

Abstract
An intricate relationship exists between gene trees and species phylogenies, due to evolutionary processes that act on the genes within and across the branches of the species phylogeny. From an analytical perspective, gene trees serve as character states for inferring accurate species phylogenies, and species phylogenies serve as a backdrop against which gene trees are contrasted for elucidating evolutionary processes and parameters. In a 1997 paper, Maddison discussed this relationship, reviewed the signatures left by three major evolutionary processes on the gene trees, and surveyed parsimony and likelihood criteria for utilizing these signatures to computationally elucidate this relationship. Here, we review progress that has been made on developing computational methods for analyses under these two criteria, and survey remaining challenges.
Multi-locus analyses and evolutionary processes
Species phylogenies and gene trees have an intricate relationship that stems from the evolutionary processes acting within, and sometimes across, species boundaries to shape the gene trees. Three major evolutionary processes are gene duplication, horizontal gene transfer, and hybridization. Gene duplication results in the creation of new copies of genes and thus plays a central role in genome evolution [1]. As these copies acquire genetic differences, their evolutionary fates might differ and result in novel gene functions [2].
In asexual species, horizontal gene transfer (HGT) shapes the genomic repertoire and imports new genes, sometimes of beneficial consequences, into the host genome [3, 4]. HGT occurs mainly through one of three mechanisms: transformation, which is the uptake of naked DNA from the environment, transduction, which is the transfer of genetic material through a plasmid or bacteriophage, and conjugation, which is the direct transfer of DNA between two cells.
In eukaryotes, the evolutionary histories of various groups of plants and animals have been shown to involve hybridization [5], which is the production of viable offspring from interspecific mating [6]. Two major outcomes of hybridization are introgression and hybrid speciation. While some parts of the genetic material contributed to the offspring in interspecific mating gets eliminated from the population in later generations, other parts are integrated into the genome, an event that is referred to as introgression. It is important to note that both HGT and introgression leave similar genomic signatures, though the former process occurs in asexual species whereas the latter occurs in sexual species. In some cases, hybridization results in hybrid lineages that become reproductively isolated from the parental species, a phenomenon known as hybrid speciation. Figure 1 illustrates gene duplication, HGT, and hybridization in three-taxon scenarios.
Figure 1. Evolutionary processes within and across species boundaries.
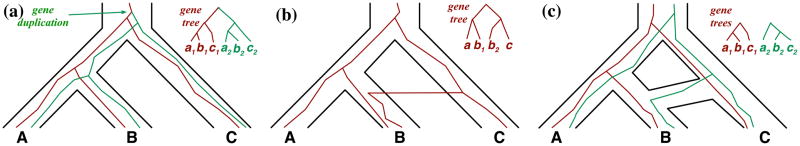
(a) A gene duplication event at the most recent common ancestor (MRCA) of all three taxa, results in two copies (red and green) of a gene within the genome, and as the genome undergoes evolution, these copies evolve, diverge, and might have different fates. (b) In prokaryotic organisms, DNA containing genes might be transferred across species boundaries, e.g., from C to B, resulting in a new gene copy. Further, a similar signature might arise in cases of introgression in sexual species. (c) Hybridization between species A and C amounts to individuals from A and B mating and producing viable offspring such that the genetic material in individuals of B can be traced back to two parental species. The gene tree in each case is shown in the inset.
Two of the main tasks of multi-locus analyses are the inference of a species phylogeny and the evolutionary processes that acted upon the individual loci. While species phylogeny inference used to be conducted almost exclusively based on a single gene sampled across species [7], it is becoming more common to use whole-genome data, or more generally, multiple loci. When gene trees have been inferred for the individual loci, the first task amounts to inferring the species phylogeny from these gene trees. The second task amounts to contrasting, or reconciling, the gene trees with the species phylogeny to elucidate the evolutionary processes that shaped the gene tree and their phenotypic consequences. Multi-locus analyses provide power, in terms of phylogenetic signal, to solve both tasks with high accuracy, yet pose new modeling and computational challenges for phylogenetic inference that mostly stem from a phenomenon known as gene tree incongruence.
Phylogenetic incongruence: A signal, rather than a problem
As illustrated in Figure 1, each of the evolutionary processes operating on a gene leaves its signature on the gene tree. These processes alone do not necessarily result in signatures in the form of incongruence between gene trees and the species phylogeny. It is often the evolutionary fates of gene copies that result in such signatures. These evolutionary fates are determined by forces such as mutation, drift, and selection. For example, in Figure 1(a), if the gene copies b1, c1, and a2 are lost, the resulting gene tree differs from the species tree. In Figure 1(b), if the HGT event results in the displacement of the b1 gene copy, then the resulting gene tree differs from the species tree. On the other hand, if the horizontally transferred gene copy, b2, is eventually lost, then the gene tree remains congruent with the species tree. In the case of hybridization, the scenario is dictated by the mode of the evolutionary process. In homoploid hybridization, the offspring has the same ploidy level, or number of chromosomes, as each of the parents in the two hybridizing species. In this case, hybridization is often followed by back-crossing, which is further mating between individuals from the hybrid population and either of the two parental populations. Repeated back-crossing, combined with drift and selection, results in unequal parental genomic contributions in the hybrid offspring and a distribution of differing gene tree topologies across the genomes. In (allo)polypoloid hybridization, the offspring gets the complete sets of chromosomes from the parents, thus having a number of chromosomes that is double that of either of the two parents. While back-crossing does not occur in cases of polyploid hybridization, drift and selection result in unequal parental genomic contributions in the hybrid offspring.
