding hdac3 can rescue the phenotype of VPA treated fish (Farooq et al., 2008), it is possible that VPA in zebrafish embryos has a more potent effect on Hdac3 than on Hdac1. Together, these data indicate that histone deacetylation is required for embryonic development and that the liver is particularly sensitive to the loss of HDAC activity.
Histone Methylation
Histone methylation marks can be repressive (i.e., H3K9me3 or H3K27me3) or activating (i.e. H3K4me3), depending on the lysine or arginine that is modified and to what degree (Fig. 1). These marks are complex and their regulation, interplay and, in some cases, even their functions are not fully understood. Moreover, some genes contain methyl marks that are both activating and repressing. Additionally, unlike the broad-action of enzymes that add or remove acetyl groups, histone methyltransferases demonstrate high specificity. The three groups of histone methylation enzymes consist of SET domain lysine methyltransferases, non-SET domain lysine methyltransferases, and arginine methyltransferases (Trievel, 2004).
Several methylated histone residues have been reported in zebrafish, including those associated with gene activation (H3K4me3 and H3K36me3) and repression (H3K9me3 and H3K27me3). Two recent genome wide studies comparing the histone methylation patterns in mammalian embryonic stem cells and in zebrafish embryos during genome activation have revealed many similarities (Lindeman et al., 2010a; Vastenhouw et al., 2010). As expected, H3K4me3 marks were primarily associated with promoters of activated genes, while H3K27me3 marks were associated with silenced genes. Additionally, many genes were marked with bivalent chromatin, possessing both activating and repressing (H3K4me3 and H3K27me3) histone marks (Lindeman et al., 2010a; Vastenhouw et al., 2010). Such genes are restrained in the off state poised for activation and are associated with transcriptional regulators and signal transducers involved in development (Lindeman et al., 2010a; Vastenhouw et al., 2010). In contrast, a collection of repressive marks accumulate on genes that become silenced during terminal differentiation. These studies add to the emergent appreciation that histone methylation marks can have different effects on gene expression depending on the cellular and developmental context. In addition, they demonstrate for the first time that the chromatin modifications in the transiently pluripotent cells of an embryo are similar to that of permanently pluripotent embryonic stem cells.
Similar to the changes in histone methylation marks during genome activation, there are massive changes in these marks during tumorigenesis. A study in zebrafish found that melanoma tumors were enriched in certain repressive histone marks (H3K9me3, HP1), when compared with nontumor tissue. Meanwhile, they found decreased levels of the activating histone marks, H3K4me3 and H4K20me2, when compared with normal skin (Anelli et al., 2009), supporting the notion that there is a global increase in repression of gene expression in tumors due to changes in chromatin marks. Approaches that manipulate the histone methyltransferases required for these marks will further elucidate their contribution to tumor onset or progression and may indicate the use of targeting these enzymes to treat melanoma.
EPIGENETICS, LIVER DEVELOPMENT, AND CANCER
It is likely that a set of genes which drive hepatocyte proliferation under physiological conditions—such as hepatic outgrowth in embryos or regeneration in adults—will be co-opted by cells as they undergo malignant transformation. Studies in zebrafish have clearly identified requirements for DNA methylation, histone deacetylation, and histone methylation during liver development (Fig. 2A; Rai et al., 2006; Sadler et al., 2007; Farooq et al., 2008; Noel et al., 2008; Anderson et al., 2009; Rai et al., 2010a). Interestingly, the phenotype in uhrf1 mutants is attributed to a combination of decreased proliferation and increased apoptosis (Sadler et al., 2007). We find the same effect in human cancer cells depleted of UHRF1 (Tien et al., 2011). Many types of cancer, including HCC express very high levels of UHRF1 (K.C. Sadler and J. Llovet, unpublished data). Thus, inhibiting UHRF1 may be an effective target for chemotherapeutic agents aimed at killing cancer cells (Bronner et al., 2007; Unoki et al., 2009).
