

Abstract
We describe here a detailed protocol for generating gene knockout rats by homologous recombination in embryonic stem (ES) cells. This protocol comprises the following procedures: derivation and expansion of rat ES cells, construction of gene-targeting vectors, generation of gene-targeted rat ES cells and, finally, production of gene-targeted rats. The major differences between this protocol and the classical mouse gene-targeting protocol include ES cell culture methods, drug selection scheme, colony picking and screening strategies. This ES cell–based gene-targeting technique allows sophisticated genetic modifications to be performed in the rat, as many laboratories have been doing in the mouse for the past two decades. Recently we used this protocol to generate Tp53 (also known as p53) gene knockout rats. The entire process requires ~1 year to complete, from derivation of ES cells to generation of knockout rats.
INTRODUCTION
Gene targeting by homologous recombination in embryonic stem (ES) cells is a powerful technique that allows any chosen gene to be genetically modified in a predetermined way1. In the past 20 years, thousands of genes have been modified in mouse ES cells by homologous recombination with gene-targeting vectors, yielding valuable research models. However, this ES cell–based gene-targeting technique was previously only available for the mouse because of the inability to establish authentic ES cells from other species. Recently, we developed a new culture system that can support the efficient derivation and maintenance of germline-competent ES cells not only from mice but also, most importantly, from different strains of rats2–4. This culture system uses a serum-free N2B27 medium supplemented with three small-molecule inhibitors (3i): CHIR99021, PD184352 and SU5402. CHIR99021 specifically inhibits glycogen synthase kinase 3, whereas PD184352 and SU5402 inhibit mitogen-activated protein kinase and fibroblast growth factor receptor tyrosine kinase, respectively2. Subsequently, we found that a more potent mitogen-activated protein kinase inhibitor, PD0325901, can be used to replace both PD184352 and SU5402 (ref. 2). Because the two-inhibitor (CHIR99021 and PD0325901), 2i medium is more effective than 3i medium for deriving and propagating rat ES cells, this protocol describes the use of 2i.
Using the 2i or 3i medium, we and others have established ES cell lines at high efficiency from different strains of rats: dark agouti (DA), Fischer 344 (F344), Sprague-Dawley, Brown Norway, Wistar, Long-Evans and spontaneously hypertensive rats (SHR; refs. 3–9 and unpublished data; Table 1). The availability of robust and germline-competent rat ES cells has opened the door for the application of ES cell–based gene targeting and related genome engineering technologies in rats. The potential utility of this technology was indicated in our recent report on the generation of Tp53 (also known as p53) gene knockout rats by homologous recombination in DA rat ES cells10. Efficient gene targeting by homologous recombination has also been reported in F344 and Sprague-Dawley rat ES cells11. These results have established a strong foundation for the development of gene-targeted rat models and the pursuit of rat functional genomics. The rat offers a complementary model choice to the mouse because rat models have been shown to more closely mimic human disease than mouse models in several areas, including neurodegenerative disease12, nephropathy13, breast cancer14 and rheumatoid arthritis15. Rats are approximately ten times larger than mice, allowing investigators to perform procedures such as nerve recordings, collection of tissue from small structures and serial blood sampling more easily.
TABLE 1.
ES cells derived from different strains of rats.
| Rat strain | ES cell derivation efficiency | Blastocyst injection
|
Gene-targeted rats | Gene-targeted ES cells | ||
|---|---|---|---|---|---|---|
| Host blastocyst | Chimaeras | Germline transmission | ||||
| DA | 33–85% (refs. 3,4 and unpublished data) | F344 (ref. 4) | Yes | Yes | Yes10 | Yes |
| Sprague-Dawley3,4 | Yes | No | No | |||
| Sprague-Dawley | 29–100% (refs. 3,4,6,11) | DA | Yes | Yes | No | Yes11 |
| F344 | 28–81% (refs. 4,6,11) | DA | No | No | N/A | Yes11 |
| Long-Evans | 11–80% (ref. 5 and unpublished data) | Wistar5 | Yes | No | N/A | N/A |
| Wistar | 13% (refs. 5,8) | Long-Evans5 | Yes | Yes | N/A | N/A |
| Wistar/DA hybrid8 | Yes | Yes | ||||
| Wistar/Long-Evans hybrid | 100% (ref. 5) | Long-Evans | Yes | Yes | N/A | N/A |
| Brown-Norway | 33–100% (refs. 6,9 and unpublished data) | Wistar9 | Yes | Yes | N/A | N/A |
| Sprague-Dawley6 | Yes | No | ||||
| SHR | 44% (Q. Qian, personal communication) | N/A | N/A | N/A | N/A | N/A |
N/A, not available.
Over the years, several technologies have been developed to modify the rat genome. These technologies include pronuclear microinjection16, lentiviral transgenesis17, N-ethyl-N-nitrosourea mutagenesis18,19, transposon mutagenesis20–22 and zinc-finger nuclease–mediated gene targeting23–27. The advantages and disadvantages of each technology are listed in Table 2, and investigators should decide which technology is most appropriate for their applications. Currently, ES cell–based gene targeting is still the most effective technology to generate rats (or mice) with genes modified by homologous recombination and to restrict the genetic modifications to a desired group of tissues or to a chosen period during the development of the animal.
TABLE 2.
Advantages and disadvantages of different methods for generating transgenic rats.
| Method | Advantages | Disadvantages |
|---|---|---|
| Pronuclear injection16 | The most straightforward method to generate transgenic rats in the absence of rat ES cells |
|
| Lentiviral transgenesis17 |
|
|
| N-ethyl-N-nitrosourea mutagenesis18,19 |
|
|
| Transposon mutagenesis20–22 |
|
|
| Zinc-finger nuclease (ZFN)-mediated gene targeting23–26 |
|
|
| ES cell-based gene targeting10,11 |
|
|
In this protocol, we describe how to generate gene knockout rats by homologous recombination in ES cells. The flowchart in Figure 1 shows the different stages of the process, which requires basic techniques in cell culture, molecular biology, rat embryology and rat surgery. Some procedures, such as karyotyping, vasectomy, embryo isolation and manipulation, microinjection and the transfer of embryos to a pseudopregnant recipient, are provided as a service by animal and transgenic core facilities in many universities and research institutes. Gene-targeting strategies involving homologous recombination have been used routinely to introduce various gene modifications into mice. However, rats are the species of choice in many areas of biomedical research, and we believe that researchers in many laboratories around the world will take advantage of the well-established ES cell–based gene-targeting technology to produce new rat models.
Figure 1.

Flowchart outlining how to generate gene knockout rats step by step.
Limitations of the protocol
The 2i or 3i medium allows us to establish ES cell lines from different strains of rats at a relatively high efficiency (Table 1). However, using the 2i or 3i medium, we and other researchers could not establish authentic ES cell lines from other species, including rabbits, pigs, cows, sheep, zebrafish and humans. Evidently, new types of ES cell media must be developed to derive ES cells from other species. ES cell lines derived from DA, Sprague-Dawley and Wistar strains of rats have been proven to be germline-competent, but the germline transmission rate is relatively low, and so far the retention of germline competency after gene targeting has only been demonstrated in DA rat ES cells3,4,8,10 (Table 1). Many factors determine the efficiency of gene targeting in rat ES cells and subsequent generation of gene-targeted rats; among these factors are the length of the homology arm, the quality of the rat ES cells and the genetic background of the host embryo chosen for the production of ES cell–rat chimeras. The protocol described here provides a basic platform for optimization of gene-targeting procedures in rats. More information about the limitations of this protocol, as compared with other technologies, is provided in Table 2.
Experimental design
Derivation and propagation of rat ES cells
Using the 2i or 3i culture system, we have been able to derive ES cell lines at a relatively high efficiency from all the rat strains that we have tested, including DA, F344, Sprague-Dawley, Brown Norway and Long-Evans (refs. 3,4; and unpublished data). Other groups have reported the derivation of ES cells from other strains of rats5–9 (Table 1). Evidence indicates that ES cells can be established from most, if not all, strains of rats by using the 2i condition. The derivation procedure is relatively simple. After the removal of the zona pellucida with Tyrode’s solution, blastocysts are transferred to a four-well plate precoated with feeders and cultured in 2i medium. ES cell lines can be established from ~50% of the rat blastocysts that had been plated. Rat ES cell derivation efficiency can be increased if the inner cell mass is isolated by immunosurgery after the removal of the zona pellucida (M. Buehr, personal communication). If a particular rat strain is characterized by low efficiency in deriving ES cells, isolation of the inner cell mass will most likely increase the chance of success. Feeders are essential for the maintenance of pluripotent rat ES cells in the 2i condition. Fibroblasts derived from either mouse or rat embryos can be used for rat ES cell culture. Before use as feeders, embryonic fibroblasts must be mitotically inactivated either by γ-irradiation or by mitomycin C treatment28. Rat ES cells grow as loosely attached or floating aggregates in the 2i condition; hence, extra care must be taken to avoid washing away the cells when changing the medium or passaging the cells. For routine passaging, we detach rat ES cells by pipetting and collect them by centrifugation, after which trypsin is added to dissociate the cell aggregates into single cells. Passaging cells in this way can also avoid the carryover of feeders, which adversely affects rat ES cell growth.
