

Abstract
Expression of HLA-B*57 and the closely related HLA-B*58:01 are associated with prolonged survival after HIV-1 infection. However, large differences in disease course are observed among HLA-B*57/58:01 patients. Escape mutations in CTL epitopes restricted by these HLA alleles come at a fitness cost and particularly the T242N mutation in the TW10 CTL epitope in Gag has been demonstrated to decrease the viral replication capacity. Additional mutations within or flanking this CTL epitope can partially restore replication fitness of CTL escape variants. Five HLA-B*57/58:01 progressors and 5 HLA-B*57/58:01 long-term nonprogressors (LTNPs) were followed longitudinally and we studied which compensatory mutations were involved in the restoration of the viral fitness of variants that escaped from HLA-B*57/58:01-restricted CTL pressure. The Sequence Harmony algorithm was used to detect homology in amino acid composition by comparing longitudinal Gag sequences obtained from HIV-1 patients positive and negative for HLA-B*57/58:01 and from HLA-B*57/58:01 progressors and LTNPs. Although virus isolates from HLA-B*57/58:01 individuals contained multiple CTL escape mutations, these escape mutations were not associated with disease progression. In sequences from HLA-B*57/58:01 progressors, 5 additional mutations in Gag were observed: S126N, L215T, H219Q, M228I and N252H. The combination of these mutations restored the replication fitness of CTL escape HIV-1 variants. Furthermore, we observed a positive correlation between the number of escape and compensatory mutations in Gag and the replication fitness of biological HIV-1 variants isolated from HLA-B*57/58:01 patients, suggesting that the replication fitness of HLA-B*57/58:01 escape variants is restored by accumulation of compensatory mutations.
Introduction
HLA class I alleles are associated with the clinical course of HIV-1 infection. HLA-B*57 and the closely related HLA-B*58:01 are overrepresented in so called long-term nonprogressors (LTNPs) [1]–[7]. Individuals carrying these HLA alleles exert strong cytotoxic T lymphocyte (CTL) responses against conserved viral regions such as Gag, resulting in immunological control of HIV-1 replication and a slower disease progression [8]–[13]. However, HIV-1 continuously adapts to the high selective pressure exerted by the immune system, resulting in escape mutations in viral epitopes and loss of recognition by CTLs [14]–[22]. Although the escape mutations allow evasion of the CTL response, viral escape is not always associated with disease progression. Some CTL escape mutations come at a fitness cost, particularly when situated in conserved viral regions, and patients may benefit from a reduced viral replication capacity despite escape from CTL-mediated killing. The T242N escape mutation in the HLA-B*57/58:01 restricted TW10 epitope in Gag impairs viral replication, which can explain the protective effect of this HLA type even after escape from CTL responses has occurred [22]–[27]. Additional mutations within or flanking the TW10 epitope can have a compensatory effect and partially restore the fitness cost associated with the T242N mutation [25]–[28]. Although HLA-B*57 and HLA-B*58:01 are overrepresented in LTNPs, the majority of patients carrying these protective HLA alleles do show a progressive disease course in the absence of antiretroviral treatment.
Previously, we studied 5 HLA-B*57/58:01 LTNPs and 5 HLA-B*57/58:01 progressors longitudinally, and observed similar frequencies of HIV-1 Gag-specific CTL responses and dynamics in escape mutations in HLA-B*57/58:01-restricted CTL epitopes [25]. This indicates that Gag specific CTL responses and the prevalence of CTL escape mutations does not relate to the differential disease course in these patients. However, an increase in replication kinetics of viral variants isolated from progressors was observed during longitudinal follow-up [25]. Furthermore, an association between disease progression and the presence of 2 or more of the compensatory mutations H219Q, I223V, M228I, N252H and G248X was observed [25].
Here, we studied which compensatory mutations were involved in the restoration of the viral fitness of variants that escaped from HLA-B*57/58:01-restricted CTL pressure. Virus isolates from HLA-B*57/58:01 individuals contained multiple CTL escape mutations, and these escape mutations were not associated with disease progression. In the HLA-B*57/58:01 progressors, 5 additional mutations were observed in the Gag protein that increased the replication rate of the HLA-B*57/58:01 CTL escape variants.
Results
Sequence variation in Gag associated with HLA-B*57/58:01
To study differences in the Gag protein of virus isolates obtained from HLA-B*57/58:01 progressors and LTNPs that may explain the differences in replication kinetics, we compared Gag sequences (positions 1-364) obtained from various time points during the course of infection (Table 1 and Table S1) using the Sequence Harmony (SH) algorithm [29]–[31]. SH detects positions within an alignment that show differences in amino acid composition between two groups of sequences and analyses the frequency distribution of amino acid variation per position [29]–[31]. Low SH-scores indicate position where the amino acid compositions are different between the two groups; a score of 0 indicates that the amino acids at a given position are completely different between both groups, while the maximum score of 1 indicates that the amino acid compositions are indistinguishable.
Table 1. Characteristics of patients positive for HLA-B*57 or HLA-B*58:01 and the Gag sequences analyzed.