The mechanisms by which interfering with epigenetic modifications halt cell proliferation or induce cell death are unknown. One possibility is that tumor suppressors and proapoptotic genes require epigenetic mechanisms for their silencing, and when DNA methylation or histone acetylation is depleted, these genes are activated. Consistent with this possibility, our preliminary data demonstrates that caspase 8 transcription increases in uhrf1 zebrafish mutants (R. Mudbhary and K.C. Sadler, unpublished data) and Caspase 8 is also required for cancer cell apoptosis caused by UHRF1 depletion (Tien et al., 2011) in human cancer cells. Alternatively, stripping the genome of DNA methylation or loading the chromatin with acetylated histones may trigger a cellular response that initiates cell death. Indeed, cells depleted of DNMT1 and thus DNA methylation undergo mitotic catastrophe (Chen et al., 2007) via a mechanism that is likely unrelated to gene transcription and supports the hypothesis that massive alterations to the epigenome may trigger a response similar to a checkpoint.
Zebrafish provide a useful whole animal vertebrate system to differentiate between these possibilities and hold the promise of identifying new players that regulate hepatocyte proliferation. The future potential of this system may extend to developing models of liver cancer that could be used for screening candidate drugs that target the epigenome.
Acknowledgments
We thank Brandon Kent for advice on the manuscript, Chinwe Ukomadu for helpful discussions, and Peter Liu for technical assistance. We are grateful for support from the NIH (5R01DK080789-02 and 3R01DK080789-01A1S1), the Breast Cancer Alliance, and the March of Dimes (to K.C.S).
References
- Akitake CM, Macurak M, Halpern ME, Goll MG. Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol. 2011;352(2):191–201. doi: 10.1016/j.ydbio.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, et al. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Bosch JA, Goll MG, et al. Loss of Dnmt1 catalytic activity reveals multiple roles for DNA methylation during pancreas development and regeneration. Dev Biol. 2009;334(1):213–223. doi: 10.1016/j.ydbio.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli V, Santoriello C, Distel M, et al. Global repression of cancer gene expression in a zebrafish model of melanoma is linked to epigenetic regulation. Zebrafish. 2009;6:417–424. doi: 10.1089/zeb.2009.0612. [DOI] [PubMed] [Google Scholar]
- Aparicio A, Weber JS. Review of the clinical experience with 5-azacytidine and 5-aza-2′-deoxycytidine in solid tumors. Curr Opin Investig Drugs. 2002;3:627–633. [PubMed] [Google Scholar]
- Arita K, Ariyoshi M, Tochio H, et al. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Blader P, Strahle U. Ethanol impairs migration of the prechordal plate in the zebrafish embryo. Dev Biol. 1998;201:185–201. doi: 10.1006/dbio.1998.8995. [DOI] [PubMed] [Google Scholar]
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M, Kim JK, Esteve PO, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- Branch S, Francis BM, Brownie CF, Chernoff N. Teratogenic effects of the demethylating agent 5-aza-2′-deoxycytidine in the Swiss Webster mouse. Toxicology. 1996;112:37–43. doi: 10.1016/0300-483x(96)88183-2. [DOI] [PubMed] [Google Scholar]
- Bronner C, Achour M, Arima Y, et al. The UHRF family: oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol Ther. 2007;115:419–434. doi: 10.1016/j.pharmthera.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Burns CE, Galloway JL, Smith AC, et al. A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence. Blood. 2009;113:5776–5782. doi: 10.1182/blood-2008-12-193607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah MS, Wallace CD, Hoffman RM. Hypomethylation of DNA in human cancer cells: a site-specific change in the c-myc oncogene. J Natl Cancer Inst. 1984;73:1057–1065. [PubMed] [Google Scholar]