Design of the targeting vector
Gene targeting by homologous recombination in ES cells has provided a powerful means to elucidate gene function and create gene knockout animal models1. The design of the gene-targeting vector is the first critical step29. Key components of a targeting vector include two homology arms with the same DNA sequences as the genomic DNA fragments flanking the region to be modified, and both a positive and a negative selection marker. Construction of the targeting vectors has been described in detail in various protocols28–32. Although the basic principle for designing a gene-targeting vector is the same for both the mouse and the rat, construction of a gene-targeting vector for the rat requires several modifications to the classic strategy. The first of these is the choice of promoter used to drive the expression of the positive selection marker. In mouse gene-targeting vectors, phosphoglycerate kinase (PGK) promoter is most often used to control the selection marker gene expression. Rat ES cells are very sensitive to drug selection, and the activity of the PGK promoter is too weak for the efficient isolation of drug-resistant colonies of rat ES cells. The cytomegalovirus early enhancer/chicken β-actin (CAG) promoter has a much stronger activity than that of the PGK promoter. The use of the CAG promoter to drive the expression of the positive selection marker has been proven to be very effective for isolating drug-resistant colonies in rat ES cells10.
The second modification to the strategy is the choice of the negative selection marker, which is the gene used to eliminate cells with targeting vectors integrated at non-homologous recombination sites. We use a gene encoding the diphtheria toxin-A chain (referred to herein as DTA) as a negative selection marker, instead of the thymidine kinase gene. Cells with random integration of targeting vectors containing the DTA negative selection gene will produce DTA that kills the cells. As a result, correctly targeted cells can be enriched without the addition of any selection drugs. We found that gancyclovir, at the working concentration of 2.5 μM, is toxic to all the rat ES cell lines that we have tested, including ES cell lines derived from DA, F344 and Sprague-Dawley rats. Gancyclovir also promotes differentiation of rat ES cells maintained in the 2i condition. Consequently, the thymidine kinase gene is not suitable for use as a negative selection marker in the rat gene-targeting vector.
The third modification to the strategy involves the source of homology arms. Currently, there is no information available for DA rat bacterial artificial chromosome end sequence. Homology arms can be amplified from DA rat genomic DNA by using PCR-based methods with high-fidelity DNA polymerases, such as the Expand High Fidelity PLUS PCR System (Roche) or Phusion DNA polymerase (New England Biolabs). The published rat genomic sequences are from the Brown Norway strain of rats33. If there are any sequence differences between the amplified DNA and the published data, it is important to determine whether these differences are polymorphisms specific to DA rat genomic DNA or mutations created by PCR. The homology arms generated by PCR are subcloned into a pUC vector, using Clontech’s infusion method, as illustrated in Figure 2a. The targeting vector is constructed by inserting the 5′ homology arm, the 3′ homology arm and the PGK-DTA-poly(A) cassette into the pCAG-EGFP-IRES-Pac plasmid (Fig. 2b).
Figure 2.

Schematic diagram showing the strategy for constructing a rat gene-targeting vector. (a) Amplification and construction of 5′ and 3′ homology arms. pUC backbone vector is amplified by PCR, using primers 1 and 2. Restriction enzyme sites SalI and SpeI are introduced into primers 1 and 2, respectively. Primers 3 and 4 are designed to amplify the 5′ or 3′ homology arm from DA rat genomic DNA. Primers 1 and 2 contain 15-bp extensions (highlighted in red) complementary to primers 3 and 4. The homology arms generated by PCR are subcloned into the pUC vector, using Clontech’s infusion method. (b) Construction of the targeting vector by sequentially inserting the 5′ homology arm, the 3′ homology arm and the PGK-DTA negative selection cassette into the pCAG-EGFP-IRES-Pac plasmid. Restriction enzyme SalI is used to linearize the targeting vector.
Introduction of targeting vectors into rat ES cells
Several methods are available for introducing foreign DNA into ES cells, including electroporation, nucleofection and chemical-based transfection. We have evaluated these methods, and our results suggest that electroporation is still the most effective means of generating correctly gene-targeted rat ES cell clones (unpublished data). The variable quality of mouse ES cell lines often hinders the development of the mouse model containing a modified gene locus34. This likely applies to the rat as well. The major factors that affect the quality of ES cells and their ability to generate germline chimeras include culture medium, quality of feeder cells, methods of passaging and the number of passages28. Under suboptimal culture conditions, ES cells progressively acquire chromosomal abnormalities as they are passaged, and this is one of the major reasons as to why ES cells might fail to contribute to the germ-line. The quality of rat ES cells should be regularly monitored by examining their karyotypes, in vitro differentiation potential and expression of pluripotency markers, and also by determining how efficiently they produce germline chimeras, which still remains the gold-standard test3,4,28. Rat ES cells grow as compact dome-shaped colonies in the 2i condition, and as the number of passages increases, some rat ES cells start attaching to the feeders and forming flat colonies. Rat ES cells with flat colony morphology should not be used for gene targeting, because they are most likely karyotypically abnormal10.
Isolation of gene-targeted rat ES cell colonies
Rat ES cells cultured in the 2i condition are very sensitive to drug selection. We administer the selection drug at half of the normal concentration used for mouse ES cells and apply a pulse selection scheme to increase the selection efficiency10. Our tests on ES cells derived from DA, F344 and Sprague-Dawley rats showed this pulse-selection scheme to be highly effective when using puromycin, G418 or hygromycin. However, it is possible that this selection scheme will not work effectively for other drugs or a particular rat ES cell line. If this is the case, the investigators should optimize the selection condition by determining the killing curve of the selection drug on the rat ES cell line that they use. As mentioned above, most of the rat ES cell colonies are either floating or loosely attached to the feeders, and extra care should be taken when changing the medium so that these colonies are not washed away. After drug selection is completed, colonies can be picked using the simple, efficient method depicted in Figure 3. Colonies are detached by pipetting and are pooled together in a sterile tube. Each colony is then placed into one drop (10 μl) of 0.025% (wt/vol) trypsin-EDTA solution to dissociate the ES colony into single cells. In this way, up to 500 colonies can be picked, dissociated and seeded in < 3 h. The dissociated colonies are transferred into duplicate 96-well plates, using a multichannel pipette.
Figure 3.

Flowchart showing a modified method for picking colonies.
Freezing and thawing of the rat ES cells cultured in the 96-well plate results in a poor recovery rate. To circumvent this problem, we split each rat ES cell colony into two 96-well plates, with one plate having three times more cells than the other. The plate with more cells is used for the initial screening by PCR, which takes 1–2 d to complete, whereas cells in the other plate are maintained in culture. Colonies that test positive in PCR screening are then expanded from the duplicate plate for confirmation by Southern blot, sequencing analysis or both. The strategies for confirmation of the targeting events in rat ES cells by Southern blot are essentially the same as in mouse ES cells28,35. The design of PCR primers and optimization of PCR conditions are critical for the initial screening. One of the PCR primer pair should be located on the genomic DNA beyond the 3′ short arm and the other located on the drug-resistance cassette (Fig. 4). If mouse embryonic fibroblasts (MEFs) are used as feeder layers, BLAST the PCR screening primer sequences against mouse genomic DNA sequences to rule out potential nonspecific amplifications. We recommend constructing a control vector containing the 3′ short homology arm with a 300-bp to 500-bp extension in which one of the PCR primer pair is located (Fig. 4). Rat ES cells transfected with the control plasmid mimic the targeting event and can therefore be used to optimize the PCR screening conditions. The genomic DNA extracted from these rat ES cells is also run in parallel with PCR primers as a positive control for the PCR screening.
Figure 4.
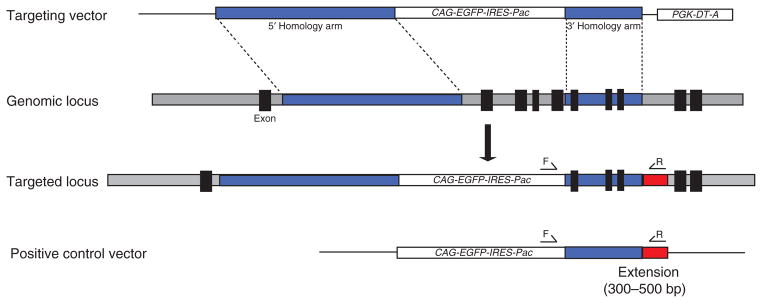
Diagram of the strategy for constructing a control vector used for optimizing PCR screening conditions. A DNA fragment containing the 3′ homology arm with a 300- to 500-bp extension (red) is subcloned into the pCAG-EGFP-IRES-Pac vector to generate a positive control vector for PCR screening. This positive control vector contains a sequence that only a correctly targeted genomic locus would have. The forward (F) and reverse (R) primers are designed to be located within the CAG-EGFP-IRES-Pac cassette and the 300- to 500-bp extension, respectively. Rat ES cells with the stably integrated positive control vector will mimic targeting events and therefore can be used to optimize the PCR screening conditions.
Strain combination
Different strains of rats have different suitabilities as models for particular human diseases. For instance, the Long-Evans rat is a good model for diet-induced obesity, and the spontaneous hypertensive rat has been widely used for the study of hypertension. The availability of ES cells from different strains of rats provides investigators the option of producing gene-targeted rat models based on a relevant genetic background. However, for the production of gene-targeted rats by ES cell-based technologies, it is essential that rat ES cells are able to contribute to the formation of chimeras and re-enter the germline. Therefore, before performing gene-targeting experiments on a rat ES cell line, the investigator must determine whether the line is germline-competent and identify the ideal strain combination that leads to its efficient germline transmission.
In the production of ES cell-mouse chimeras, it has been shown that the genetic background of the host embryos is one factor that determines the efficiency with which the ES cell genome is transmitted to the progeny. One of the early findings during the development of gene targeting in 129 mouse ES cells was that the proportion of chimeras that resulted in germline transmission was determined by the genetic background of the host blastocyst. Schwartzberg et al.36 used out-bred MF-1, outbred CD-1 and inbred C57BL/6 mouse blastocysts to produce chimeras with CCE 129 mouse ES cells. All three blastocyst donor mouse strains produced ES cell-mouse chimeras. However, only C57BL/6 host blastocysts produced chimeras that transmitted the genetically modified ES cell genome through the germline. The same result has been observed for the mouse C57BL/6 ES cell lines. The efficiency of germline transmission is increased when the host blastocyst that is used to make ES cell chimeras with C57BL/6 ES cells is derived from C57BL/6-Tyrc-2J/J mice instead of BALB/c, SWR, FVB/N, C57BL/6-Tyrcbrd or 129/Sv mice37–41.