From an inference perspective, these signatures can then be utilized as phylogenetic signal to recover population parameters, evolutionary processes, and the species phylogeny itself [8]. However, it is important to keep in mind several issues that make the inference task very challenging in practice. First, incomplete sampling of gene copies by the practitioner might give rise to artificial signatures that mislead or confound inference tasks. For example, if the practitioner samples only copes a1, b1, and c1 in the scenario given in Figure 1(a), the occurrence of a gene duplication event might not be recovered. Second, multiple occurrences of the same evolutionary process might cancel out or complicate the signature. For example, assuming displacement of gene copy b1 by the HGT event in the scenario of Figure 1(b), a subsequent HGT event from B to A, involving gene copy b2 and the displacement of the original copy of the gene in A, results in a gene tree that is congruent with the species tree (in terms of topology, but not branch lengths). Third, the occurrence of an evolutionary process might not leave a signature on the gene tree topologies. For example, an HGT between two sister taxa does not result in incongruence between the gene and species trees. Fourth, the signature left by an evolutionary process might not be unique to that process [9]. For example, if gene copies b1, c1, and a2 are lost in the scenarios of Figures 1(a) and 1(c), and the gene copy b1 is lost in the scenario of Figure 1(b), then we end up with the same gene tree topology in all three cases. Further, as we discussed above, HGT and introgression might give rise to identical genomic signatures, though they occur in different groups of species. It is crucial that these issues are kept in mind when applying inference methods, developing new ones, or interpreting the results thereof.
It is probably due to these issues, and others, that several genomic studies that are mainly aimed at obtaining the species phylogeny mask signatures by selecting few loci that satisfy stringent criteria so as to eliminate the possibility of incongruence and other studies have strived to do phylogenetic inference despite incongruence. This review, on the other hand, takes the position that incongruence is a powerful phylogenetic signal that is “desirable, as it often illuminates previously poorly understood evolutionary phenomena” [9]. Fields such as molecular population genetics and phylogenetics have long relied on polymorphism and divergence at the sequence level as signal for inference, and in the post-genomic era, phylogenomics relies on phylogenetic incongruence as the major signal for inference. Therefore, phylogenetic incongruence should not be viewed as a problem to be masked or despite which inference should be made; rather, it should be viewed as a powerful character with a rich set of states to reconstruct and understand evolutionary phenomena, while accounting for the aforementioned issues.
For example, in 1979, Goodman et al. proposed a parsimony-based approach for fitting a gene tree onto a species tree to elucidate gene duplication and loss events from a set of globin sequences [10]. In 1997, Maddison proposed to count the minimum number of branch moves need to convert the species tree into the gene tree, where branch moves do not violate the temporal constraints provided by the trees, as a proxy for the number of HGT or hybridization events [11]. Indeed, if these methods were applied to the scenarios in Figure 1, the true evolutionary events would be uncovered. While these two approaches mainly reveal information about the evolutionary processes themselves, model-based approaches would help elucidate, in addition, knowledge about parameters such as population sizes, divergence times, duplication rates, etc. Further, these reconciliation approaches can be turned into species phylogeny inference approaches by seeking a species phylogeny that, when all gene trees are reconciled with it, achieves some optimality score. In 1997, Maddison surveyed phylogenetic incongruence, and described parsimony and likelihood criteria for various reconciliation and inference problems [11]. Much progress has been made since 1997 on developing mathematical models and computational methods for these problems, and the goal of this review is to revisit the two criteria and provide an update on this progress. We use Maddison’s article as an organizing principle for the remainder of this review.
Phylogenetic incongruence: Maddison’s 1997 survey
In [11], Maddison discussed phylogenetic incongruence and the two computational problems of reconciliation and inference. The reconciliation problem seeks a fitting of a given gene tree within, or across, the branches of a given species tree assuming a source of incongruence. That is, every leaf in the gene tree is mapped to a leaf in the species phylogeny, and then internal nodes (which correspond to events of coalescence, duplication, HGT, etc.) in the gene tree are mapped to the branches of the species phylogeny. In this way, the reconciliation reveals the evolutionary processes that acting on the gene, and when model-based approaches are, the reconciliation also reveals information about the timing of these processes, as well as parameters such as population sizes, duplication rates, etc. The inference problem seeks the species tree, given a collection of loci sampled from a set of species. In traditional phylogenetics, the inference problem amounts to estimating a phylogenetic tree from a molecular sequence alignment, often assuming only base-pair mutations. Analogously, in phylogenetic analyses involving multiple loci, the inference problem amounts to estimating a species phylogeny from a collection of gene trees, assuming some of the evolutionary processes discussed above.
For the reconciliation problem, Maddison discussed parsimony approaches for the cases where gene duplication and loss (DL) are both at play and when HGT is at play. In the case of DL, Goodman et al. had already proposed a parsimony-based approach for fitting a gene tree onto a species tree to minimize the number of duplications [10]. In this approach, a node x in the gene tree is mapped to the most recent common ancestor (MRCA) of the set of species that contain gene copies descended from node x; see Figure 2(a) for an illustration. In the case of HGT, Maddison proposed to count the minimum number of branch moves need to convert the species tree into the gene tree, where branch moves do not violate the temporal constraints provided by the trees. This number would constitute a lower bound on the number of HGT events needed to explain the incongruence between the species tree and gene tree; see Figure 2(b) for an illustration. Given the imbalance in the parental genetic contributions to hybrid offspring, parsimonious detections of hybridization events can be carried out in a similar fashion to that of HGT. In other words, while HGT and hybridization are very different biological processes, their inference under parsimony is very similar, and the same can be said of meiotic recombination. In addition the aforementioned evolutionary processes, Maddison discussed the role that random genetic drift plays in phylogenetic incongruence, a phenomenon we now introduce. Gene trees might disagree with each other, as well as with the species tree, due to random genetic drift acting within the populations, a phenomenon known as incomplete lineage sorting, or ILS [7, 12]; see Figure 3 for an illustration. Unlike gene duplication and loss (DL), HGT, and hybridization, ILS does not introduce new genetic material into genomes; instead, it is a reflection of the inherent stochasticity associated with neutral evolution. Maddison proposed that the same mapping of gene tree nodes to species tree nodes as that employed by [10] would result in a parsimonious reconciliation (one that minimizes the number of “extra” gene lineages) assuming ILS as the source of incongruence. The reconciliations of the gene tree and species tree given in Figure 2 are shown under ILS, DL, and HGT in Figure 4.