| Sequences | ||||||||
| Patient | Seroconversion (S) or HIV+ Entry (E) date | HLA A alleles | HLA B alleles | Δ32 CCR5 genotype | Time after seroconversion or study entry (months) | Number of sequences | ||
| L5 | 06-11-1984 (E) | A*26 | A*68:01 | B*57 | B*07:02 | Wild type | 89 | 6 |
| 177 | 6 | |||||||
| L6 | 09-02-1988 (E) | A3 | A11 | B*58:01 | B7 | Heterozygous | 17 | 5 |
| 114 | 15 | |||||||
| L7 | 03-12-1984 (E) | A*01 | A*02 | B*57:01 | B*050:1 | Heterozygous | 26 | 5 |
| 78 | 6 | |||||||
| 102 | 8 | |||||||
| 136 | 12 | |||||||
| L8 | 06-02-1990 (S) | A*02 | A*02 | B*57:01 | B*37:01 | Wild type | 70 | 2 |
| 91 | 9 | |||||||
| 137 | 6 | |||||||
| L9 | 04-10-1985 (S) | A*01 | A*68:02 | B*57:01 | B*14:02 | Wild type | 42 | 1 |
| 59 | 1 | |||||||
| 77 | 6 | |||||||
| P9 | 20-01-1985 (E) | A2 | A3 | B*57:01 | B*07:02 | Wild type | 9 | 8 |
| 78 | 3 | |||||||
| P10 | 01-01-1984 (E) | A*02 | A*26 | B*57:01 | B*41:02 | Wild type | 69 | 7 |
| 84 | 7 | |||||||
| 123 | 9 | |||||||
| P11 | 15-05-1990 (S) | A1 | A11 | B*57:01 | B*08:01 | Wild type | 3 | 2 |
| 25 | 9 | |||||||
| 32 | 23 | |||||||
| 69 | 13 | |||||||
| P12 | 24-08-1992 (E) | A*01 | A*26 | B*57:01 | B*38:01 | Wild type | 0.4 | 5 |
| 48 | 11 | |||||||
| 74 | 12 | |||||||
| P13 | 22-01-1985 (E) | A*01 | A*01 | B*57:01 | B*08:01 | Wild type | 3 | 6 |
| 46 | 10 |
L: long term non-progressor; P: progressor. S: date of seroconversion for HIV-1 antibodies during active follow-up in the cohort. E: HIV+ entry into the cohort.
First, we identified amino acid variation in Gag specific for patients carrying HLA-B*57 or HLA-B*58:01 irrespective of their disease course. Therefore, we compared longitudinally obtained sequences from HLA-B*57/58:01 patients (n = 10: 5 progressors and 5 LTNPs, 221 sequences in total; Table 1 and Table S1) to longitudinally obtained sequences from patients negative for HLA-B*57 and HLA-B*58:01 alleles (n = 19, 152 sequences) using the SH algorithm with a cut-off value of 0.90. In this way, amino acid positions were considered to be different between the two groups when there was more than an approximate 10% difference in amino acid distribution at this position. In total, 6 amino acid positions in Gag were identified that showed significant variation in the dominant amino acid between sequences from individuals with or without HLA-B*57/58:01. Among these HLA-B*57/58:01 specific amino acid mutations were the HLA-B*57/58:01 associated CTL escape mutations T242N and G248A located in the TW10 epitope and the I147L mutation in the IW9 epitope. Additionally, the S173T adjacent to epitope KF11, and mutations V159I and T280V were associated with the presence of HLA-B*57/58:01 (Figure 1A, Table 2 and Table S1).
Figure 1. Sequence variation in Gag affects viral replication fitness.

A. Sequence Harmony analysis detecting amino acid changes associated with the presence of HLA-B*57/58:01, irrespective of disease course. Longitudinal Gag sequences obtained from HLA-B*57/58:01 progressors and LTNPs (n = 10) were compared to Gag sequences obtained from patients negative for HLA-B*57/58:01 (n = 19). An SH value of 0.90 was used as cut-off. Six amino acid changes were identified to be specifically associated with the presence of HLA-B*57/58:01: I147L, V159I, S173T, T242N, G248A and T280V. Amino acid positions in capsid associated with HLA-B*57/58:01 are shown in red. B. Sequence Harmony analysis detecting amino acid changes associated with disease progression in HLA-B*57/58:01 patients. Gag sequences obtained from late in infection of HLA-B*57/58:01 progressors (n = 5) and HLA-B*57/58:01 LTNPs (n = 5) were compared. An SH value of 0.90 was used as cut-off. Five amino acid changes were identified to be associated with HLA-B*57/58:01 disease progression: S126N, L215T, H219Q, M228I and N252H. Amino acid positions in capsid associated with HLA-B*57/58:01 disease progression are shown in red. C. Replication kinetics (days 2–17) of constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 (I147L, V159I, S173T, T242N, G248A and T280V; red), in combination with mutation S126N (orange), L215T (yellow), H219Q (purple), M228I (green), L215T and H219Q (grey), or a combination of all 5 compensatory mutations (S126N, L215T, H219Q, M228I and N252H; blue). Mean p24 concentrations (ng/ml) are given. Error bars represent the standard error of the mean. Data from one representative experiment are shown. D. Replication kinetics of NL4-3.Ba-L and constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 (I147L, V159I, S173T, T242N, G248A and T280V) in the absence or presence of compensatory mutations. Normalized mean p24 values on day 11 were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001. Error bars represent 2.5 – 97.5 percentiles. Data from one representative experiment are shown. E. Replication kinetics of NL4-3.Ba-L and constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 (I147L, V159I, S173T, T242N, G248A and T280V) in the absence or presence of compensatory mutations described by Brockman et al. Normalized mean p24 values on day 11 were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001. Error bars represent 2.5–97.5 percentiles. Data from one representative experiment are shown.
Table 2. Amino acid variation in Gag p17 and p24 specific for HLA-B*57/58:01.
| AA | Comparison | AA variation*** | ||||
| Position | SH | HLA-B*57/58:01 | Non-B*57/58:01 | |||
| SH value* | Z score** | Dominant | Minor | Dominant | Minor | |
| I147 | 0.44 | −120 | L | MI | I | L |
| V159 | 0.80 | −54 | IV | V | I | |
| S173 | 0.68 | −65 | ST | S | AT | |
| T242 | 0.05 | −400 | N | TS | T | |
| G248 | 0.78 | −47 | GA | T | G | A |
| T280 | 0.88 | −19 | TV | SIA | T | VSA |
*The Sequence Harmony (SH) score represents overlap in amino acid composition between the two groups analyzed (HLA-B*57/58:01 versus non-HLA-B*57/58:01). An SH value of 0 indicates no overlap (largest possible differences) while a value of 1 indicates complete overlap (no differences).