The strain combination is another controlling factor in the germ-line transmission of rat ES cells. Our results suggest that DA rat ES cells can transmit through the germline when they are injected into F344 rat blastocysts, but not when they are injected into Sprague-Dawley rat blastocysts4. ES cells derived from Sprague-Dawley and Wistar rats have also been shown to be germline-competent3,5,8. Another factor to be considered when choosing the strain combination is the coat color genetic background. The identification of chimerism and germline transmission can be facilitated by combining rat strains that have distinct coat colors (Fig. 5). Identification of the ideal strain combination will facilitate the broad application of ES cell–based technology for the production of gene-targeted rat models. In this protocol, we describe gene targeting performed in DA rat ES cells. We have deposited several germline-competent DA rat ES cell lines at the Rat Resource and Research Center in Columbia, Missouri, USA (http://www.rrrc.us/), which has begun distributing the rat ES cell lines to the research community.
Figure 5.

ES cell–rat chimera and its germline pups. The ES cell–rat chimera (right) was generated by injection of DA rat ES cells into a Fischer 344 blastocyst and subsequent transfer of the embryo to a recipient Sprague-Dawley rat. The agouti coat color denotes the presence of DA ES cell–derived cells in the albino Fischer 344 host. The germline transmission of the DA ES cell genome in the offspring can be easily identified by the appearance of agouti coat color when the DA ES cell–rat chimera is mated with albino Sprague-Dawley rats.
MATERIALS
REAGENTS
PCR primers and plasmids
Custom PCR primers for amplifying homology arms (see REAGENT SETUP)
Custom PCR primers for amplifying pUC cloning vector (see REAGENT SETUP)
Custom PCR primers for screening correctly targeted cells (see REAGENT SETUP)
pUC19 vector (from Zero Blunt TOPO PCR Cloning kit, Invitrogen, cat. no. K2830-20)
PGKneoF2L2DTA vector containing the PGK-DTA cassette (Addgene, cat. no. 13445)
pCAG-EGFP-IRES-Pac (provided by Austin Smith at University of Cambridge)
Targeting vector: prepared in advance as described in Box 1
QIAprep Spin Miniprep kit (Qiagen, cat. no. 27106)
QIAGEN Plasmid Maxi kit (Qiagen, cat. no. 12163)
QIAquick Gel Extraction kit (Qiagen, cat. no. 28706)
QIAquick PCR Purification kit (Qiagen, cat. no. 28104)
ZR-96 Quick-gDNA kit (Zymo Research, cat. no. D3010)
Paq5000 DNA polymerase (Agilent Technologies, cat. no. 600684)
Puregene Tissue Core kit A (Qiagen, cat. no. 158622)
Expand High Fidelity PCR System (Roche, cat. no. 11732650001)
DIG High Prime DNA Labeling and Detection Starter Kit (Roche, cat. no. 11745832910)
Infusion cloning kit (Clontech, cat. no. 639602)
Appropriate restriction enzyme for linearizing targeting vectors (New England Biolabs)
BOX 1. CONSTRUCTION OF RAT GENE-TARGETING VECTORS ● TIMING 30 D.
Extract genomic DNA from isogenic rat tissues or rat ES cells.
Set up and run two separate PCR reactions to amplify the 5′ and 3′ homology arms using the Expand High Fidelity PCR System.
Set up and run two separate PCR reactions to amplify pUC (one for cloning the 5′ homology arm and one for cloning the 3′ homology arm).
Separate the PCR products for the two homology arms (from Step 2) and the modified pUC cloning vectors (from Step 3) by electrophoresis on 1% (wt/vol) agarose gels. Purify the PCR products from the gel, using the QIAquick Gel Extraction Kit.
Set up two separate infusion cloning reactions between each of the two homology arms and the relevant modified pUC cloning vector, according to the manufacturer’s instructions. Digestion reaction with a restriction enzyme is not required in this step.
Prepare mini-preps of the recombinant pUC vectors containing the homology arm and confirm the sequence of the homology arms by DNA sequencing.
Digest the relevant plasmid for one homology arm with appropriate restriction enzymes and subclone the homology arm into the targeting vector backbone plasmid containing a CAG-driven positive selection cassette.
Prepare mini-preps of the modified targeting vector containing one homology arm. Repeat Step 7 to insert the other homology arm and the PGK-DTA negative selection cassette into the backbone plasmid (Fig. 2).
Prepare a maxi-prep of the final targeting vector. Prepare 100 μg of the linearized targeting vector by cutting with an appropriate restriction enzyme. This restriction enzyme should cut the targeting vector at a single site located between the upstream site of the 5′ homology arm and the downstream site of the PGK-DTA cassette.
Animals
Dark Agouti inbred male and female rats (DA/OlaHsd; Harlan Laboratories). They are used to obtain blastocysts for generating ES cells in this protocol.
Fischer 344 inbred male and female rats (F344/NHsd; Harlan Laboratories). They are used to obtain blastocysts for microinjection in this protocol.
Sprague-Dawley outbred vasectomized male rats (Hsd:Sprague-Dawley SD; Harlan Laboratories). The samples are prepared as described in Box 2.
Sprague-Dawley outbred pseudo-pregnant female rats (Hsd:Sprague-Dawley SD; Harlan Laboratories). The samples are prepared as described in Box 2.
Sprague-Dawley outbred female rats (Hsd:Sprague-Dawley SD; Harlan Laboratories). They are used for crossing to chimeric males in this protocol.
CF-1 mouse (Charles River Laboratory, Stain Code 023). It is used for obtaining MEFs.
DR4 mouse (Tg-DR4/1Jae/J; Jackson Laboratory, stock number 003208). It is used for preparing MEFs. ! CAUTION Animal experiments should be performed according to the investigator’s protocols approved by the international and institutional regulations.
BOX 2. PREPARATION OF VASECTOMIZED MALE RATS AND PSEUDO-PREGNANT FEMALE RATS ● TIMING 60 D.
Preparing vasectomized males
Restrain the male rat when it is ~5 weeks old and inject it with ketamine (80–100 mg kg−1) and xylazine (5–10 mg kg−1) intraperitoneally. We use Sprague-Dawley males.
After anesthesia, sanitize the shaved lower abdomen by swabbing with 70% (vol/vol) alcohol. Make a vertical incision on the testis membrane close to the left side of the midline wall.
Carefully push the testis to the left, using blunt forceps, and isolate the vas deferens from the surrounding tissue and cut off a part in the loop.
Clean the wound and suture the skin with stitches.
Repeat steps 3 and 4 on the other testis.
Keep the rat warm until it has fully recovered from the anesthesia. Place the rat back into a clean cage and house it as described in EQUIPMENT SETUP.
At 3–4 weeks after surgery, set up matings between the vasectomized male and two females to confirm the success of the vasectomy. We use Sprague-Dawley females. Pregnancy is normally apparent within 2 weeks; if pregnancy is not visible after this time, the male can be used for inducing pseudo-pregnant females.
Preparing pseudo-pregnant females
Set up matings between female Sprague-Dawley rats and vasectomized Sprague-Dawley males 4 d before the planned injection date.
Check for mating by the presence of copulation plugs the following morning.
Cage the plugged females individually for uterine transfer on the injection date.
Cell culture
β-mercaptoethanol (Sigma, cat. no. M7522; see REAGENT SETUP)
GMEM medium (Sigma, cat. no. G5154)
DMEM/F12 (Sigma, cat. no. D6421)
Neurobasal medium (Invitrogen, cat. no. 21103-049)
B-27 supplement (50×; Invitrogen, cat. no. 17504-044)
Heat-inactivated FBS (Hyclone)
L-glutamine (200 mM, Invitrogen, cat. no. 25030-081)
Gelatin (Sigma, cat. no. G1890; see REAGENT SETUP)
Penicillin-streptomycin (Invitrogen, cat. no. 15140-122)
Trypsin (2.5% (wt/vol), Invitrogen, cat. no. 15090-046; see REAGENT SETUP)
PBS (pH 7.2; Invitrogen, cat. no. 20012-050)
Non-essential amino acids solution (10 mM; Invitrogen, cat. no. 11140-050)
Sodium pyruvate solution (100 mM; Invitrogen, cat. no. 11360-070)
BSA fraction V (75 mg ml−1 in PBS; Invitrogen, cat. no. 15260-037)
EDTA (Invitrogen, cat. no. 15575-020)
DMSO (Sigma, cat. no. D2438) ! CAUTION It is flammable; harmful if swallowed; toxic when in contact with skin and eye; and use protective gloves and safety glasses when handling.
Chicken serum (Sigma, cat. no. C5405)
M2 medium (Sigma, cat. no. M7167)
M16 medium (Sigma, cat. no. M7292)
Insulin (Sigma, cat. no. I1882; see REAGENT SETUP)
Apo-transferrin (Sigma, cat. no. T1147; see REAGENT SETUP)
Progesterone (Sigma, cat. no. P8783; see REAGENT SETUP)
Putrescine (Sigma, cat. no. P5780; see REAGENT SETUP)
Sodium selenite (Sigma, cat. no. S5261; see REAGENT SETUP)
CHIR99021 (Axon Medchem BV, cat. no. Axon 1386; see REAGENT SETUP)
PD0325901 (Axon Medchem BV, cat. no. Axon 1408; see REAGENT SETUP)
Tyrode’s solution (Sigma, cat. no. T1788)
Colcemid solution (10 μg ml−1 in HBSS, Sigma, cat. no. D1925) ! CAUTION It is flammable; harmful if swallowed; toxic when in contact with skin and eye; and use protective gloves and safety glasses when handling.