Figure 2. Fitting a gene tree onto a species tree.
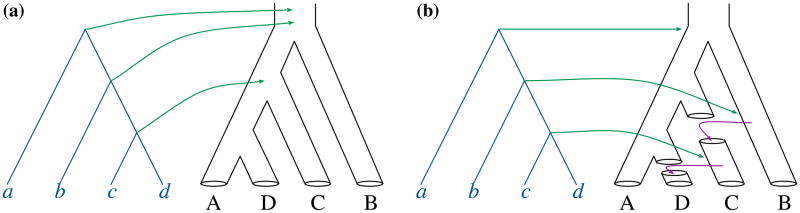
Gene trees are drawn with solid lines, and species trees are drawn with tubes. (a) In the case of DL (and ILS), each node x in the gene tree is mapped to (denoted by the green arrows) the most recent common ancestor (MRCA) of the species that contain gene copies descended from node x. (b) In the cases of HGT and hybridization, a smallest set of branch moves (denoted by the purple arrows) that makes the species tree identical to the gene tree and do not violate “a linear time order” is a parsimonious set of HGT or hybridization events that explain the difference between the species tree and gene tree.
Figure 3. Incomplete lineage sorting.

As the evolution of three sampled alleles (blue solid circles at the bottom) is traced backward in time, alleles from A and B might fail to coalesce in the ancestral population. This results in all three alleles entering the ancestral population of all three species, and the alleles from B and C coalescing first, by chance, giving rise to a gene tree that is incongruent with the species tree. The probability of this event happening in this scenario is a function of the branch length, t, as measured in coalescent units (one coalescent unit equals 2N generations, where N is the population size).
Figure 4. Reconciliation of a gene tree with a species tree.
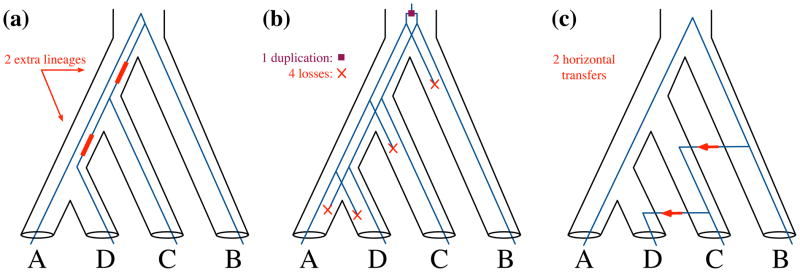
(a) Reconciliation assuming ILS results in two extra lineages, highlighted with thick red lines. (b) Reconciliation assuming DL results in a single duplication event and four losses. (c) Reconciliation assuming HGT (or hybridization) results in two horizontal transfer events, highlighted with red arrows.
These parsimony-based approaches to reconciliation naturally give rise to three parsimony-based criteria for species tree inference: Of all the possible species tree candidates, seek one that minimizes the total number of “extra” gene lineages, duplication events, or HGT events, respectively, when all gene trees in the sample are reconciled with it. Maddison further proposed a maximum likelihood (ML) formulation for the inference problem. However, unlike the case of the parsimony formulations, Maddison considered only deep coalescence (equivalently, ILS) in the case of ML, the reason being that the coalescent theory from population genetics already provided a mechanism for computing the probability of a gene tree, whereas no similar theory existed for computing gene tree probabilities when DL, HGT or hybridization were involved. The ML formulation proposed in [11] assumes a given collection of sequence alignments, each for a sampled locus, and seeks a tree that maximizes the probability of observing these alignments by accounting for mutations within each locus and incongruence across loci. In the next section, I review progress that has been made on Maddison’s proposals for reconciliation and inference under the assumption of an individual evolutionary process being at play, and then discuss in the following section progress that has been made on unification of processes within integrated frameworks.
Progress on methods that deal with individual processes
As Maddison mentioned, a reconciliation of a gene tree with a species tree under ILS or DL, using the mapping described above, is efficiently computable. Several algorithms have been introduced to compute reconciliations under ILS [13] and DL [14]. In terms of mathematical characterizations, Zhang [15] showed that when the species tree and gene tree have exactly the same leaf-set (i.e., exactly one gene copy from each species is used to infer the gene tree), then the number of extra lineages required to reconcile the trees assuming only ILS equals the number of losses minus twice the number of duplications required to reconcile the same trees assuming only DL. For example, in Figure 4, the number of extra lineages in panel (a) is 2, the number of losses and duplications in panel (b) are 4 and 1 respectively, and we have 2 = 4 − (2 · 1). The formula relating the three quantities becomes slightly more involved when the two trees do not necessarily have the same leaf-set [15]. For the inference problem, Maddison and Knowles [16] proposed a heuristic for searching for the species tree that minimizes the number of extra lineages assuming ILS is the sole cause of incongruence. Than and Nakhleh [13] later devised exact algorithms for the problem, including for cases where multiple alleles are sampled [17]. Than and Rosenberg proved that this parsimony criterion of minimizing the number of extra lineages is in fact statistically inconsistent (that is, inference under this criterion might converge on the wrong species tree, even as the number of gene trees used in the inference increases) [18]. Bayzid et al. devised exact algorithms for inferring a species tree that minimizes the number of duplications and losses [19].