**All Z scores represent P values <0.0001 with correction for multiple testing (450 alignment positions).
***The most frequent amino acids found at a certain position are depicted as “Dominant”, whereas the “Minor” amino acids are present at least 2 times less frequent than the most frequently occurring amino acid at this position. Amino acids are ordered from highest to lowest frequency.
To identify sequence variation within Gag associated with disease progression, we compared sequences from viral variants obtained late in infection from 5 HLA-B*57/58:01 progressors (43 sequences; Table 1 and Table S1) and 5 HLA-B*57/58:01 LTNPs (45 sequences; Table 1 and Table S1) with SH, using a cut-off value of 0.90. Frequent variation in amino acid composition was observed at 5 positions within Gag (S126N, L215T, H219Q, M228I and N252H) in viral sequences obtained from progressors, whereas these amino acids were absent or only present at low frequency in sequences obtained from LTNPs (Table 3, Figure 1B and Table S1). When we repeated the SH analysis correcting for the number of sequences per patient to give each patient equal weight, the same amino acid residues showed significant variation between sequences obtained from LTNPs and progressors (data not shown).
Table 3. Amino acid variation in Gag p17 and p24 associated with HLA-B*57/58:01 disease progression.
| AA | Comparison | AA variation*** | ||||
| Position | SH | Progressors | LTNPs | |||
| SH value* | Z score** | Dominant | Minor | Dominant | Minor | |
| S126 | 0.83 | −10.7 | SK | N | S | N |
| L215 | 0.61 | −27.8 | LT | I | L | M |
*The Sequence Harmony (SH) score represents overlap in amino acid composition between the two groups analyzed (HLA-B*57/58:01 progressors versus HLA-B*57/58:01 LTNPs). An SH value of 0 indicates no overlap (largest possible differences) while a value of 1 indicates complete overlap (no differences).
**All Z scores represent P values <0.0001 with correction for multiple testing (450 alignment positions).
***The most frequent amino acids found at a certain position are depicted as “Dominant”, whereas the “Minor” amino acids are present at least 2 times less frequent than the most frequently occurring amino acid at this position. Amino acids are ordered from highest to lowest frequency.
Mutations associated with disease progression in HLA-B*57/58:01 patients increase the viral replication kinetics in vitro
We hypothesized that the amino acid variation observed within Gag of viral variants obtained from HLA-B*57/58:01 progressors are compensatory mutations that increase the replication capacity of viral variants containing the CTL escape mutations. In order to study the effects of the sequence variation found, the mutations associated with disease progression (alone or in combination) were placed in the NL4-3.Ba-L molecular clone together with the 6 mutations specifically associated with the presence of HLA-B*57/58:01 (T242N, G248A, I147L, S173T, V159I and T280V) (Tables 2 and 3). The NL4-3.Ba-L molecular clone backbone already contains N252H, and this amino acid was not changed in the constructed viruses. The replication kinetics of the obtained viruses were analyzed on PHA stimulated PBMCs for 17 days (Figure 1C). An increase in viral replication was observed after introduction of the L215T and H219Q mutation (p = 0.002 and p = 0.0002 respectively, Figure 1D; Figure S1A), whereas mutations S126N and M228I did not alter replication kinetics (Figure1D and Figure S1A). The highest replication capacity was observed when the combination of all compensatory mutations was introduced in NL4-3.Ba-L carrying the HLA-B*57/58:01-associated mutations (p<0.0001, Figure 1D; Figure S1A). As mutations L215T and H219Q show the highest increase in replication capacity analyzing the single mutations, we also introduced a combination of these two mutations in the mutant virus carrying the 6 HLA-B*57/58:01-specific mutations. The combination of these two compensatory mutations, however, did not significantly increase viral replication kinetics compared to that HLA-B*57/58:01 background virus (Figure 1D and Figure S1A).
A previous study by Brockman et al. reported that the compensatory mutations H219Q, I223V, M228I, G248A and N252H partially restored replication kinetics of NL4-3 virus carrying the T242N escape mutation [27]. Three of these mutations (H219Q, M228I, N252H) were also associated with disease progression in our analysis, while the G248A mutation was found in all HLA-B*57/58:01 patients irrespective of their disease course. The I223V mutation was not observed to be different between LTNPs and progressors in our analysis, nor was it associated with the presence of HLA-B*57/58:01. To compare the effect of the compensatory mutations described by Brockman et al. and our present analysis on the replication fitness of the virus, we placed the described mutations in the NL4-3.Ba-L carrying the HLA-B*57/58:01-specific mutations (Table 2). The I223V mutation alone had no significant effect on the replication kinetics of NL4-3.Ba-L carrying the HLA-B*57/58:01 specific mutations (Figure 1E and Figure S1B). The combination of mutations described by Brockman et al. did indeed increase the replication kinetics of the NL4-3.Ba-L carrying the 6 HLA-B*57/58:01-specific mutations (p = 0.0012, Figure 1E; Figure S1B). However, the replication capacity of this virus was significantly lower than that observed for the virus carrying all of the mutations associated with HLA-B*57/58:01 disease progression identified in our present study (p = 0.0082, Figure 1E; Figure S1B). These findings suggest an additive effect of additional compensatory mutations on viral replication.
The total number of escape and compensatory mutations in Gag correlates with the replication kinetics of biological HIV-1 isolates obtained from HLA-B*57/58:01 patients
During the course of infection, mutations accumulate in the virus as a result of the continuous selection for the fittest variant. Our results may suggest that compensatory mutations restore viral fitness in a cumulative manner. Therefore, we analysed whether there is an association between the total number of escape and compensatory mutations in Gag that we identified and the replication capacity of biological HIV-1 isolates obtained during the course of infection from our HLA-B*57/58:01 progressors and LTNPs (previously described in [25]). We observed a correlation between the number of mutations and an increasing replication capacity (R = 0.33, p = 0.04, Figure 2). This supports the idea that compensation of the fitness cost associated with HLA-B*57/58:01 CTL escape is the result of the accumulation of multiple mutations that increase viral fitness.