MEF feeder cells (see REAGENT SETUP)
EQUIPMENT
Tissue culture dish (35 mm; Falcon, cat. no. 353001)
Tissue culture dish (60 mm; Falcon, cat. no. 353002)
Tissue culture dish (100 mm; Falcon, cat. no. 353003)
Tissue culture plate (6 well; Falcon, cat. no. 353046)
Tissue culture plate (24 well; Falcon, cat. no. 353047)
Tissue culture plate (96 well; Falcon, cat. no. 351172)
Conical tube (15 ml; Falcon, cat. no. 352196)
Conical tube (50 ml; Falcon, cat. no. 352070)
Plastic disposable pipette (1 ml; Falcon, cat. no. 357520)
Plastic disposable pipette (5 ml; Falcon, cat. no. 357543)
Plastic disposable pipette (10 ml; Falcon, cat. no. 357551)
Plastic disposable pipette (25 ml; Falcon, cat. no. 357525)
Center-well organ-culture dishes (Falcon, cat. no. 353653)
Eppendorf tubes (1.5 ml; Axygen, cat. no. MCT-150-C)
Glass pipettes (9 inch; VWR, cat. no. 14672-380)
Embryo-handling pipettes: pull a 9-inch glass pipette over a flame to create a narrow opening. Assemble the pipette with an aspirator mouthpiece and tubing as previously described28.
Humidified tissue culture incubator at 37 °C, 5% CO2 (NuAire AUTOFLOW 5510, NuAire)
Laminar flow tissue culture hood (SterilGARD e3, The Baker Company)
Inverted microscope (OLYMPUS CKX31, Olympus)
Dissecting microscope (OLYMPUS SZX10, Olympus)
Centrifuge (Centrifuge 5702, Eppendorf)
Gene Pulser XCell and Gene Pulser Cuvette (Bio-Rad)
Water bath (37 °C; Barnstead Lab-line, Barnstead)
Hemocytometer (Hausser Scientific)
Pipette pump (Pipet Aid)
Nunc CryoTube vial (1.0 ml; Nunc, cat. no. 375353)
NALGENE Cryofreezing container (NALGENE, cat. no. 5100-0001)
Liquid nitrogen tank
PCR thermocycler (Eppendorf Mastercycler, Eppendorf, cat. no. 5345)
HB-1000 hybridization oven (UVP laboratory product, cat. no. UVP-95-0030-01)
UV crosslinker (Stratalinker, Stratagene, Model 2400)
Clone Manager Basic 9 (Sci-Ed Software)
PowerPac Basic Power Supply (Bio-Rad, cat. no. 164-5050)
Surgical blades, forceps and scissors for dissection ! CAUTION Sterilize by autoclave.
Serrefine clamp (Biomedical Research Instruments, cat. no. 34-2800)
REAGENT SETUP
β-mercaptoethanol
Prepare 0.1 M stock solution by diluting 100-μl β-mercaptoethanol with 14.1 ml of distilled H2O. Sterilize through a 0.2-μ filter and store at 4 °C for up to 1 month. ! CAUTION It is flammable; harmful if swallowed; toxic when in contact with skin and eye; and use protective gloves and safety glasses when handling.
Gelatin 10× stock solution
Dissolve 5 g in 500 ml of distilled H2O to produce 1% (wt/vol) gelatin stock. Autoclave and store in 50-ml aliquots at 4 °C for up to 3 months.
Gelatin 1× solution
Add 50 ml of prewarmed 1% (wt/vol) gelatin to 450 ml of PBS. Store at 4 °C for up to 1 month.
Insulin
Dissolve in sterile 0.01 M HCl overnight at 4 °C to produce a 10 mg ml−1 stock solution. Store in 1-ml aliquots at −20 °C. ▴ CRITICAL Insulin does not dissolve readily; hence, ensure that the suspension is mixed well before aliquotting.
Apo-transferrin
Dissolve in sterile distilled H2O to produce a 100 mg ml−1 stock solution. Store in 1-ml aliquots at −20 °C.
Progesterone
Dissolve 3 mg in 5-ml ethanol to produce a 0.6 mg ml−1 stock solution. Sterilize through a 0.2-μm filter. Store in 0.5-ml aliquots at −20 °C. It is good for at least 5 years. Freezing and thawing is fine.
Putrescine
Dissolve 1.6 g in 10-ml distilled H2O to produce a 160 mg ml−1 stock. Sterilize through a 0.2-μm filter. Store in 1-ml aliquots at −20 °C. It is good for at least 5 years. Freezing and thawing is fine.
Sodium selenite
Dissolve 2.59 mg in 5-ml distilled H2O to produce a 3 mM stock. Sterilize through a 0.2-μm filter. Store in 0.5-ml aliquots at −20 °C. It is good for at least 5 years. Freezing and thawing is fine.
CHIR99021
Dissolve 4 mg of CHIR99021 in 860-μl DMSO to produce a 10 mM stock. Store in 100-μl aliquots at −20 °C. It can be stored for at least 3 years.
PD0325901
Dissolve 4 mg of PD0325901 in 830-μl DMSO to produce a 10 mM stock. Store in 50-μl aliquots at −20 °C. It can be stored for at least 3 years.
N2 100× stock (10 ml)
To 7.187-ml DMEM/F12 medium, add 0.67 ml of 75 mg ml−1 BSA, 33 μl of 0.6 mg ml−1 progesterone solution, 100 μl of 160 mg ml−1 putrescine solution, 10 μl of 3 mM sodium selenite solution, 1 ml of 100 mg ml−1 apo-transferrin and 1 ml of 10 mg ml−1 insulin. Mix well by pipetting and store in 1 ml aliquots at −20 °C. It can be stored for up to 6 months.
DMEM/F12-N2 medium
To 100 ml of DMEM/F12, add 1 ml of N2 100× stock solution. The final concentration of each component of N2 in the DMEM/F12-N2 medium is as follows: insulin 10 μg ml−1, transferrin 100 μg ml−1, progesterone 20 ng ml−1, putrescine 16 μg ml−1, sodium selenite 30 nM and BSA 50 μg ml−1.
Neurobasal/B27 medium
To 100 ml of neurobasal medium, add 2 ml of B27 and 0.5 ml of 200 mM L-glutamine.
N2B27 medium
Mix DMEM/F12-N2 medium with neurobasal/B27 medium at a ratio of 1:1. To 200 ml of N2B27 medium, add 200 μl of 0.1 M β-mercaptoethanol. The final concentration of β-mercaptoethanol in N2B27 medium is 0.1 mM. Store at 4 °C for up to 1 month.
2i medium
To 100 ml of N2B27 medium, add 30 μl of 10 mM CHIR99021 and 10 μl of 10 mM PD0325901. The final concentrations of CHIR99021 and PD0325901 in the 2i medium are 3 and 1 μM, respectively. Store at 4 °C for up to 1 month.
Trypsin-EDTA solution (0.025% (wt/vol))
Add 5 ml of 2.5% (wt/vol) trypsin, 5 ml of chicken serum and 0.5 ml of 0.5 M EDTA to 500 ml of sterile PBS. Mix well and store in 30 ml aliquots at −20 °C. It can be stored for up to 6 months.
MEF medium
Add 50 ml of heat-inactivated fetal bovine serum, 5 ml of 200 mM L-glutamine solution and 5 ml of penicillin-streptomycin solution to 500 ml of GMEM medium. Store at 4 °C for up to 1 month.
MEF feeders
We use standard methods to prepare MEF feeders28. Mitotically inactivate MEFs either by γ-irradiation or mitomycin-C treatment. Plate mitotically inactivated MEFs into gelatin-coated dishes at a density of 2 × 104 to 3 × 104 cells per cm2 and culture in MEF medium. Prepare MEF feeder-coated plates in advance and use within 1 week. MEFs derived from the CF1 mouse strain are used for routine rat ES cell culture. MEFs prepared from other strains of mice should also work for rat ES cell culture. When rat ES cells are under drug selection, they are cultured on drug-resistant MEFs, such as DR4 MEFs, which are resistant to G418, hygromycin, puromycin and 6-thioguanine10.
Freezing medium
Dissolve 10% (vol/vol) DMSO in MEF medium. Prepare just before use.
Primer design for amplifying homology arms
Access the University of California Santa Cruz (UCSC) genome browser website and find the genomic DNA sequence of the gene of interest for performing gene targeting. Retrieve the whole gene sequence and ~15 kb of upstream and ~15 kb of downstream sequences. Import the DNA sequence into sequence analysis software, such as Clone Manager (Scientific & Educational Software). Decide on the location and size of short and long homology arms based on the structure and function of the gene, information of exons and introns, percentages of repetitive sequence and the restriction enzyme sites. The minimal length of the homology arms required for successful gene targeting in rat ES cells has not been determined yet. From our experience, ~6 kb of the long homology arm and ~1.5 kb of the short homology arm are sufficient for gene targeting by homologous recombination in rat ES cells. Design a PCR primer pair to individually amplify the 5′ and 3′ homology arms from genomic DNA extracted from isogenic rat tissues or rat ES cells. The minimum requirements for the PCR primers are described as follows: length, 23–30 bp; annealing temperature, 60–68 °C; and G + C content, 40–60%.
Primer design for amplifying pUC vector
Each primer in this pair contains a 15-bp sequence complementary to the 5′ end of the primer used for amplifying either the 5′ or the 3′ homology arm from genomic DNA. A restriction enzyme site is also introduced into each primer to facilitate the subcloning of the homology arm into the targeting vector backbone plasmid (Box 1 and Fig. 2).