As for HGT, the field has evolved rapidly so as to deal with complexities not discussed in [11]. The reconciliation problem assuming HGT is very hard algorithmically [20, 21, 22] and, several methods for reconciling a pair of trees were devised (see [23] for a review); these methods vary in the assumptions and restrictions they make about the trees and reconciliations. Perhaps the issue that challenges Maddison’s original proposal most is the concept of a species tree when HGT, or other reticulate evolutionary events, occur. While a species tree in the case of ILS and DL can fit within its branches the evolutionary histories of all genes within the genomes under consideration, that structure would fail to capture adequately the evolutionary histories of genes that are exchanged horizontally. To accommodate reticulate evolutionary histories, phylogenetic networks were introduced as a model of evolutionary histories that capture both vertical and horizontal descent of genetic material [24, 25, 23, 26, 27]; see Figure 5. A phylogenetic network extends the notion of phylogenetic trees by allowing for nodes with more than one parent—reticulation nodes. Assuming no ILS or DL, the evolutionary history of each gene in a set of species whose evolutionary history is given by a phylogenetic network N is captured by one of the trees displayed (or, induced) by the phylogenetic network N. A tree is induced by phylogenetic network N if it can be obtained by removing all but one of the parents for each of the reticulation nodes in the network. For example, the four trees induced by the network in Figure 5 are (((A,D),C),B), (A,(B,(C,D))), ((A,(C,D)),B), ((A,D),(B,C)). Reconciling a gene tree with a phylogenetic network, excluding ILS and DL, is related to testing whether the gene tree is one of the trees induced by the network, which has been shown to be computationally a very hard problem [28].
Figure 5. Gene trees within the branches of a phylogenetic network.

The phylogenetic network, drawn with tubes, fits the evolutionary histories of all genes, including those that evolve vertically (e.g., the gene tree drawn with green lines) and those that involved horizontal transfer (e.g., the gene tree drawn with blue lines, and HGT or introgression events highlighted with red arrows).
Not only do phylogenetic networks provide a more adequate model than trees for capturing reticulate evolutionary histories, but they also allow extending Maddison’s original proposal from reconciling a pair of trees to a collection of trees. Indeed, in today’s phylogenomic analyses, multiple loci are sequenced and multiple gene trees need to be reconciled. Maddison’s proposal for reconciling a gene tree with a species tree does not carry over cleanly to a set of gene trees for the inference problem. Introducing the notion of a phylogenetic network, the parsimony version of the inference problem under HGT becomes: find a phylogenetic network with the minimum number of reticulation nodes needed to display all of the gene trees. Several methods have been proposed recently for solving versions of this problem [29, 30, 31, 32, 33, 34, 35]. The progress on likelihood approaches for dealing with gene tree incongruence has been much greater for ILS than the other evolutionary processes, owing mainly to the mature theoretical foundations of the coalescent model that deal with ILS naturally. As we discussed above, the maximum likelihood formulation given in [11] was proposed in the context of ILS alone. Based on that formulation, the likelihood of a species tree is
While Maddison used the summation over gene trees, this is to be treated as integration when branch lengths of the gene trees are also considered. The first probability of observing a set of (aligned) sequences given a gene tree depends on the model of sequence evolution and can be computed efficiently [36]. The second probability of observing a gene tree given a species tree is derived from coalescent theory, and methods have been devised for computing it when the gene tree is given only by its topology [37] and when the gene tree is given by its topology and branch lengths [38]. Likelihood methods have been proposed for inference based on this formulation [39, 40]. Advances have been made recently on methods for computing the second probability when only DL is at play was given in [41, 42, 43]. When only reticulation is at play and a parameterized species phylogenetic network is provided, computing the probability of a gene tree is straightforward [23].
Unifying processes and accounting for error
The fact that much progress has been made on methods that deal with each of the evolutionary processes individually is not to be construed as a statement that these processes do indeed occur in a mutually exclusive manner. As phylogenomic analyses grow in scope to involve more species, individuals, and loci, accounting simultaneously for multiple evolutionary processes becomes essential. Indeed, several studies have highlighted this issue in various groups of organisms. For example, while introgression was hypothesized between Neanderthals and humans [44], this hypothesis was later dismissed in favor of ILS [45]. Simultaneous patterns of introgression and ILS were reported in 2012 in the house mouse (Mus musculus) genome [46], in the butterfly (Heliconius melpomene) genome [47], in sunflower (Helianthus) genomes [48], and in yeast genomes [49]. Simultaneous patterns of ILS and DL were recently reported in a multi-locus analysis of a group of fungi [50]. Further, simultaneous patterns of DL and reticulation have been reported [51]. Maddison [11] pointed to two challenges facing the development of a “mixed method” that allows all three processes to occur: the algorithmic challenge of conducting reconciliation and inference under multiple processes, and the challenge associated with weighting the three different processes (e.g., should one HGT event be counted as equal to one duplication event?). While the weighting relates mostly to parsimony approaches, its counterpart in a likelihood approach is setting the rates of the various processes (e.g., the rates of duplication, loss, etc.).