Figure 2. Accumulation of mutations in Gag correlates with a higher replication fitness.
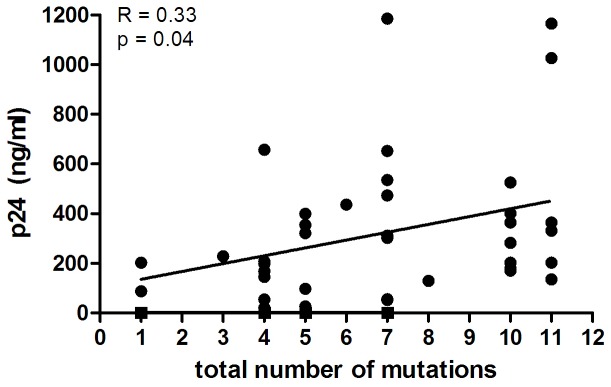
Spearman correlation analysis of the total number of escape and compensatory mutations associated with HLA-B*57/58:01 and the maximum p24 value observed for biological HIV-1 variants obtained from HLA-B*57/58:01 progressors and LTNPs in an in vitro replication assay.
Discussion
HLA-B*57 and HLA-B*58:01 are overrepresented in HIV-1 long-term nonprogressors, however, large differences in clinical disease course are observed among HLA-B*57/58:01 patients. Previously, we have shown that HLA-B*57/58:01 LTNPs and progressors have similar frequencies of HIV-1 Gag specific CTL responses and similar dynamics in escape mutations in HLA-B*57/58:01-restricted CTL epitopes [25]. Although escape mutations allow for evasion of CTL responses, the prevalence of escape mutations does not explain the differential disease course observed in these patients. CTL escape mutations in the HLA-B*57/58:01-restricted epitopes situated in Gag often impair viral replication and this fitness loss can be partially compensated for by additional mutations within or flanking these CTL epitopes [22]–[28]. We here studied whether the variation in clinical course of HIV-1 infection of HLA-B*57/58:01-positive individuals may be explained by differences in the number of mutations that restore fitness of viral variants that escaped from HLA-B*57/58:01-restricted CTL pressure.
The SH algorithm was used to study differences in Gag viral sequences obtained from HLA-B*57/58:01 negative and positive individuals, and from HLA-B*57/58:01 progressors and LTNPs. An important advantage of this approach is that it is unbiased toward certain amino acid positions that have previously been reported to be associated with HLA-B*57/58:01 or with a progressive disease course. Furthermore, the SH algorithm also allowed detection of very small differences in the frequency of amino acids between the two groups of sequences, whereas other specificity site detection methods would discard these due to lack of within-group conservation. Particularly in the case of the highly conserved Gag region the detection of very small differences in amino acid composition is crucial.
We observed that virus isolates from HLA-B*57/58:01 individuals, irrespective of their disease course, contained CTL escape mutations in the TW10 epitope (T242N and G248A) and the IW9 epitope (I147L), and a mutation adjacent to the KF11 epitope (S173T). Additionally, two other mutations (V159I and T280V) were observed specifically in viral variants from HLA-B*57/58:01 positive individuals; however, these mutations could not be related to escape from CTL pressure restricted by HLA-B*57/58:01. When comparing viral Gag sequences obtained late in infection from HLA-B*57/58:01 progressors and HLA-B*57/58:01 LTNPs, frequent changes in the amino acid sequence were observed for progressors at 5 positions: S126N, L215T, H219Q, M228I, N252H. An increase in replication kinetics was observed after introduction of two single mutations (L215T and H219Q) in the NL4-3.Ba-L virus containing the HLA-B*57/58:01-specific mutations. Nonetheless, the combination of all mutations associated with HLA-B*57/58:01 disease progression resulted in an even higher increase in replication kinetics. Introduction of a combination of mutations L215T and H219Q did not result in a significant increase in replication kinetics compared to the NL4-3.Ba-L virus containing the HLA-B*57/58:01-specific mutations, indicating that the combination of these two mutations does not account for the increase in replication kinetics observed for the virus containing the HLA-B*57/58:01-specific mutations. These findings suggest that multiple mutations are required to restore viral fitness after escape from HLA-B*57/58:01-restricted CTL responses [32]. To our knowledge, the S126N and L215T mutations have not been associated with an increase in viral fitness in HLA-B*57/58:01 CTL escape variants before, and we are the first to show that these mutations serve as compensatory mutations.
A previous study by Brockman et al. reported a higher frequency of mutations H219Q, I223V, M228I, G248A and N252H in combination with the HLA-B*57 T242N CTL escape mutation in sequences obtained from HLA-B*57 progressors than seen in sequences from HLA-B*57 non-progressors [27]. We observed that these mutations were able to increase the replication of NL4-3.Ba-L virus carrying the HLA-B*57/58:01-specific mutations; however, the replication capacity was still impaired as compared to the virus carrying all of the mutations associated with HLA-B*57/58:01 disease progression identified in our present study.
Our results and previous observations by others [32] suggest that compensatory mutations restore viral fitness in a cumulative manner. In sequences obtained from LTNPs late in the course of infection 1 or 2 compensatory mutations were present, whereas a higher number of mutations was usually observed in sequences obtained from progressors. The presence of a low number of compensatory mutations likely results in only a partial restoration of viral fitness. In line with this, our previous results show that biological viral variants from LTNPs obtained early and late in infection do not differ in their replication kinetics, whereas in HLA-B*57/58:01 progressors viral fitness increased over the course of infection [25]. Moreover, we here show a positive correlation between the number of HLA-B*57/58:01 escape and compensatory mutations and the replication capacity of HIV-1 biological viral variants obtained from HLA-B*57/58:01 patients, again suggesting there is a cumulative effect of compensatory mutations on viral replication fitness. Introduction of single mutations H219Q and L215T showed a significant increase in replication capacity, which may suggest that these mutations play a more prominent role in restoration of viral fitness.