Primer design for PCR screening
Design a PCR primer pair for the initial screening of correctly targeted rat ES cells. As shown in Figure 4, one of the PCR primer pair should be located on the genomic DNA beyond the 3′ short arm and the other located on the drug resistance cassette. The minimum requirements for the PCR primers are described as follows: length, 20–25 bp; annealing temperature, 58–65 °C; and G + C content, 40–60%.
EQUIPMENT SETUP
Animal housing
Mice and rats are housed in the vivarium and maintained under routine husbandry practices, according to the guidelines approved by the Institutional Animal Care and Use Committee. Rats undergoing surgical procedures are housed solitarily for the first 1–2 weeks after surgery. Rat pups are weaned at the age of 4–5 weeks.
PROCEDURE
Derivation and expansion of ES cells from rat blastocysts ● TIMING 30 d
-
1|
Set up matings of rats and examine the plug the following morning. The morning when the plug is observed is defined as 0.5-d postcoitum (d.p.c.). Kill the pregnant rat at 4.5 d.p.c by CO2 asphyxiation, followed by thoracotomy. Timed-pregnant rats can also be ordered from Harlan Laboratories or Charles River Laboratories International. We use DA rats for preparing ES cells.
! CAUTION Animal experiments should be performed according to the investigator’s protocols approved by international and institutional regulations.
-
2|
Perform surgery to isolate the uterus and flush out the blastocysts using N2B27 medium. The procedures are the same as those described in the mouse28,42. First, lay the rat on its back and cleanse the abdominal skin and fur with 70% (vol/vol) alcohol. Expose the uterus by making an incision along the midsection with scissors. Cut out the uterus and transfer it to a 6-cm dish prefilled with 3 ml of PBS. Remove the mesometrium of the uterus with fine scissors. Finally, flush the blastocysts from the uterus with a 27-gauge needle using N2B27 medium.
-
3|
Collect blastocysts using an embryo-handling pipette and wash them using two drops of acidic Tyrode’s solution.
-
4|
Transfer blastocysts to a fresh drop of acidic Tyrode’s solution and monitor them under a microscope; when the zona pellucida has dissolved, immediately wash blastocysts with two drops of fresh N2B27 medium.
-
5|
Replace the MEF medium with 500 μl of 2i medium in each well of the four-well plate preseeded with MEF feeders. MEF feeders need to be prepared in advance (see REAGENT SETUP). Transfer up to six blastocysts to each well, and culture at 37 °C with 5% CO2 for 4–5 d. It is not necessary to change the medium during this period.
-
6|
Detach and transfer the outgrowth of each blastocyst into a 1.5-ml Eppendorf tube containing 200 μl of 0.025% (wt/vol) trypsin-EDTA solution, using a mouth-controlled glass pipette. Incubate at 37 °C for 3 min.
▴ CRITICAL STEP Use a glass pipette, the diameter of which at the tip is just a little bigger than the outgrowth. Each outgrowth should be handled separately.
-
7|
Add 1 ml of MEF medium to the tube and gently pipette up and down four to six times to break up the outgrowth.
-
8|
Centrifuge at 200g for 3 min. Aspirate the supernatant and resuspend the pellet in 500 μl of N2B27 medium. Centrifuge again at 200g for 3 min and aspirate the supernatant. Resuspend the pellet in 500 μl of 2i medium and transfer the cells to a new four-well plate preseeded with MEF feeders (see REAGENT SETUP).
▴ CRITICAL STEP Rat ES cells maintained in serum-free 2i medium are very sensitive to even a small amount of residual serum or trypsin. Take extra care to remove any residual serum and trypsin, especially when using the conventional method to disaggregate the outgrowth42.
-
9|
Incubate at 37 °C in a humidified 5% CO2 incubator. Add 250 μl of fresh 2i medium to each well on day 2. ES cell colonies emerge after 3–5 d of incubation. It is not necessary to change the medium during this period.
? TROUBLESHOOTING
-
10|
Approximately 3–5 d after the first disaggregation, check each well of the four-well plate under a microscope for the appearance of ES cell colonies.
-
11|
Detach ES cell colonies by pipetting up and down several times with a 1-ml pipette tip; the contents of each well should be handled separately. Rat ES cell colonies cultured in the 2i medium attach loosely to the feeders and are easily detached by pipetting.
-
12|
Transfer the colonies to a 1.5-ml Eppendorf tube and centrifuge at 200g for 3 min. Carefully aspirate off the supernatant and resuspend the pellet in 200 μl of 0.025% (wt/vol) trypsin-EDTA solution. Incubate at 37 °C for 3 min.
-
13|
Add 1 ml of MEF medium to the tube and pipette up and down four to six times with a 1-ml pipette tip to dissociate the colonies into single cells. Centrifuge at 200g for 3 min and carefully aspirate off the supernatant.
-
14|
Dissociate the cell pellet with 500 μl of 2i medium and transfer the cells to a new four-well plate preseeded with MEF feeders (see REAGENT SETUP). Incubate at 37 °C in a humidified 5% CO2 incubator for 2–3 d (passage 1).
-
15|
Passage the cells every 2–3 d, as described in Steps 11–14. Gradually increase the size of the well, according to the number of cells that are present. It is not necessary to change the medium during the 2- to 3-d period between passaging. However, if the medium turns yellow, add some fresh 2i medium to the well. For example, if there is 1 ml of 2i medium in the well, add 0.5 ml of fresh 2i medium. When the total cell number of an individual rat ES cell clone is more than 0.5 million, the cells can be passaged or frozen, as described in Box 3, or used directly in Step 16.
BOX 3. ROUTINE PASSAGING AND FREEZING OF RAT ES CELLS.
Routine passaging of rat ES cells. ● TIMING 1 h
The passaging method is, in principle, the same as described in Steps 11–15 of the main procedure, only on a larger scale.
-
Use a 5- or 10-ml serological pipette to detach the rat cells by pipetting. Transfer the rat ES cells to a 15-ml conical tube and centrifuge at 200g for 3 min.
▴ CRITICAL STEP Do not aspirate off the medium directly from the culture dish. Good rat ES cells tend to grow as floating colonies or loosely attach to the feeders in the 2i culture condition.
Carefully aspirate off the supernatant, and resuspend the cell pellet in 0.5–1 ml of 0.025% (wt/vol) trypsin-EDTA solution. Incubate at 37 °C for 3 min.
Add 5 ml of MEF medium to neutralize the trypsin. Pipette up and down four to six times with a 5- or 10-ml serological pipette to dissociate the cell pellet into single cells. Centrifuge at 200g for 3 min and carefully aspirate off the supernatant.
Resuspend the cell pellet with 2i medium and plate the cells onto a six-well plate or a 10-cm culture dish preseeded with MEF feeders (see REAGENT SETUP). Incubate at 37 °C in a humidified 5% CO2 incubator.
Passage the cells every 2–3 d.
Freezing rat ES cells. ● TIMING 1 h
Collect and dissociate rat ES cells as described in the above steps 1–3.
Resuspend the cell pellet in the freshly prepared freezing medium at a concentration of 0.5–1.5 × 106 cells per ml.
Aliquot 0.5 ml of the cells to each 1.0-ml cryotube vial. If the total cell number is <5 × 105, resuspend all the cells in 200- to 500-μl of freezing medium and transfer them to one 1.0-ml cryotube vial. Rat ES cells recover well from freezing and thawing.
Transfer the vial to a NALGENE Cryofreezing container and store in a −80 °C freezer overnight.
Transfer the vial to a liquid nitrogen tank for long-term storage.
Thawing rat ES cells ● TIMING 30 min
Remove a vial of rat ES cells from the liquid nitrogen tank and thaw the vial rapidly in a 37 °C water bath.
Transfer the cells into a sterile 15-ml conical tube with 10 ml of MEF or N2B27 medium.
Centrifuge at 200g for 3 min.
Aspirate off the supernatant and resuspend the cell pellet in 2i medium.
Transfer the cells into a culture dish preseeded with feeders and culture at 37 °C in a humidified 5% CO2 incubator.
Introduction of targeting vector into rat ES cells by electroporation ● TIMING 4 h
-
16|
Grow and collect the rat ES cells as described in Steps 11–15 and in Box 3. A total of 0.5–1 × 107 rat ES cells are usually required for each transfection.
▴ CRITICAL STEP Use the rat ES cells that have previously been proven to be germline competent.
-
17|
Wash the rat ES cells twice with 10 ml of PBS. Collect the cells by centrifugation at 200g for 3 min. Resuspend the cell pellet in 0.7 ml of PBS.
-
18|
Dissolve 50–100 μg of linearized gene-targeting vector (from step 9 in Box 1) in 0.1 ml of PBS. Add the linearized plasmid DNA to the cell suspension and mix by gently inverting the tube.
-
19|
Transfer DNA/rat ES cell mixture to a Gene Pulser cuvette (4-mm gap). Perform electroporation at a pulse voltage of 200 V, 500 μF on Bio-Rad Gene Pulser XCell.
-
20|
Transfer the electroporated rat ES cells to three 10-cm tissue culture dishes preseeded with MEFs (see REAGENT SETUP) and culture in 2i medium at 37 °C in a humidified 5% CO2 incubator.
▴ CRITICAL STEP The MEFs should be resistant to the drug used to select transfected rat ES cells. We use DR4 MEFs for the culture of rat ES cells that are under selection with puromycin, G418 or hygromycin.
? TROUBLESHOOTING
Selection of drug-resistant colonies ● TIMING 9 d
-
21|
One day after electroporation, replace the medium with fresh 2i medium supplemented with the appropriate selection drug (e.g., 0.5 μg ml−1 of puromycin).