As we discussed above, a phylogenetic network provides a more appropriate model of evolutionary relationships than trees when reticulation is involved. It is important to note that a phylogenetic network not only accommodates HGT and hybridization, but treelike evolutionary processes, such as ILS and DL, can be modeled within its branches. For example, Figure 6 illustrates how a phylogenetic network simultaneously models hybridization between species and ILS involving gene trees. It further illustrates the generality of the model in terms of accommodating multiple individuals sampled per species or population. Therefore, while Maddison did not discuss phylogenetic networks in his original survey, this review takes the position that for unification of all evolutionary processes, a species phylogeny in the form of a network is more appropriate than a tree. In fact, when recombination occurs within a locus, even the gene tree is better modeled using a network that is often referred to in the population genetics literature as an ancestral recombination graph [8]. This position is not to be interpreted as invoking reticulation in every analysis; rather, it is advocates the development of mathematical models and computational methods that utilize the more general model, which is a network rather than a tree, and account for the possibility that in some, or maybe most, cases the inferred network could be a special case that is a tree. The other approach of utilizing a tree as the topological model would exclude the possibility of a reticulate evolutionary history, merely by definition of the model used. Progress on parsimonious reconciliation and inference methods that assume more than a single source of incongruence has been made. Bansal et al. [52] recently introduced an efficient algorithm for reconciling a gene tree with a species tree assuming both DL and HGT. Yu et al. [53, 54] introduced methods that assume both hybridization and ILS. In particular, the work in [54] provides algorithms for reconciliation as well as search heuristics that explore the space of phylogenetic networks to solve the inference problem. Recently, Stolzer et al. introduced a method for reconciling a gene tree with a species tree under DL, HGT, and ILS [55]. While a natural way for integrating all evolutionary processes within a parsimony framework is to optimize a weighted sum of the numbers of events detected, a likelihood approach requires probabilistic models of these processes. While the coalescent model has provided a natural framework for thinking about ILS, recent studies are beginning to shed light on how to probabilistically model processes including HGT [56, 57, 58] and DL [59, 2, 43]. For integrative likelihood approaches, a method for computing the probability of a gene tree given a species tree assuming both ILS and DL was given in [50], assuming DL and HGT was given in [60], and assuming ILS and HGT in a special case was given in [61]. Methods have been developed for computing the probabilities of gene trees under hybridization and ILS in special, limited cases [62, 63, 53, 64], and then for computing the probabilities in general cases [49]. Marcussen et al. [65] recently developed a method for inferring phylogenetic networks in the presence of ILS that is aimed at modeling polyploid hybridization. A salient feature of all phylogenetic analyses, whether they involve a single locus or multiple loci, is the fact that gene trees are estimated from molecular sequences and, consequently, they are likely to be inaccurate. Maddison [11] wrote: “I assume through most of this discussion that the true gene trees are known without error. Of course, there will be errors in practice, and these errors will mean that reconstructed gene trees and species trees will have additional sources of discord.” Indeed, Hahn [66] recently showed the effect of error in gene tree estimates on the computed reconciliations and Yang and Warnow [67] showed that methods that explicitly account for error in the gene trees outperform others. While incongruence caused by evolutionary processes provides a signal for inferring the processes themselves and the species phylogeny, incongruence due to error in the inferred gene trees is a confounding factor that must be accounted for carefully, as it produces topological signatures in the gene trees that can cancel out true evolutionary signals or masquerade as ones. One way to deal with error in the estimates of gene tree topologies is to contract all branches with low support (e.g., as measured by a bootstrap analysis, or posterior probabilities from a Bayesian analysis), and develop methods that can handle non-binary, or multifurcating, trees. In the parsimony setup, a natural way to define the reconciliation of a non-binary gene tree with a species tree is to find the refinement of the polytomies in the gene tree that results in the most parsimonious reconciliation over all possible reconciliations (Figure 7). Indeed, this refinement concept was used in [68, 69, 70] for reconciling non-binary gene and species trees. While the number of refinements is exponential in the degrees of the polytomies (the number of children of a node), Yu et al. recently devised polynomial-time, exact algorithms for finding the refinement that results in an optimal reconciliation under ILS [71, 72]. Further, the same ideas were extended to the problem of parsimonious reconciliation of a non-binary gene tree with a phylogenetic network [54]. Under the likelihood approach, it is less clear how to deal with non-binary gene trees. Should the gene tree be refined in a way that maximizes the probability of observing the (binary) gene tree (e.g., as implemented for inference under ILS in [73])? Or, should the probability of the non-binary gene tree be computed as the average probability of all binary refinements? A different method to handle error in the gene trees is to directly make use of the support values. The major challenge facing this approach is in translating support values from gene tree branches to support values of reconciliations. Nonetheless, some heuristics were introduced recently based on this approach for reconciliation under HGT [74, 75]. Further, Yu et al. incorporated posterior probabilities in methods for reconciling a gene tree with a phylogenetic network under both likelihood [49] and parsimony [54]. Of course, methods that work directly from the sequence alignments of the multiple loci, rather than from estimated gene trees, account implicitly for error. The Bayesian methods of [76, 77] for inference under ILS, and the parsimony and likelihood methods of [78, 79] for inferring HGT events follow this approach.
Figure 6. Simultaneous modeling of hybridization and ILS with a phylogenetic networks.

Two individuals are sampled per species, and there is a hybridization event that involves species B and C. Further, ILS patterns complicate the gene genealogy, giving rise to the gene-tree topology shown in the inset. For example, the gene copies in green coalesce with the ancestral copy of the genes in red from A and B, before the latter one coalesces with the copy from C.
Figure 7. Parsimonious reconciliation of a non-binary gene tree.
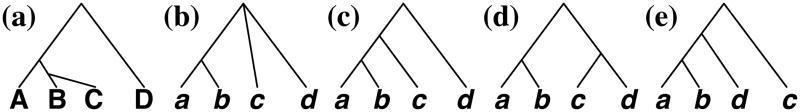
(a) A binary species tree. (b) A non-binary gene tree. (c–e) The three possible refinements of the non-binary gene tree. Under parsimony, the refinement in (c) results in the best reconciliation with the species tree, as it results, assuming ILS, DL, or HGT, in 1 extra lineage, 1 duplication and 3 losses, and 1 HGT, respectively.