The Gag protein is highly conserved and amino acid variation observed in Gag seems to be limited to specific positions, whereas other positions remain unchanged due to structural constraints. As a result, a high degree of conformity was observed in the amino acid positions described here and by previous studies [22], . This suggests that there is only a limited amount of positions within Gag that are capable of changing without a substantial fitness cost. Variation at these sites may serve as a general “coping” mechanism, and allow adaptation to changes in the capsid structure and capsid stability. Most of the compensatory mutations are located in one of three loops on the outer surface of capsid and are not involved in the formation of p24 hexamers and pentamers that make up the capsid lattice structure. The outer surface loops interact with host cellular proteins, like cyclophilin A (CypA) and restriction factor TRIM5α. The compensatory mutations, in particular H219Q, influence viral sensitivity to TRIM5α and reduce binding of capsid to CypA. This suggests that the introduction of compensatory mutations may influence HIV-1 replication through mechanisms affecting host factor dependency and sensitivity to intrinsic immunity [33]–[36].
In conclusion, we here identified 5 positions in the Gag region of HIV-1 that are more frequently mutated in sequences from HLA-B*57/58:01 progressors. The combination of these mutations can restore the replication fitness of CTL escape HIV-1 variants, which may explain the differences in disease progression. Furthermore, we observed that the total number of escape and compensatory mutations correlated with the replication fitness of biological HIV-1 isolates obtained from HLA-B*57/58:01 patients, suggesting that the replication fitness of HLA-B*57/58:01 escape variants is restored by accumulation of compensatory mutations.
Materials and Methods
Patients
Previously, we reported on 22 participants from the Amsterdam Cohort Studies (ACS) on HIV infection carrying the protective HLA-B*57 or HLA-B*58:01 allele who showed large differences in clinical disease course [25]. Ten patients who were followed longitudinally and from whom clonal HIV-1 viral sequences from at least two time points during follow-up were available were selected for the present study (Table 1). Seven individuals were HIV-1 infected (seroprevalent) at the moment of entry and three individuals seroconverted for HIV antibodies during active follow-up. All cohort participants had routine 3 monthly visits for blood donation and physical examination. Individuals who developed AIDS or who started with antiviral therapy within nine years after seroconversion or seroprevalent entry in the Amsterdam Cohort Studies were called progressors (n = 5; ACH19567 [P9], ACH18932 [P10], ACH18887 [P11], ACH13879 [P12], ACH11679 [P13]). Long-term non-progressors (LTNP) were those participants who had stable CD4+ T cell counts that were still above 400 cells/µl blood in the 10th year after SC or seroprevalent entry in the Amsterdam Cohort Studies or who had a CD4+ T cell decline less than 40*109cells/l per year over a period of at least 10 years (n = 5; ACH19933 [L5], ACH19922 [L6], ACH19789 [L7], ACH19784 [L8], ACH19291 [L9]).
Two participants (L6 and L7) were heterozygous for the 32 base pair deletion in the CCR5 gene, whereas all other participants had a CCR5 wild type genotype. Typing of the HLA A and B alleles of all participants (Table 1) did not reveal other HLA types that were associated with a difference in disease progression.
Ethics statement
The ACS has been conducted in accordance with the ethical principles set out in the declaration of Helsinki and written informed consent is obtained prior to data collection. This study was approved by the Amsterdam Medical Center institutional medical ethics committee.
Viruses
Five HLA-B*57/58:01 progressors and five HLA-B*57/58:01 LTNPs were followed longitudinally. Clonal HIV-1 variants were isolated from patient PBMCs that were obtained early after seroconversion or study entry, at a time point as late as possible in the course of infection before the start of therapy and from PBMCs isolated at an in between time point (Table 1) [25]. From some patients virus isolation from PBMCs that were obtained early in the course of infection failed due to low viral loads at these time points. Clonal viral variants were obtained from single productively infected cells by cocultivation of PHA-stimulated PBMCs from healthy donors and serial dilutions with patient PBMC as described previously [37]. In brief, increasing numbers of patient PBMC were cocultivated with PHA-stimulated healthy donor PBMC in 96-well microtiter plates with four parallel microcultures per patient cell number. Every week, culture supernatants were tested for the presence of p24 antigen by an in-house p24 ELISA [38]. At the same time, one-third of the culture volume was transferred to a new 96-well plate and fresh PHA-stimulated PBMC from a healthy donor were added to propagate the culture. From the wells positive in the p24 antigen ELISA, virus stocks were grown and cryopreserved.
DNA isolation, PCR amplification and sequencing
Viral DNA was isolated from cryopreserved PBMC that were infected with one clonal HIV-1 variant, using the L6 isolation method [39]. Gag DNA (positions 1−364) was amplified by a nested polymerase chain reaction (PCR) with outer primers Gag forward (5′-CGACGCAGGACTCGGCTTGCTG-3′; HXB2 nucleotides 684−705) and Gag outer reversed (5′-GCCTGTCTCTCAGTAC-3′; HXB2 nucleotides 2065−2080) and 2 different inner PCR primer combinations: Gag BssHII fw (5′-TGCTGAAGCGCCCGCACGGC-3′; HXB2 nucleotides 701−720) or Gag ClaI fw (5′-GGGAGAATTAGATCGATGGG-3′; HXB2 nucleotides 818−837) in combination with Gag p17 rev (5′-CAAAACTCTTGCCTTATGG-3′; HXB2 nucleotides 1859−1880) and Gag p17 fw (5′-TGCTAAACACAGTGGGGGGACAT-3′; HXB2 nucleotides 1349−1371) in combination with Gag ApaI rev (5′-TTCCTAGGGGCCCTGCAA-3′; HXB2 nucleotides 2000−2017).