- Chen T, Hevi S, Gay F, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump JG, Swartz ME, Eberhart JK, Kimmel CB. Moz-dependent Hox expression controls segment-specific fate maps of skeletal precursors in the face. Development. 2006;133:2661–2669. doi: 10.1242/dev.02435. [DOI] [PubMed] [Google Scholar]
- Cunliffe VT. Histone deacetylase 1 is required to repress Notch target gene expression during zebrafish neurogenesis and to maintain the production of motoneurones in response to hedgehog signalling. Development. 2004;131:2983–2995. doi: 10.1242/dev.01166. [DOI] [PubMed] [Google Scholar]
- Cunliffe VT, Casaccia-Bonnefil P. Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mech Dev. 2006;123:24–30. doi: 10.1016/j.mod.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Davison JM, Woo Park S, Rhee JM, Leach SD. Characterization of Kras-mediated pancreatic tumorigenesis in zebrafish. Methods Enzymol. 2008;438:391–417. doi: 10.1016/S0076-6879(07)38027-0. [DOI] [PubMed] [Google Scholar]
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- Farooq M, Sulochana KN, Pan X, et al. Histone deacetylase 3 (hdac3) is specifically required for liver development in zebrafish. Dev Biol. 2008;317(1):336–353. doi: 10.1016/j.ydbio.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010a;107(19):8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010b;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn EJ, 3rd, Trent CM, Rawls JF. Ontogeny and nutritional control of adipogenesis in zebrafish (Danio rerio) J Lipid Res. 2009;50:1641–1652. doi: 10.1194/jlr.M800590-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley JE, Yeh JR, Maeder ML, et al. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN) PLoS One. 2009;4:e4348. doi: 10.1371/journal.pone.0004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll MG, Anderson R, Stainier DY, et al. Transcriptional silencing and reactivation in transgenic zebrafish. Genetics. 2009;182:747–755. doi: 10.1534/genetics.109.102079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gross JM, Perkins BD, Amsterdam A, et al. Identification of zebrafish insertional mutants with defects in visual system development and function. Genetics. 2005;170:245–261. doi: 10.1534/genetics.104.039727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Berman MG, Wittner BS, et al. Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J. 2005;19:1166–1168. doi: 10.1096/fj.04-3425fje. [DOI] [PubMed] [Google Scholar]
- Harrison MR, Georgiou AS, Spaink HP, Cunliffe VT. The epigenetic regulator Histone Deacetylase 1 promotes transcription of a core neurogenic programme in zebrafish embryos. BMC Genomics. 2011;12:24. doi: 10.1186/1471-2164-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Horton JR, Zhang X, et al. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrie D, Sadler KC. White adipose tissue development in zebrafish is regulated by both developmental time and fish size. Dev Dyn. 2010;239:3013–3023. doi: 10.1002/dvdy.22443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL. IGF2 loss of imprinting: a potential heritable risk factor for colorectal cancer. Gastroenterology. 2004;126:1190–1193. doi: 10.1053/j.gastro.2004.02.026. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Ko E, Kim Y, Kim SJ, et al. Promoter hypermethylation of the p16 gene is associated with poor prognosis in recurrent early-stage hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev. 2008;17:2260–2267. doi: 10.1158/1055-9965.EPI-08-0236. [DOI] [PubMed] [Google Scholar]
- Lam SH, Wu YL, Vega VB, et al. Conservation of gene expression signatures between zebrafish and human liver tumors and tumor progression. Nat Biotechnol. 2006;24:73–75. doi: 10.1038/nbt1169. [DOI] [PubMed] [Google Scholar]