Other approaches
Several approaches that do not fit within Maddison’s parsimony and likelihood formulations have been proposed. Concatenation is an approach in which the sequences from multiple loci are concatenated, thus resulting in a “super gene,” and a phylogenetic tree is inferred from the super gene. This approach was used for example in inferring a phylogenetic tree of a set of yeast species [80]. There are at least three issues with this approach. First, the approach is applicable to loci for which exactly one copy per species is sampled. However, even then, the phylogeny estimated from the concatenated alignment might be wrong [81]. Second, this approach yields, by definition, a phylogenetic tree. Therefore, it masks any signal of reticulate evolution if it exists. Third, the method does not allow for inference of the evolutionary processes. The democratic vote approach amounts to taking the gene tree with the highest frequency as a proxy for the species tree [82]. Applicability of this approach when a small sample of loci is used or when duplication and loss events are involved is questionable. Even when a large number of loci is used and each has exactly a single copy sampled per species, this method produces a misleading phylogeny in the “anomaly zone” [7]. Finally, it is not clear how to interpret the gene tree with the highest frequency when the species evolutionary history is reticulate. The majority-rule consensus is a third approach to produce a phylogenetic tree given a set of conflicting gene tree topologies. This approach often results in a phylogenetic tree with a very low degree of resolution. Further, the approach is not well defined for cases where the incongruence is due to DL. Third, like the previous two approaches, this approach always produces a tree, even when the evolutionary history is reticulate.
Mossel and Roch [83] recently introduced a distance-based method for inferring a species tree from pairwise distances computed from multiple loci. This method requires accurate estimates of the distances, and is applicable to neither DL nor reticulate evolutionary events. Bayesian approaches for inferring species trees under ILS were also recently introduced [84, 77]. These methods differ from all the methods discussed above in that they work directly with sequence alignments and perform simultaneous inference of gene and species trees. They have been shown to produce very good results, yet to be inefficient computationally. Further, these Bayesian approaches currently do not handle DL or reticulate evolutionary events. Methods for inferring HGT events based on an assumed species tree and sequence alignments of genes were proposed based on the maximum parsimony and maximum likelihood criteria [23]. These methods do not account for ILS or DL, and assume knowledge of an underlying species tree. Joly et al. showed how to use coalescent-based simulations to detect hybridization [85]; however, their approach was presented for a pair of species only. Last but not least, Holland et al. [86, 85] demonstrated how to use consensus networks to detect hybridization in the presence of ILS.
Summary and future directions
In 1997, Wayne Maddison discussed the intricate relationship between a species tree and the gene trees that grow with, and across, its branches. Further, he discussed parsimony and likelihood approaches to reconciling a gene tree with a species tree, and for the inference of species trees from collections of gene trees. Solving these two tasks would shed light on central issues in evolutionary and molecular biology, including speciation, evolutionary processes acting within and across population, the evolution of morphological characters, and genotype-phenotype relationships. Sixteen years later, the significance of understanding this relationship cannot be overstated, given the ability to sequence hundreds of prokaryotic genomes in a day, and eukaryotic genomes in slightly longer timeframes. Indeed, in less than two decades, the computational biology and bioinformatics communities responded to Maddison’s proposal by making significant inroads in establishing mathematical results and devising computational methods for detecting, resolving, and ameliorating incongruence that arise in phylogenomic studies. Still, much more is needed in terms of mathematical and computational developments. While models of incongruence and methods for reconciliation and inference have been developed, computational requirements are still a major bottleneck. Most, if not all, of the methods described above are limited to small- or medium-sized data sets. High-performance computing approaches will definitely be needed if it is desired that these methods apply to thousands of loci and hundreds to thousands of taxa. Currently, these data sets are beyond the capabilities of existing tools. Maddison explicitly stated in [11] that his formulations assumed no recombination within a locus. But what happens if this assumption is violated? Recent work has accounted for recombination within phylogenetic networks [87]. A recent study showed ignoring recombination within loci might not have a significant effect on the quality of the inferred species tree under ILS [88]. Similar studies do not exist for cases of DL and HGT. Nevertheless, more generally, the availability of whole-genome data allows for defining gene trees as the genealogies built from non-recombining regions, which include coding and non-coding DNA. However, potentially more challenging than recombination are the findings of rearrangements at the sub-level, such as gene fission and fusion, which seem to be ubiquitous in prokaryotic genomes [89] and even in eukaryotic genomes [90]. These findings not only complicate the species-gene evolutionary relationships, but also raise broader questions about orthology, gene families, and the “cloudiness” [11] of the species phylogeny.
Further, Maddison assumed the loci are unlinked and, hence, the fact that gene trees can be reconciled with a species phylogeny independently. Indeed, all methods described above assume unlinked loci and it is currently incumbent upon the practitioner to sample loci from the genomes in such a way that ensures this assumption holds (or that violation thereof is minimal). However, to make full use of whole-genome data, models that incorporate linkage across loci, including functional linkage [91], must be devised, and methods for inference under such models much be developed. A mathematical model for two linked loci was introduced in [92]. More recently, approaches for modeling ILS while accounting for linkage across loci were introduced [93, 94]. These approaches do not account for DL or reticulation at the species level (they do account for recombination). Further, these methods have been applied to three-species data sets and it would be challenging to achieve scalability of these methods to large data sets. This further emphasizes the need for high-performance computing approaches, as modeling dependence only makes the problem harder. While many methods have been developed for reconciliation, the relative performance of these methods is yet to be investigated thoroughly. This is especially important as the practitioner is faced with a wide array of methods that differ in terms of the assumptions they make and the computational resources they need. While studies are beginning to emerge [95, 67, 96, 88, 97], more comprehensive studies are still needed. In particular, most performance studies focus on ILS, probably due to the fact that the coalescent theory provides a clean generative model for simulating synthetic data, whereas no such theory exists for DL or HGT. Further, while some methods perform poorly under certain conditions, they might perform well under other conditions. Full characterization of conditions under which a method performs well would be of utmost help to practitioners. Most importantly, measures that reflect these characterizations from real data are needed. For example, the anomaly zone has been established for several methods [7, 18]. However, the question that the practitioners face is: Do their data fall within an anomaly zone for a specific method?