PCR products were purified and sequenced with the ABI prism BigDye Terminator sequencing kit (Perkin Elmer, Froster City, California, USA) on an ABI 3130 XL DNA sequencer according to the manufacturer's protocol using the same PCR primers that were used for the nested PCR. DNA sequences were analyzed with Seqman software (DNAStar software package; Lasergene, Madison, Wisconsin, USA). The nucleotide sequences of the gag region were translated and edited with the BioEdit program (BioEdit v 7.0.5, Tom Hall, Ibis Therapeutics, Carlsbad, California, USA).
Comparison of viral sequences with Sequence Harmony
To evaluate amino acid differences in Gag between patients positive and negative for HLA-B*57/58:01 and between HLA-B*57/58:01 positive LTNPs and HLA-B*57/58:01 progressors, the gag sequences obtained from these patients were analyzed with Sequence Harmony (SH) [29]–[31]. The SH algorithm is an entropy-based method, that detects positions with compositional differences within a multiple sequence protein alignment, as previously described [30] and in the online documentation on the web server (www.ibi.vu.nl/programs/seqharmwww). SH measures the overlap in distribution of amino acid types between two subgroups (A and B), in this case gag sequences obtained from HLA-B*57/58:01 and non- HLA-B*57/58:01 patients or HLA-B*57/58:01 LTNPs and HLA-B*57/58:01 progressors, at a certain position (i) in the sequence alignment as follows:
where 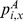 indicates the observed frequency in group A for amino acid type x at position i in the sequence and
indicates the observed frequency in group A for amino acid type x at position i in the sequence and 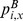 analogously for amino acid frequencies observed in group B sequences. The final SH score is calculated by
analogously for amino acid frequencies observed in group B sequences. The final SH score is calculated by 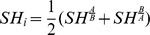 . Therefore, an SH score of 0 indicates amino acid positions that are specific for one of the sequence groups, whereas an SH score of 1 indicates an identical amino acid composition (i.e. complete overlap) at this alignment position between the two groups. To account for the unbalanced numbers of sequences available per patient (due to differing numbers of sample time points and numbers of sequences retreived per sample), we have also performed the SH analysis where we weighted each sequences by 1/Np, where Np is the number of sequences for patient p in the calculation of the SH score, such that each patient in total has equal weight in the SH analysis.
. Therefore, an SH score of 0 indicates amino acid positions that are specific for one of the sequence groups, whereas an SH score of 1 indicates an identical amino acid composition (i.e. complete overlap) at this alignment position between the two groups. To account for the unbalanced numbers of sequences available per patient (due to differing numbers of sample time points and numbers of sequences retreived per sample), we have also performed the SH analysis where we weighted each sequences by 1/Np, where Np is the number of sequences for patient p in the calculation of the SH score, such that each patient in total has equal weight in the SH analysis.
In the SH analyses performed here, a cut-off value of 0.90 was used to determine amino acid positions that are different between two groups of sequences. As the SH values are in a logarithmic scale, one could interpret the 0.90 cut-off as an approximate 10% difference in amino acid distribution. The high cut-off allows for detection of relatively small differences against a background of overall high conservation. Note that conservation overall or within a group of sequences is not explicitly taken into account in the SH analysis. The cut-off of 0.9 for the SH score implies that sites are already of interest when the frequency of occurrence of some amino acid types at a certain position changes. Positions where the difference between the two groups of sequences was located at a minor amino acid for both groups, or where the aberrant amino acid was identical to our NL4-3.Ba-L backbone were excluded from the analysis.
Z-scores were calculated based on random SH scores from 100-fold random shuffling of sequence labels (i.e. random re-distribution of sequences over two groups of the same size as the patient groups in the analysis); we assumed a normal distributions of these random SH scores based on previous tests [31]. P values were estimated based on the Z-scores and correction for multiple testing was applied based on the number of columns in the multiple sequence alignment, which was around 450. In this calculation, Z<−6 (absolute value) corresponds to an uncorrected p value of <1e-9, therefore a Z<−10 (absolute value) corresponds to a corrected p-value of <1e-9. In the discussion of the results, we will for simplicity state p<0.0001 for Z<−10 (absolute value).
Cells
293T cells (ATCC) were cultured in Dulbecco Modified Eagle's Medium (DMEM) supplemented with 10% inactivated fetal calf serum (FCS; Hyclone, Logan, Utah, USA), 100 U/ml penicillin (Gibco, Paisly, Scotland, UK) and 100 µg/ml streptomycin (Gibco) in a humidified 10% CO2 incubator at 37°C.
PBMC from 10 healthy donors were isolated from buffy coats by Ficoll density centrifugation, pooled and cryopreserved. After thawing, the PBMC pool was stimulated for three days in Iscoves' Modified Dulbecco's Medium (IMDM) supplemented with 10% FCS, 100 U/ml penicillin, 100 µg/ml streptomycin, 5 µg/ml ciproxin (Bayer, Mijdrecht, the Netherlands) and 1 µg/ml phytohaemagglutinin (PHA; Remel Europe, Dartford, England, UK) at a cell density of 5×106 cells/ml in a humidified 10% CO2 incubator at 37°C. PBMC cultures were continued in IMDM supplemented with 10% FCS, 20 U/ml recombinant interleukin 2 (rIL2; Chiron Benelux, Amsterdam, the Netherlands, 5 µg/ml polybrene (Sigma, Zwijndrecht, the Netherlands), 100 U/ml penicillin and 100 µg/ml streptomycin at a cell density of 1×106/ml in a humidified 10% CO2 incubator at 37°C.
Construction of replication competent molecular viral clones
For the construction of HIV-1 variants that contain mutations in the Gag protein, the gag region of the molecular clone NL4-3.Ba-L was removed using restriction enzymes BssHII and ApaI and then cloned into the pGEM T easy vector (Promega, Madison, Wisconsin, USA). Mutations in the gag region were introduced using site directed mutagenesis as described by the manufacturer (Quick exchange kit, Stratagene). The gag insert was sequenced to confirm successful mutagenesis using primers T7 (5′-TAATACGACTCACTATAGGG-3′) and SP6 (5′-GATTTAGGTGACACTATAG-3′).