▴ CRITICAL STEP Rat ES cells loosely attach to feeders or grow as floating colonies; hence, do not aspirate off the medium directly from the culture dish. Instead, transfer the medium from the 10-cm culture dish to a sterile 15-ml conical tube and immediately add fresh 2i medium with the selection drug back into the culture dish. Collect rat ES cells in the culture medium by centrifugation and add them back into the culture.
-
22|
After incubation in the presence of selection drug for 2 d, replace the medium with fresh 2i medium and culture the cells in the absence of selection drug for 1 d. This represents one selection cycle. Repeat this selection cycle two more times.
▴ CRITICAL STEP Rat ES cells are very sensitive to drug selection. Do not add the drug continuously, as doing so will kill rat ES cells even if they are transfected. It is important to determine the killing curve of each drug for the rat ES cell line used for gene transfection. This will help to optimize the concentration for drug selection.
? TROUBLESHOOTING
Picking drug-resistant colonies ● TIMING 4 h
-
23|
Gently detach rat ES cell colonies from feeders by pipetting. Transfer the medium containing ES colonies into a sterile 15-ml conical tube.
-
24|
Spin down to pool the colonies at 100g for 3 min. Aspirate off the supernatant and resuspend the colonies in 1 ml of 2i medium. Transfer the colonies to a center-well organ-culture dish or a 6-cm Petri dish.
-
25|
Dispense drops of 0.025% (wt/vol) trypsin-EDTA solution (10 μl per drop) to a 150-mm Petri dish using a 10- to 100-μl multichannel pipette.
-
26|
Transfer the rat ES colonies into drops of trypsin/EDTA solution (one colony per drop), using a mouth-controlled glass pipette, as illustrated in Figure 3. Incubate at room temperature (25 °C) for 3 min.
-
27|
Transfer the trypsin/EDTA-treated ES colonies into a 96-well plate precoated with MEF feeders (see REAGENT SETUP). Each well contains 200 μl of 2i medium supplemented with 1% (vol/vol) FBS. Addition of FBS to the 2i medium is necessary to neutralize the trypsin.
-
28|
Pipette up and down several times to dissociate the rat ES cell colonies into single cells. Transfer 50 μl of the cell suspension from each well into a duplicate 96-well plate preseeded with MEF feeders. Add another 50 μl of fresh 2i medium to each well. Culture at 37 °C in a humidified 5% CO2 incubator.
▴ CRITICAL STEP This duplicate plate with fewer cells is used to retrieve positive clones at Step 35 after PCR screening (Steps 31–34).
-
29|
Repeat Steps 23–28 until a sufficient number of colonies is processed. The next morning, carefully aspirate off the medium and add 100 μl of fresh 2i medium to each well, using a multichannel pipette.
▴ CRITICAL STEP Rat ES cells are very sensitive to serum and trypsin; hence, it is critical to replace the serum and trypsin–containing medium with fresh 2i medium within 24 h of plating.
-
30|
Add 50–100 μl of fresh 2i medium to each well every 2–3 d. After 6–8 d of plating, ES cell colonies will usually appear in most of the wells.
Screening clones by PCR ● TIMING 2 d
-
31|
Aspirate off the medium from each well of the 96-well plate used for PCR screening (Step 30). Add 100 μl of the genomic lysis buffer (provided in the ZR-96 Quick-gDNA kit) to each well and extract genomic DNA from each rat ES cell clone, according to the manufacturer’s instructions; elute DNA in 30 μl of distilled water.
-
32|
Set up a PCR mix as tabulated below.
▴ CRITICAL STEP The optimum PCR screening conditions likely vary among different gene-targeting vectors; therefore, it is important to optimize the PCR screening condition for each targeting vector before performing gene-targeting experiments. To optimize the PCR conditions, first construct a positive control vector as shown in Figure 4. Next, generate and expand rat ES cells with the positive control vector integrated stably. Finally, use genomic DNA extracted from these rat ES cells to optimize the PCR conditions. This genomic DNA can also be run with PCR screening primers as a positive control.
Reagent Volume per 50 μl reaction (μl) Final Paq5000 Reaction buffer (10×) 5 1× dNTP mix (100 mM; 25 mM each dNTP) 0.4 0.8 mM dNTP Forward primer (10 μM) 1 0.2 μM Reverse primer (10 μM) 1 0.2 μM Genomic DNA 100 ng 100 ng Paq5000 DNA Polymerase (5 U μl−1) 0.5 0.05 U μl−1 ddH2O Up to final 50 μl -
33|
Amplify DNA, using the following cycling conditions.
Cycle Denature Anneal Extend Hold 1 95 °C, 2 min — — — 2–41 95 °C, 20 s 60 °C, 20 s 72 °C, 1 min — 42 72 °C, 5 min — 43 4 °C -
34|
Separate the PCR product by electrophoresis on a 1% (wt/vol) agarose gel and analyze the results. A PCR product with the expected size should be amplified from gene-targeted rat ES cell clones.
Confirmation of gene targeting by Southern blot analysis ● TIMING 15 d
-
35|
Expand positive rat ES cell clones identified in Step 34 from the duplicated 96-well plate, as described in Steps 11–15. Next, freeze a portion of the cells as described in Box 3.
-
36|
Extract genomic DNA from each clone, e.g., using Puregene Tissue Core Kit A, according to the manufacturer’s instructions.
-
37|
Digest genomic DNA samples with appropriate restriction enzymes and separate on 1% (wt/vol) agarose gels.
-
38|
Denature, neutralize and blot the DNA on a membrane, using standard methods35.
▴ CRITICAL STEP Southern blot strategies for the verification of gene-targeting events in rat ES cells are essentially the same, as described in mouse ES cells29,43. For the confirmation of accurate gene targeting, genomic DNA samples should be analyzed by Southern blot, using 5′, 3′ and internal probes.
-
39|
Synthesize the digoxigenin-labeled hybridization probes by PCR, e.g., using the Expand High Fidelity PCR System. Set up reactions as described in the table below.
Reagent Volume per 100 μl reaction (μl) Final Expand PCR buffer (10×) 10 1× MgCl2 (25 mM) 2 0.5 mM dATP (10 mM) 2 0.2 mM dGTP (10 mM) 2 0.2 mM dCTP (10 mM) 2 0.2 mM dTTP (10 mM) 1.7 0.17 mM Dig-dUTP (10 mM) 3 0.3 mM Forward primer (10 μM) 4 0.4 μM Reverse primer (10 μM) 4 0.4 μM DNA template 20 ng 20 ng High-fidelity enzyme (5 U μl−1) 1.5 0.075 U μl−1 ddH2O Up to final 100 μl -
40|
Run the PCR, using the following conditions.
Cycle Denature Anneal Extend Hold 1 95 °C, 2 min — — — 2–41 95 °C, 30 s 60 °C, 30 s 72 °C, 45 s — 42 72 °C, 7 min — 43 4 °C -
41|
Separate the PCR product by electrophoresis on a 1% (wt/vol) agarose gel. Purify the fragment using the QIAquick Gel Extraction Kit (Qiagen) and dissolve it in 20 μl of distilled water.
-
42|
Fix the DNA membrane and hybridize with the probe. Apply anti-digoxigenin-AP (from DIG High Prime DNA Labeling and Detection Starter Kit) to develop a film, according to the manufacturer’s instructions. A typical example of analysis by Southern blot and the outcome are shown in Figure 6a,b.
-
43|
Identify positive clones and use these for producing rat chimeras (Step 44). Alternatively, carry out the subcloning and karyotyping procedure described in Box 4 before proceeding to Step 44; this may improve germline competency.
Figure 6.

Confirmation of p53 gene targeting in rat ES cells and rats by Southern blot and genotyping. (a) Schematic diagram showing the structures of the wild-type (WT) rat p53 allele, the rat p53 gene-targeting vector and the predicted rat p53 gene-targeted allele. H, HindIII; S, SpeI; black squares indicate exons. (b) Southern blot analysis of WT (DAc8) and p53 gene-targeted (DAc8-p53) rat ES cells. For Southern blot analysis with 5′ or internal probes, genomic DNA from rat ES cells was digested with SpeI. For Southern blot analysis using 3′ probe, rat ES-cell genomic DNA was digested with HindIII. (c) Diagrams showing the strategy for identifying p53 gene-targeted rats by PCR genotyping. F1, F2 and R are PCR primers designed to amplify fragments of different sizes from genomic DNA extracted from WT and p53 gene-targeted rats. (d) Identification of the p53 gene-targeted allele by PCR genotyping. M, 100-bp DNA ladder; (1) DAc8 rat ES cells; (2) DAc8-p53 rat ES cells; (3) WT rat; and (4) p53 gene-targeted heterozygote rat. The expected sizes of PCR products for WT and p53 gene-targeted alleles are 309 and 498 bp, respectively. (e) Southern blot analysis, using 5′ probe as shown in a. (1) p53 gene-targeted heterozygote rat and (2) WT rat. The figures are reproduced from the original p53 gene-targeting rat paper10 with permission from Nature.
BOX 4. REFINEMENT OF TARGETED CLONES: SUBCLONING AND KARYOTYPING (OPTIONAL) ● TIMING 30 D.
Low-efficiency contribution of rat ES cells to the germline is often caused by chromosomal abnormalities in the contributing ES cells. We have frequently observed a high percentage of cells with an abnormal karyotype in gene-targeted rat ES cell clones, despite their parental cell lines having normal karyotypes. Rat ES cells with a normal karyotype can be isolated from these clones by subcloning. This subcloning step may improve the germline competency of gene-targeted rat ES cells.
Plate 1 × 103 rat ES cells onto a 10-cm culture dish preseeded with MEF feeders (see REAGENT SETUP) and culture in 2i medium.
Replace the medium every other day as described in Step 21 of the main protocol.