Last but not least, when the evolutionary history is reticulate, it is more appropriate to speak of a phylogenetic, or species, network, rather than a species tree. In the population genetics literature, this issue has long been recognized, and ancestral recombination graphs—a class of phylogenetic networks—have been adopted for modeling genealogies that include recombination [98]. Using phylogenetic networks, and more generally, networks, might help uncover hypotheses that would be undetected otherwise [99]. The different flavors in which phylogenetic networks come might have been confusing to the community of practitioners and, consequently, limited their applicability. Recent monograms have been written to clarify the similarities, differences, and applications of the various types of networks [23, 26, 27]. Developments to address the issues above should be applicable to phylogenetic networks.
Highlights.
Evolutionary processes acting on genes within and across the branches of a species phylogeny leave their signatures on the gene tree.
These signatures can be utilized to elucidate information about the processes and infer accurate phylogenetic relationships.
W. Maddison had reviewed the three major evolutionary events that cause incongruence and proposed approaches for dealing with them.
We review progress on Maddison’s proposal.
Even though very good progress has been made, much is still needed.
Acknowledgments
The author would like to acknowledge the three anonymous reviewers and Dr. Paul Craze for extensive and thorough comments on the first revision of this manuscript which helped improve it significantly in terms of contents and organization. Further, the author acknowledges Mr. Siavash Mirarab, and Drs. Noah Roseberg and Tandy Warnow for comments on the first revision. This work was supported in part by NSF grants DBI-1062463 and CCF-130217, grant R01LM009494 from the National Library of Medicine, an Alfred P. Sloan Research Fellowship, and a Guggenheim Fellowship to L.N. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NSF, National Library of Medicine, the National Institutes of Health, the Alfred P. Sloan Foundation, or the John Simon Guggenheim Memorial Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dittmar K, Liberles D, editors. Evolution after Gene Duplication. Wiley-Blackwell; Hoboken, New Jersey: 2010. [Google Scholar]
- 2.Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nature Reviews Genetics. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 3.Lerat E, et al. Evolutionary origins of genomic repertoires in bacteria. PLoS Biology. 2005;3:0807–0814. doi: 10.1371/journal.pbio.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boto L. Horizontal gene transfer in evolution: facts and challenges. Proceedings of the Royal Society B. 2010;277:819–827. doi: 10.1098/rspb.2009.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbott R, Rieseberg L. eLS. 2012. Hybrid speciation. [Google Scholar]
- 6.Baack E, Rieseberg L. A genomic view of introgression and hybrid speciation. Current Opinion in Genetics and Development. 2007;17:513–518. doi: 10.1016/j.gde.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degnan J, Rosenberg N. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology and Evolution. 2009;24:332–340. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Siepel A. Phylogenomics of primates and their ancestral populations. Genome Research. 2009;19:1929–1941. doi: 10.1101/gr.084228.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendel JF, Doyle JJ. Phylogenetic incongruence: window into genome history and molecular evolution. In: Soltis D, Soltis P, Doyle J, editors. Molecular Systematics of Plants II. Springer; 1998. pp. 265–296. [Google Scholar]
- 10.Goodman M, et al., editors. Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Syst Zool. 1979;28:132–163. [Google Scholar]
- 11.Maddison W. Gene trees in species trees. Systematic Biology. 1997;46:523–536. [Google Scholar]
- 12.Knowles L, Kubatko L, editors. Estimating species trees: Practical and theoretical aspects. Wiley-Blackwell; Hoboken, New Jersey: 2010. [Google Scholar]
- 13.Than C, Nakhleh L. Species tree inference by minimizing deep coalescences. PLoS Computational Biology. 2009;5:e1000501. doi: 10.1371/journal.pcbi.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eulenstein O, et al. Reconciling phylogenetic trees. In: Dittmar K, Liberles D, editors. Evolution after gene duplication. Wiley-Blackwell; Hoboken, New Jersey: 2010. pp. 185–206. [Google Scholar]
- 15.Zhang L. From gene trees to species trees II: Species tree inference by minimizing deep coalescent events. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2011;8:1685–1691. doi: 10.1109/TCBB.2011.83. [DOI] [PubMed] [Google Scholar]
- 16.Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Systematic Biology. 2006;55:21–30. doi: 10.1080/10635150500354928. [DOI] [PubMed] [Google Scholar]
- 17.Than C, Nakhleh L. Inference of parsimonious species phylogenies from multi-locus data by minimizing deep coalescences. In: Knowles L, Kubatko L, editors. Estimating Species Trees: Practical and Theoretical Aspects. Wiley-VCH; 2010. pp. 79–98. [Google Scholar]
- 18.Than CV, Rosenberg NA. Consistency properties of species tree inference by minimizing deep coalescences. Journal of Computational Biology. 2011;17:1–15. doi: 10.1089/cmb.2010.0102. [DOI] [PubMed] [Google Scholar]
- 19.Bayzid M, et al. Inferring optimal species trees under gene duplication and loss. Pacific Symposium on Biocomputing. 2013;18:250–261. doi: 10.1142/9789814447973_0025. [DOI] [PubMed] [Google Scholar]
- 20.Bordewich M, Semple C. On the computational complexity of the rooted subtree prune and regraft distance. Annals of Combinatorics. 2004;8:409–423. [Google Scholar]
- 21.Bordewich M, Semple C. Computing the minimum number of hybridization events for a consistent evolutionary history. Discrete Applied Mathematics. 2007;155:914–928. [Google Scholar]
- 22.Humphries P, et al. On the complexity of computing the temporal hybridization number for two phylogenies. Discrete Applied Mathematics. 2013;161:871–880. [Google Scholar]
- 23.Nakhleh L. Evolutionary phylogenetic networks: models and issues. In: Heath L, Ramakrishnan N, editors. The Problem Solving Handbook for Computational Biology and Bioinformatics. Springer; New York: 2010. pp. 125–158. [Google Scholar]