- Langenau DM, Feng H, Berghmans S, et al. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2005;102:6068–6073. doi: 10.1073/pnas.0408708102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee HJ, Kim JH, et al. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371–1378. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lindeman LC, Reiner AH, Mathavan S, et al. Tiling histone H3 lysine 4 and 27 methylation in zebrafish using high-density microarrays. PLoS One. 2010a;5:e15651. doi: 10.1371/journal.pone.0015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman LC, Winata CL, Aanes H, et al. Chromatin states of developmentally-regulated genes revealed by DNA and histone methylation patterns in zebrafish embryos. Int J Dev Biol. 2010b;54:803–813. doi: 10.1387/ijdb.103081ll. [DOI] [PubMed] [Google Scholar]
- Martin CC, Laforest L, Akimenko MA, Ekker M. A role for DNA methylation in gastrulation and somite patterning. Dev Biol. 1999;206:189–205. doi: 10.1006/dbio.1998.9105. [DOI] [PubMed] [Google Scholar]
- Menegola E, Di Renzo F, Broccia ML, Giavini E. Inhibition of histone deacetylase as a new mechanism of teratogenesis. Birth Defects Res C Embryo Today. 2006;78:345–353. doi: 10.1002/bdrc.20082. [DOI] [PubMed] [Google Scholar]
- Miller CT, Maves L, Kimmel CB. moz regulates Hox expression and pharyngeal segmental identity in zebrafish. Development. 2004;131:2443–2461. doi: 10.1242/dev.01134. [DOI] [PubMed] [Google Scholar]
- Mirbahai L, Williams TD, Zhan H, et al. Comprehensive profiling of zebrafish hepatic proximal promoter CpG island methylation and its modification during chemical carcinogenesis. BMC Genomics. 2011;12:3. doi: 10.1186/1471-2164-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens CB, Donn TM, Wolf-Saxon ER, Ma TP. Reverse genetics in zebrafish by TILLING. Brief Funct Genomic Proteomic. 2008;7:454–459. doi: 10.1093/bfgp/eln046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto M, Kanari Y, Kubo E, et al. Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J Biol Chem. 2002;277:34549–34555. doi: 10.1074/jbc.M205189200. [DOI] [PubMed] [Google Scholar]
- Nambiar RM, Ignatius MS, Henion PD. Zebrafish colgate/hdac1 functions in the non-canonical Wnt pathway during axial extension and in Wnt-independent branchiomotor neuron migration. Mech Dev. 2007;124:682–698. doi: 10.1016/j.mod.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel ES, Casal-Sueiro A, Busch-Nentwich E, et al. Organ-specific requirements for Hdac1 in liver and pancreas formation. Dev Biol. 2008;322:237–250. doi: 10.1016/j.ydbio.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Oki Y, Aoki E, Issa JP. Decitabine—bedside to bench. Crit Rev Oncol Hematol. 2007;61:140–152. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Parichy DM, Elizondo MR, Mills MG, et al. Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev Dyn. 2009;238:2975–3015. doi: 10.1002/dvdy.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeri MJ, Cinaroglu A, Gao C, Sadler KC. Hepatic steatosis in response to acute alcohol exposure in zebrafish requires sterol regulatory element binding protein activation. Hepatology. 2009;49:443–452. doi: 10.1002/hep.22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O’Carroll D, Scherthan H, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Pillai R, Coverdale LE, Dubey G, Martin CC. Histone deacetylase 1 (HDAC-1) required for the normal formation of craniofacial cartilage and pectoral fins of the zebrafish. Dev Dyn. 2004;231:647–654. doi: 10.1002/dvdy.20168. [DOI] [PubMed] [Google Scholar]
- Postlethwait JH, Woods IG, Ngo-Hazelett P, et al. Zebrafish comparative genomics and the origins of vertebrate chromosomes. Genome Res. 2000;10:1890–1902. doi: 10.1101/gr.164800. [DOI] [PubMed] [Google Scholar]