Eight days after plating, isolate colonies with compact dome-shaped morphology under a dissecting microscope. Use a 20-μl pipette to transfer each colony to one drop (10 μl) of 0.025% (wt/vol) trypsin-EDTA solution and incubate at room temperature for 3 min. Normal rat ES cells form compact dome-shaped colonies under 2i condition, as shown in Figure 7a.
Pipette up and down to dissociate the colonies into single cells; transfer them into a 96-well plate (one colony per well) preseeded with MEF feeders and culture in 2i medium supplemented with 1% (vol/vol) FBS.
Replace the medium with fresh 2i medium the next day.
Expand and freeze the cells when appropriate, as described in Steps 11–15 and Box 3.
For karyotyping, plate 1 × 106 cells per well into a six-well plate preseeded with MEF feeders and culture in 2i medium. On the next day, add colcemid solution with a final concentration of 100 ng ml−1 to the culture and incubate for 2 h at 37 °C in a humidified 5% CO2 incubator.
Perform karyotyping using standard methods28. Use karyotypically normal rat ES cell subclones for subsequent experiments. In our experience, a rat ES cell population in which >80% of the cells have a normal karyotype is suitable for proceeding to the next step, that is, production of chimeras by blastocyst injection (Step 44 in the main procedure).
Preparation of rat ES cells for injection ● TIMING 1 h
-
44|
One week before the scheduled day of microinjection, thaw rat ES cells (from Step 35 or step 8 in Box 4) stored in liquid nitrogen and expand them as described in Steps 11–15 and Box 3. Male DA rat ES cell lines (as determined by karyotyping or PCR) are used for the production of ES cell–rat chimeras.
-
45|
One day before microinjection, split the cells to ensure 50–70% confluence by the next day.
-
46|
On the day of injection, collect the cells as described in Steps 11–15 and Box 3 and resuspend them in 0.5–1.0 ml of N2B27 medium.
▴ CRITICAL STEP Ensure that the cell aggregates are dissociated into single cells.
-
47|
Transfer the cell suspension into a sterile 1.5-ml Eppendorf tube and place it on ice while preparing for microinjection.
Preparation of blastocysts ● TIMING 4 h
-
48|
Collect blastocysts from timed-pregnant F344 rats at 4.5 d.p.c., as described in Steps 1–2.
-
49|
Wash the blastocysts with several drops of M2 medium to rinse off the debris.
-
50|
Transfer the blastocysts to a culture dish containing a few drops of M16 medium.
-
51|
Incubate for 1–4 h at 37 °C in a humidified 5% CO2 incubator to allow the expansion of the blastocyst cavity.
Microinjection of rat ES cells ● TIMING 2 h
-
52|
Assemble the microinjection setup. We use a standard setup as previously described28.
-
53|
Pick up well-expanded blastocysts and immobilize them with a holding pipette.
-
54|
Inject 12–15 ES cells into the blastocyst cavity.
-
55|
Place the injected blastocysts back into the incubator until all blastocysts are injected and ready for transfer to pseudo-pregnant surrogate mothers.
Transfer of blastocysts to recipients ● TIMING 1 h
-
56|
Anesthetize the pseudo-pregnant Sprague-Dawley rat (see Box 2) at 3.5 d.p.c. by injection of ketamine (80–100 mg kg−1) and xylazine (5–10 mg kg−1) intraperitoneally.
-
57|
Wipe the back of the rat with 70% (vol/vol) ethanol.
-
58|
Make an incision in the skin right above the ovary, as well as in the peritoneal wall over the left oviduct, using fine dissection scissors.
-
59|
Locate the fat pad attached to the ovary, pull it out and secure it with a Serrafine clamp to exteriorize the ovary, the oviduct and the upper part of the uterus.
-
60|
Hold the top of the uterus gently with blunt forceps. Using a 27-gauge needle, make a hole in the uterus a few millimeters down from the uterotubal junction.
-
61|
Transfer 8–10 injected blastocysts (from Step 55) into the uterus, using the glass capillary, through the hole made by the needle.
-
62|
Place the uterus, oviduct and ovary back inside the body cavity. Sew up the muscle wall and the skin.
-
63|
At the end of the procedure, place the rat in a clean cage and maintain it warm until it recovers from anesthesia. Cage the rat by itself (see EQUIPMENT SETUP). The pups will be born 18–19 d after the transfer of blastocysts into the uterus of the surrogate female rat.
▴ CRITICAL STEP Maintain the rat warm during and after the surgery. Wait for the rat to fully recover from anesthesia before returning it to the vivarium.
Production of heterozygote mutant rats ● TIMING 90 d
-
64|
Breed male rats exhibiting chimerism (produced in Step 63) with Sprague-Dawley female rats to test germline transmission of the ES cell genome. DA rat ES cells have an agouti (A/A) coat color genetic background, which is dominant to albino (c/c) of the F344 and Sprague-Dawley rats. The genetically determined coat color distinctions provide a convenient indicator of chimerism and germline transmission of the DA rat ES cell genome.
? TROUBLESHOOTING
-
65|
Identify heterozygote rats by genotyping and Southern blot analysis on tail biopsies using standard methods28,43. A typical example of analysis by genotyping and Southern blot and the outcome are shown in Figure 6c–e.
Production of homozygote knockout rats ● TIMING 90 d
-
66|
Intercross male and female heterozygote rats identified in Step 65 to generate homozygote mutant rats.
-
67|
Identify homozygote mutant rats by genotyping tail biopsies using standard methods28.
-
68|
Confirm the homozygosity of the rats by northern blot and western blot analyses, using standard methods35.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Steps | Problem | Possible reason | Solution |
|---|---|---|---|
| 9 | No colonies or typical rat ES cells appear | Bad batch of N2B27 medium | Always test each batch of N2B27 medium before use in critical experiments. Try adding insulin directly to the final DMEM/F12-N2 medium instead of adding it to the N2 100× stock. Both N2 supplement and ready-to-use N2B27 medium are commercially available. But we found that they are usually inferior to in-house made N2 or N2B27 medium |
| Residual serum or trypsin in the culture | Take extra care to remove all the trypsin and serum after trypsinization | ||
| Poor quality of feeders28 | Use freshly prepared MEF feeders. Use MEFs within six passages | ||
| Nonpermissive strains of rats | Isolate ICM by immunosurgery | ||
| 20 | Low transfection efficiency | Linearized DNA is not well dissolved | Dissolve plasmid in PBS for up to 6 h; tap tube occasionally to ensure dissolution |
| Cell death | Use good batch of N2B27 medium. Immediately seed electroporated ES cells onto dishes with prewarmed medium | ||
| 22 | No or few positive colonies appear | Drug concentration is too high | Determine the killing curve of the drug and optimize selection scheme |
| Feeder cell death under drug selection | Use drug-resistant feeders, such as DR-4 feeders, which are resistant to G418, puromycin, hygromycin and 6-thioguanine. Add extra feeder cells after each cycle of drug selection | ||
| 64 | Failure of germline transmission of the gene-targeted allele | The cell line used for injection is not a pure gene-targeted clone | Take extra care to avoid cross-contamination when picking up colonies. Isolate pure gene-targeted rat ES cells through subcloning |
| The quality of rat ES cells is poor | Use early passage- and germline-competent rat ES cells for gene-targeting experiments. Karyotype gene-targeted rat ES cell clones and select the ones with the highest proportion of normal-karyotype cells for blastocyst injection. Isolate karyotypically normal rat ES cells from gene-targeted clones through subcloning | ||
| Cell culture contamination | Test for mycoplasma contamination. If contaminated, discard the cells and use a new batch of cells. Rat ES cells that have previously been contaminated with bacteria or other microorganisms should not be used for gene-targeting experiments. We advise against the use of penicillin-streptomycin or any other antibiotics in routine rat ES cell culture, as the use of antibiotics prevents the immediate detection of cell culture contamination |
● TIMING
Steps 1–15, Derivation and expansion of rat ES cells: 30 d
Steps 16–43, Generation of gene-targeted rat ES cells: 65 d
Steps 44–68, Production of gene knockout rats: 8 months
Box 1, Construction of rat gene-targeting vectors: 30 d
Box 2, Preparation of vasectomized male rats and pseudo-pregnant female rats: 60 d
Box 3, Routine passaging and freezing of rat ES cells: 1 h
Box 3, Freezing rat ES cells: 1 h
Box 3, Thawing rat ES cells: 30 min
Box 4, Refinement of targeted clones: subcloning and karyotyping (optional): 30 d
ANTICIPATED RESULTS
Rat ES cell lines can be established from ~50% of the rat blastocysts that are plated under 2i culture conditions. Similar to mouse ES cells, rat ES cells are routinely passaged using trypsin and can be frozen and thawed using conventional methods with typical recovery rates of over 90%. Rat ES cells grow as compact dome-shaped colonies (Fig. 7a) and express pluripotency markers (Fig. 7b–e). Gene-targeted rat ES cells can be routinely generated by homologous recombination. Gene-targeting efficiency in rat ES cells is similar to that in mouse ES cells. Rat ES cells are very sensitive to drug selection and they cannot survive if the selection drug is added continuously, even if they are transfected. To overcome this problem, we add selection drugs at half the concentration used for mouse ES cells and apply a ‘pulsed’ drug selection regimen, as described in the procedure. The majority of the colonies that emerge after applying the ‘pulsed’ drug selection regimen are transfected (Fig. 8). On average, 300–500 drug-resistant colonies can be recovered from 7 × 106 rat ES cells after electroporation with targeting vectors.
Figure 7.