For the production of replicating NL4-3.Ba-L clones containing the desired mutations, the mutated gag insert was ligated into the full length molecular NL4-3.Ba-L clone using BssHII and ApaI restriction sites. The full-length NL4-3.Ba-L molecular clones were then transfected into 293T cells using the calcium phosphate method [40]. Virus production from the 293T cells was analyzed at day 3 and day 7 after transfection using an in house p24 ELISA [38]. Virus stocks were grown on PHA-stimulated PBMC and virus titers were determined by 50% tissue culture infectious dose (TCID50) as previously described [37]. The gag regions of obtained viruses were sequenced as described above to confirm the introduction of mutations.
Viral replication assay
To determine the viral replication rates of the different HIV-1 molecular clones and biological variants, 2×106 PHA-stimulated pooled PBMC were inoculated with 100 TCID50 per virus variant for 2 hours at 37°C in a shaking water bath in a total volume of 1.5 ml. Subsequently, the inoculated PBMC were washed with 5 ml of IMDM supplemented with 10% FCS, and cultured in IMDM supplemented with 10% FCS, 20 U/ml rIL2, 5 µg/ml polybrene, 100 U/ml penicillin and 100 µg/ml streptomycin at a cell density of 1×106 per ml in a humidified 10% CO2 incubator at 37°C. On day 5, 8, 11 and 14 after inoculation, 1×106 fresh PHA-stimulated pooled PBMC in 1 ml of IMDM culture medium supplemented with 10% FCS, 20 U/ml rIL2, 5 µg/ml polybrene, 100 U/ml penicillin and 100 µg/ml streptomycin were added to the cultures. Samples for determination of p24 antigen production (75 µl) were harvested every day after inoculation. All samples were tested for p24 antigen production simultaneously at the end of the experiment using an in-house p24 ELISA [38].
On the last day of the replication experiments, DNA from every well was isolated using the L6 method [39] and the gag region of all samples was sequenced as described above to confirm the presence of mutations. The replication rates of all viruses were tested at least in twofold and in 2 independent experiments for which the same pool of healthy donor PBMC was used. P24 values were normalized to the mean p24 values observed for the constructed NL4-3.Ba-L virus carrying mutations associated with the presence of HLA-B*57/58:01. The area under the curve for the replication curves spanning day 2−17 was calculated and normalized for the constructed NL4-3.Ba-L virus carrying mutations associated with the presence of HLA-B*57/58:01. Statistical significance of differences in the p24 production on day 11 and the area under the curve were tested with the unpaired Student's T test. In Figure 1 and Figure S1 data from 1 representative experiment are shown.
To test the correlation between the number of escape and compensatory mutations in Gag with the replication fitness of biological HIV-1 variants obtained from HLA-B*57/58:01 progressors and LTNPs, a Spearman correlation test was performed. P values <0.05 were considered significant. Statistical analyses were preformed using Graphpad Prism version 5 and SPSS version 19.
Supporting Information
Sequence variation in Gag affects viral replication fitness. A. Replication kinetics of constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 in the absence or presence of compensatory mutations. The area under the curve (day 2–17) was calculated and normalized mean AUCs were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001. Error bars represent 2.5–97.5 percentiles. Data from one representative experiment are shown. B. Replication kinetics of constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 in the absence or presence of compensatory mutations described by Brockman et al. The area under the curve (day 2–17) was calculated and normalized mean AUCs were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001.Error bars represent 2.5 – 97.5 percentiles. Data from one representative experiment are shown.
(TIF)
Amino acid sequence variation in Gag. A. Sequence variation within Gag located at amino acid positions associated with the presence of HLA-B*57/5801 or disease progression as identified with SH in sequences obtained from LTNPs from various time points during the course of HIV-1 infection. B. Sequence variation within Gag at amino acid positions associated with the presence of HLA- B*57/5801 or disease progression as identified with SH in sequences obtained from progressors from various time points during the course of HIV-1 infection.
(DOC)
Acknowledgments
The Amsterdam Cohort Studies on HIV infection and AIDS, a collaboration between the Amsterdam Health Service, the Academic Medical Center of the University of Amsterdam, Sanquin Blood Supply Foundation, the University Medical Center Utrecht, and the Jan van Goyen Clinic are part of the Netherlands HIV Monitoring Foundation and financially supported by the Center for Infectious Disease Control of the Netherlands National Institute for Public Health and the Environment. We are indebted to all participants for their continuous participation in the study. The authors thank Brigitte Boeser-Nunnink for excellent technical assistance.