- Qian C, Li S, Jakoncic J, et al. Structure and hemimethylated CpG binding of the SRA domain from human UHRF1. J Biol Chem. 2008;283:34490–34494. doi: 10.1074/jbc.C800169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Huggins IJ, James SR, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Jafri IF, Chidester S, et al. Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J Biol Chem. 2010a;285:4110–4121. doi: 10.1074/jbc.M109.073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Nadauld LD, Chidester S, et al. Zebra fish Dnmt1 and Suv39h1 regulate organ-specific terminal differentiation during development. Mol Cell Biol. 2006;26:7077–7085. doi: 10.1128/MCB.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Sarkar S, Broadbent TJ, et al. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell. 2010b;142:930–942. doi: 10.1016/j.cell.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Chernoff N. 5-Aza-2′-deoxycytidine-induced cytotoxicity and limb reduction defects in the mouse. Teratology. 2002;65:180–190. doi: 10.1002/tera.10029. [DOI] [PubMed] [Google Scholar]
- Sadler KC, Amsterdam A, Soroka C, et al. A genetic screen in zebrafish identifies the mutants vps18, nf2 and foie gras as models of liver disease. Development. 2005;132:3561–3572. doi: 10.1242/dev.01918. [DOI] [PubMed] [Google Scholar]
- Sadler KC, Krahn KN, Gaur NA, Ukomadu C. Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, uhrf1. Proc Natl Acad Sci USA. 2007;104:1570–1575. doi: 10.1073/pnas.0610774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabason JE, Tofilon PJ, Camphausen K. HDAC inhibitors in cancer care. Oncology (Williston Park) 2010;24:180–185. [PMC free article] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi S, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Shimoda N, Yamakoshi K, Miyake A, Takeda H. Identification of a gene required for de novo DNA methylation of the zebrafish no tail gene. Dev Dyn. 2005;233:1509–1516. doi: 10.1002/dvdy.20455. [DOI] [PubMed] [Google Scholar]
- Stadler JA, Shkumatava A, Norton WH, et al. Histone deacetylase 1 is required for cell cycle exit and differentiation in the zebrafish retina. Dev Dyn. 2005;233:883–889. doi: 10.1002/dvdy.20427. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- Tien AL, Senbanerjee S, Kulkarni A, et al. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem J. 2011;435:175–185. doi: 10.1042/BJ20100840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14:1741–1748. doi: 10.3748/wjg.14.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittle RK, Sze R, Ng A, et al. Uhrf1 and Dnmt1 are required for development and maintenance of the zebrafish lens. Dev Biol. 2011;350:50–63. doi: 10.1016/j.ydbio.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trievel RC. Structure and function of histone methyltransferases. Crit Rev Eukaryot Gene Expr. 2004;14:147–169. doi: 10.1615/critreveukaryotgeneexpr.v14.i3.10. [DOI] [PubMed] [Google Scholar]
- Unoki M, Brunet J, Mousli M. Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharmacol. 2009;78(10):1279–1288. doi: 10.1016/j.bcp.2009.05.035. [DOI] [PubMed] [Google Scholar]
- Urasaki A, Kawakami K. Analysis of genes and genome by the tol2-mediated gene and enhancer trap methods. Methods Mol Biol. 2009;546:85–102. doi: 10.1007/978-1-60327-977-2_6. [DOI] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, et al. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464:922–926. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigna E, Recchia AG, Madeo A, et al. Epigenetic regulation in myelodysplastic syndromes: implications for therapy. Expert Opin Investig Drugs. 2011;20:465–493. doi: 10.1517/13543784.2011.559164. [DOI] [PubMed] [Google Scholar]
- Wardle FC, Odom DT, Bell GW, et al. Zebrafish promoter microarrays identify actively transcribed embryonic genes. Genome Biol. 2006;7:R71. doi: 10.1186/gb-2006-7-8-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Tonou-Fujimori N, Komori A, et al. Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development. 2005;132:3027–3043. doi: 10.1242/dev.01881. [DOI] [PubMed] [Google Scholar]
- Yang HW, Kutok JL, Lee NH, et al. Targeted expression of human MYCN selectively causes pancreatic neuroendocrine tumors in transgenic zebrafish. Cancer Res. 2004;64:7256–7262. doi: 10.1158/0008-5472.CAN-04-0931. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Matsubara K, Qian GS, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet. 2001;28:29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]