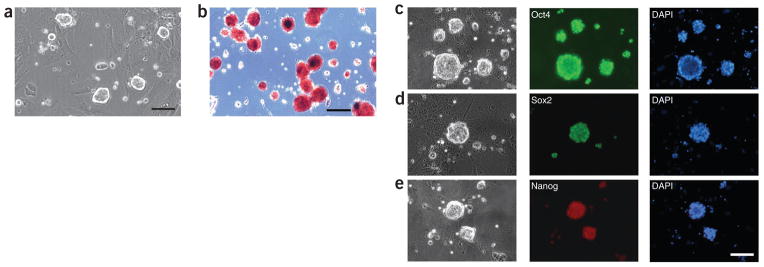
Rat ES cells derived and maintained in the 2i condition. (a) DA rat ES cells cultured on feeders in 2i medium. Scale bar, 50 μm. (b) DA rat ES cells stained with alkaline phosphatase. Scale bar, 50 μm. (c–e) DA rat ES cells stained for pluripotency markers Oct4 (c), Sox2 (d) and Nanog (e). Scale bar, 50 μm.
Figure 8.
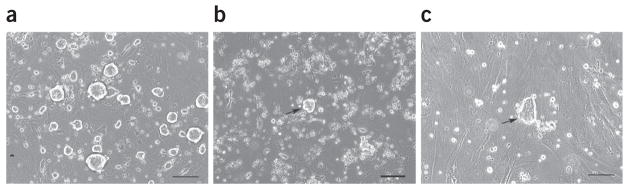
DA rat ES colonies formed after electroporation (a–c). DA rat ES cells were transfected with the p53 gene-targeting vector by electroporation and were replated onto 10-cm dishes at a density of 1 × 106 cells per dish. The pictures show the cells 7 d after electroporation, without (a) or with (b,c) drug selection. Round domed-shaped colonies (arrow in b), as opposed to flat colonies (arrow in c), should be picked and expanded for verification of correctly targeted events. Scale bar, 50 μm.
The targeted mutations in rat ES cells can transmit through the germline to produce gene-targeted animals10. The quality of rat ES cells is the major factor affecting their germline competency. Therefore, it is important to karyotype rat ES cells before microinjecting them into recipient blastocysts. The genetic background of the host embryo chosen for the production of ES cell–rat chimeras has a dramatic effect on the successful production of chimeric animals that transmit the ES cell haplotype. Germline transmission of the rat ES cell genome can be achieved when DA rat ES cells are injected into F344 rat blastocysts but not when injected into Sprague-Dawley rat blastocysts4,10. The resultant male chimeric rats are crossed with Sprague-Dawley or F344 rats to verify the germline transmission of targeted DA rat ES cells.
Acknowledgments
We thank N. Wu and Y. Yan for blastocyst injection; G. Chester for ordering rats; R. Montano and colleagues for rat husbandry; and T. Saunders for scientific input. This work was funded by a US National Institutes of Health National Center for Research Resouces grant (R01 RR025881).
Footnotes
AUTHOR CONTRIBUTIONS C.T. and Q.-L.Y. designed the study. C.T., G.H. and P.L. conducted the experiments. G.H. and C.T. wrote a draft of the paper. C.A. and Q.-L.Y. proofread and finalized the paper.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests (see the HTML version of this article for details).
References
- 1.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 2.Ying QL, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buehr M, et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell. 2008;135:1287–1298. doi: 10.1016/j.cell.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Li P, et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell. 2008;135:1299–1310. doi: 10.1016/j.cell.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawamata M, Ochiya T. Generation of genetically modified rats from embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:14223–14228. doi: 10.1073/pnas.1009582107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, et al. Derivation of embryonic stem cells from Brown Norway rats blastocysts. J Genet Genomics. 2010;37:467–473. doi: 10.1016/S1673-8527(09)60066-7. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi T, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142:787–799. doi: 10.1016/j.cell.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 8.Hirabayashi M, et al. Establishment of rat embryonic stem cell lines that can participate in germline chimerae at high efficiency. Mol Reprod Dev. 2010;77:94. doi: 10.1002/mrd.21123. [DOI] [PubMed] [Google Scholar]
- 9.Hirabayashi M, et al. Rat transgenesis via embryonic stem cells electroporated with the Kusabira-orange gene. Mol Reprod Dev. 2010;77:474. doi: 10.1002/mrd.21181. [DOI] [PubMed] [Google Scholar]
- 10.Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213. doi: 10.1038/nature09368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meek S, et al. Efficient gene targeting by homologous recombination in rat embryonic stem cells. PLoS One. 2010;5:e14225. doi: 10.1371/journal.pone.0014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen HP, et al. Behavioral abnormalities precede neuropathological markers in rats transgenic for Huntington’s disease. Hum Mol Genet. 2006;15:3177–3194. doi: 10.1093/hmg/ddl394. [DOI] [PubMed] [Google Scholar]
- 13.Wharram BL, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 14.Smits BM, Cotroneo MS, Haag JD, Gould MN. Genetically engineered rat models for breast cancer. Breast Dis. 2007;28:53–61. doi: 10.3233/bd-2007-28106. [DOI] [PubMed] [Google Scholar]
- 15.Holmdahl R, et al. Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev. 2001;184:184–202. doi: 10.1034/j.1600-065x.2001.1840117.x. [DOI] [PubMed] [Google Scholar]
- 16.Cozzi J, et al. Methods in Molecular Biology. Vol. 561. Humana Press; 2009. Pronuclear DNA injection for the production of transgenic rats; pp. 73–88. [DOI] [PubMed] [Google Scholar]
- 17.Dann CT, Alvarado AL, Hammer RE, Garbers DL. Heritable and stable gene knockdown in rats. Proc Natl Acad Sci USA. 2006;103:11246–11251. doi: 10.1073/pnas.0604657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Boxtel R, Gould MN, Cuppen E, Smits BM. Methods in Molecular Biology. Vol. 597. Humana; 2010. ENU mutagenesis to generate genetically modified rat models; pp. 151–167. [DOI] [PubMed] [Google Scholar]
- 19.Zan Y, et al. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nat Biotechnol. 2003;21:645–651. doi: 10.1038/nbt830. [DOI] [PubMed] [Google Scholar]
- 20.Izsvak Z, et al. Generating knockout rats by transposon mutagenesis in spermatogonial stem cells. Nat Methods. 2010;7:443–445. doi: 10.1038/nmeth.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitada K, et al. Transposon-tagged mutagenesis in the rat. Nat Methods. 2007;4:131–133. doi: 10.1038/nmeth1002. [DOI] [PubMed] [Google Scholar]
- 22.Kitada K, Keng VW, Takeda J, Horie K. Generating mutant rats using the Sleeping Beauty transposon system. Methods. 2009;49:236–242. doi: 10.1016/j.ymeth.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Mashimo T, et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One. 2010;5:e8870. doi: 10.1371/journal.pone.0008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menoret S, et al. Characterization of immunoglobulin heavy chain knockout rats. Eur J Immunol. 2010;40:2932–2941. doi: 10.1002/eji.201040939. [DOI] [PubMed] [Google Scholar]
- 25.Geurts AM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui X, et al. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol. 2011;29:64–67. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 27.Meyer M, de Angelis MH, Wurst W, Kuhn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci USA. 2010;107:15022–15026. doi: 10.1073/pnas.1009424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo, A Laboratory Manual. 3. Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- 29.Wu S, Ying G, Wu Q, Capecchi MR. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat Protoc. 2008;3:1056–1076. doi: 10.1038/nprot.2008.70. [DOI] [PubMed] [Google Scholar]
- 30.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cotta-de-Almeida V, Schonhoff S, Shibata T, Leiter A, Snapper SB. A new method for rapidly generating gene-targeting vectors by engineering BACs through homologous recombination in bacteria. Genome Res. 2003;13:2190–2194. doi: 10.1101/gr.1356503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbs RA, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, et al. Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev Dyn. 1997;209:85–91. doi: 10.1002/(SICI)1097-0177(199705)209:1<85::AID-AJA8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 36.Schwartzberg PL, Goff SP, Robertson EJ. Germ-line transmission of a c-abl mutation produced by targeted gene disruption in ES cells. Science. 1989;246:799–803. doi: 10.1126/science.2554496. [DOI] [PubMed] [Google Scholar]
- 37.Auerbach W, et al. Establishment and chimera analysis of 129/SvEv- and C57BL/6-derived mouse embryonic stem cell lines. Biotechniques. 2000;29:1024–1028. 1030–1032. doi: 10.2144/00295st04. [DOI] [PubMed] [Google Scholar]
- 38.Lemckert FA, Sedgwick JD, Korner H. Gene targeting in C57BL/6 ES cells. Successful germ line transmission using recipient BALB/c blastocysts developmentally matured in vitro. Nucleic Acids Res. 1997;25:917–918. doi: 10.1093/nar/25.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuster-Gossler K, et al. Use of coisogenic host blastocysts for efficient establishment of germline chimeras with C57BL/6J ES cell lines. Biotechniques. 2001;31:1022–1024. 1026. doi: 10.2144/01315st01. [DOI] [PubMed] [Google Scholar]
- 40.Seong E, Saunders TL, Stewart CL, Burmeister M. To knockout in 129 or in C57BL/6: that is the question. Trends Genet. 2004;20:59–62. doi: 10.1016/j.tig.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Ledermann B, Burki K. Establishment of a germ-line competent C57BL/6 embryonic stem cell line. Exp Cell Res. 1991;197:254–258. doi: 10.1016/0014-4827(91)90430-3. [DOI] [PubMed] [Google Scholar]
- 42.Bryja V, Bonilla S, Arenas E. Derivation of mouse embryonic stem cells. Nature Protoc. 2006;1:2082–2087. doi: 10.1038/nprot.2006.355. [DOI] [PubMed] [Google Scholar]
- 43.Kuhn R, Wurst W. Methods in Molecular Biology. Vol. 530. Humana Press; 2009. Gene knockout protocols. [Google Scholar]