Funding Statement
This work was supported by the Aids Fonds Netherlands [grant 2008023]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gao X, Bashirova A, Iversen AK, Phair J, Goedert JJ, et al. (2005) AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med 11: 1290–1292. [DOI] [PubMed] [Google Scholar]
- 2. Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, et al. (2003) Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17: 2581–2591. [DOI] [PubMed] [Google Scholar]
- 3. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, et al. (2010) The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330: 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrington M, O'Brien SJ (2003) The influence of HLA genotype on AIDS. Annu Rev Med 54: 535–551. [DOI] [PubMed] [Google Scholar]
- 5. Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, et al. (2000) HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A 97: 2709–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaslow RA, Carrington M, Apple R, Park L, Munoz A, et al. (1996) Influence of combinations of human major histocompatibility complex genes in the course of HIV-1 infection. Nature Medicine 2: 405–411. [DOI] [PubMed] [Google Scholar]
- 7. Deeks SG, Walker BD (2007) Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27: 406–416. [DOI] [PubMed] [Google Scholar]
- 8. Klein MR, van Baalen CA, Holwerda AM, Kerkhof GardeSR, Bende RJ, et al. (1995) Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J Exp Med 181: 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MBA (1994) Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol 68: 6103–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, et al. (1999) Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283: 857–860. [DOI] [PubMed] [Google Scholar]
- 11. Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, et al. (1994) Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol 68: 4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kent SJ, Woodward A, Zhao A (1997) Human immunodeficiency virus type 1 (HIV-1)-specific T cell responses correlate with control of acute HIV-1 infection in macaques. J Infect Dis 176: 1188–1197. [DOI] [PubMed] [Google Scholar]
- 13.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, et al. (2006) HLA Alleles Associated with Delayed Progression to AIDS Contribute Strongly to the Initial CD8(+) T Cell Response against HIV-1. PLoS Med 3: : e403- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goulder P, Price D, Nowak M, Rowland-Jones S, Phillips R, et al. (1997) Co-evolution of human immunodeficiency virus and cytotoxic T-lymphocyte responses. Immunol Rev 159: 17–29. [DOI] [PubMed] [Google Scholar]
- 15. Wang YE, Li B, Carlson JM, Streeck H, Gladden AD, et al. (2009) Protective HLA class I alleles that restrict acute-phase CD8+ T-cell responses are associated with viral escape mutations located in highly conserved regions of human immunodeficiency virus type 1. J Virol 83: 1845–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore CB, John M, James IR, Christiansen FT, Witt CS, et al. (2002) Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296: 1439–1443. [DOI] [PubMed] [Google Scholar]
- 17. Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, et al. (2009) The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 206: 1253–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, et al. (1997) Antiviral pressure exerted by HIV-1 specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nature Med 3: : 205- [DOI] [PubMed] [Google Scholar]
- 19. Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, et al. (1991) Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354: 453–459. [DOI] [PubMed] [Google Scholar]
- 20. Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, et al. (2000) Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407: 386–390. [DOI] [PubMed] [Google Scholar]
- 21. O'Connor DH, Allen TM, Vogel TU, Jing P, DeSouza IP, et al. (2002) Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med 8: 493–499. [DOI] [PubMed] [Google Scholar]
- 22. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, et al. (2004) HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10: 282–289. [DOI] [PubMed] [Google Scholar]
- 23.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, et al. (2008) Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog 4: : e1000033- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, et al. (2008) Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med 205: 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, et al. (2006) Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80: 3617–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, et al. (2007) Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 81: 12608–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crawford H, Prado JG, Leslie A, Hue S, Honeyborne I, et al. (2007) Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J Virol 81: 8346–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feenstra KA, Pirovano W, Krab K, Heringa J (2007) Sequence harmony: detecting functional specificity from alignments. Nucleic Acids Res 35: W495–W498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pirovano W, Feenstra KA, Heringa J (2006) Sequence comparison by sequence harmony identifies subtype-specific functional sites. Nucleic Acids Res 34: 6540–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brandt BW, Feenstra KA, Heringa J (2010) Multi-Harmony: detecting functional specificity from sequence alignment. Nucleic Acids Res 38: W35–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, et al. (2010) Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84: 11937–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Battivelli E, Migraine J, Lecossier D, Yeni P, Clavel F, et al. (2011) Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J Virol 85: 11846–11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battivelli E, Lecossier D, Clavel F, Hance AJ (2013) Delaying reverse transcription does not increase sensitivity of HIV-1 to human TRIM5alpha. PLoS ONE 8: : e52434- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crawford H, Matthews PC, Schaefer M, Carlson JM, Leslie A, et al. (2011) The hypervariable HIV-1 capsid protein residues comprise HLA-driven CD8+ T-cell escape mutations and covarying HLA-independent polymorphisms. J Virol 85: 1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kootstra NA, Navis M, Beugeling C, van Dort KA, Schuitemaker H (2007) The presence of the Trim5alpha escape mutation H87Q in the capsid of late stage HIV-1 variants is preceded by a prolonged asymptomatic infection phase. AIDS 21: 2015–2023. [DOI] [PubMed] [Google Scholar]
- 37. van 't Wout AB, Schuitemaker H, Kootstra NA (2008) Isolation and propagation of HIV-1 on peripheral blood mononuclear cells. Nat Protoc 3: 363–370. [DOI] [PubMed] [Google Scholar]
- 38. Tersmette M, Winkel IN, Groenink M, Gruters RA, Spence P, et al. (1989) Detection and subtyping of HIV-1 isolates with a panel of characterized monoclonal antibodies to HIV-p24 gag. Virology 171: 149–155. [DOI] [PubMed] [Google Scholar]
- 39. Boom R, Sol CJA, Salimans MMM, Jansen CL, Wertheim-van Dillen PME, et al. (1991) A rapid and simple method for purification of nucleic acids. J Clin Microbiol 28: 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen C, Okayama H (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7: 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence variation in Gag affects viral replication fitness. A. Replication kinetics of constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 in the absence or presence of compensatory mutations. The area under the curve (day 2–17) was calculated and normalized mean AUCs were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001. Error bars represent 2.5–97.5 percentiles. Data from one representative experiment are shown. B. Replication kinetics of constructed NL4-3.Ba-L viral variants containing mutations associated with HLA-B*57/58:01 in the absence or presence of compensatory mutations described by Brockman et al. The area under the curve (day 2–17) was calculated and normalized mean AUCs were compared using the unpaired Student's T test. Statistical significance compared to the mutant virus carrying the mutations associated with the presence of HLA-B*57/58:01 are denoted in red, and significance compared to the virus carrying all mutations associated with the presence of HLA-B*57/58:01 and with disease progression is shown in blue. Statistical significance is indicated as follows: * p<0.05, ** p<0.01, *** p<0.0001.Error bars represent 2.5 – 97.5 percentiles. Data from one representative experiment are shown.
(TIF)
Amino acid sequence variation in Gag. A. Sequence variation within Gag located at amino acid positions associated with the presence of HLA-B*57/5801 or disease progression as identified with SH in sequences obtained from LTNPs from various time points during the course of HIV-1 infection. B. Sequence variation within Gag at amino acid positions associated with the presence of HLA- B*57/5801 or disease progression as identified with SH in sequences obtained from progressors from various time points during the course of HIV-1 infection.
(DOC)