- 24.Morrison DDA. Networks in phylogenetic analysis: new tools for population biology. International Journal of Parasitology. 2005;35:567–582. doi: 10.1016/j.ijpara.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Huson D, Bryant D. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 26.Huson D, et al. Phylogenetic networks: concepts, algorithms, and applications. Cambridge University Press; New York: 2011. [Google Scholar]
- 27.Morrison D. Introduction to phylogenetic networks. RJR Productions; 2011. [Google Scholar]
- 28.Kanj I, et al. Seeing the trees and their branches in the network is hard. Theoretical Computer Science. 2008;401:153–164. [Google Scholar]
- 29.Huson D, Rupp R. Summarizing multiple gene trees using cluster networks. In: Crandall K, Lagergren J, editors. Proceedings of the Workshop on Algorithms in Bioinformatics, vol. 5251 of Lecture Notes in Bioinformatics. 2008. pp. 296–305. [Google Scholar]
- 30.Beiko R, Ragan M. Untangling hybrid phylogenetic signals: Horizontal gene transfer and artifacts of phylogenetic reconstruction. Methods Mol Biol. 2009;532:241–256. doi: 10.1007/978-1-60327-853-9_14. [DOI] [PubMed] [Google Scholar]
- 31.van Iersel L, et al. Phylogenetic networks do not need to be complex: using fewer reticulations to represent conflicting clusters. Bioinformatics. 2010;26:i124–i131. doi: 10.1093/bioinformatics/btq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y. Close lower and upper bounds for the minimum reticulate network of multiple phylogenetic trees. Bioinformatics. 2010;26:140–148. doi: 10.1093/bioinformatics/btq198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park H, et al. Algorithmic strategies for estimating the amount of reticulation from a collection of gene trees. Proceedings of the Ninth Annual International Conference on Computational Systems Biology. 2010:114–123. [Google Scholar]
- 34.Park H, Nakhleh L. MURPAR: A fast heuristic for inferring parsimonious phylogenetic networks from multiple gene trees. Proceedings of the International Symposium on Bioinformatics Research and Applications (ISBRA 12), vol. 7292 of Lecture Notes in Bioinformatics; 2012. pp. 213–224. [Google Scholar]
- 35.Wu Y. An algorithm for constructing parsimonious hybridization networks with multiple phylogenetic trees. The 17th Annual International Conference on Research in Computational Molecular Biology (RECOMB), vol. 7821 of LNCS; 2013. pp. 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. Journal of Molecular Evolution. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 37.Degnan JH, Salter LA. Gene tree distributions under the coalescent process. Evolution. 2005;59:24–37. [PubMed] [Google Scholar]
- 38.Rannala B, Yang Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubatko L, et al. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics. 2009;25:971–973. doi: 10.1093/bioinformatics/btp079. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y. Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood. Evolution. 2012;66:763–775. doi: 10.1111/j.1558-5646.2011.01476.x. [DOI] [PubMed] [Google Scholar]
- 41.Akerborg O, et al. Simultaneous Bayesian gene tree reconstruction and reconciliation analysis. Proceedings of the National Academy of Sciences. 2009;106:5714–5719. doi: 10.1073/pnas.0806251106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Górecki P, et al. Maximum likelihood models and algorithms for gene tree evolution with duplications and losses. BMC Bioinformatics. 2011;12:S15. doi: 10.1186/1471-2105-12-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konrad A, et al. Toward a general model for the evolutionary dynamics of gene duplicates. Genome biology and evolution. 2011;3:1197. doi: 10.1093/gbe/evr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green RE, et al. A draft sequence of the neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eriksson A, Manica A. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominids. Proceedings of the National Academy of Sciences. 2012;109:13956–13960. doi: 10.1073/pnas.1200567109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staubach F, et al. Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus) PLoS Genetics. 2012;8:e1002891. doi: 10.1371/journal.pgen.1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Consortium THG. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moody M, Rieseberg L. Sorting through the chaff, nDNA gene trees for phylogenetic inference and hybrid identification of annual sunflowers (Helianthus sect Helianthus) Molecular Phylogenetics And Evolution. 2012;64:145–155. doi: 10.1016/j.ympev.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 49.Yu Y, et al. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genetics. 2012;8:e1002660. doi: 10.1371/journal.pgen.1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasmussen M, Kellis M. Unified modeling of gene duplication, loss, and coalescence using a locus tree. Genome Research. 2012;22:755–765. doi: 10.1101/gr.123901.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamneva OK, et al. Analysis of genome content evolution in PVC bacterial super-phylum: Assessment of candidate genes associated with cellular organization and lifestyle. Genome biology and evolution. 2012;4:1375–1390. doi: 10.1093/gbe/evs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bansal M, et al. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics. 2012;28:i283–i291. doi: 10.1093/bioinformatics/bts225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Y, et al. Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Systematic Biology. 2011;60:138–149. doi: 10.1093/sysbio/syq084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu Y, et al. Parsimonious inference of hybridization in the presence of incomplete lineage sorting. Systematic Biology To appear. 2013 doi: 10.1093/sysbio/syt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stolzer M, et al. Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics. 2012;28:i409–i415. doi: 10.1093/bioinformatics/bts386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain R, et al. Horizontal gene transfer accelerates genome innovation and evolution. Molecular Biology and Evolution. 2003;20:1598–1602. doi: 10.1093/molbev/msg154. [DOI] [PubMed] [Google Scholar]
- 57.Cohen O, et al. The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to horizontal gene transfer. Molecular biology and evolution. 2011;28:1481–1489. doi: 10.1093/molbev/msq333. [DOI] [PubMed] [Google Scholar]
- 58.Stiller JW. Experimental design and statistical rigor in phylogenomics of horizontal and endosymbiotic gene transfer. BMC evolutionary biology. 2011;11:259. doi: 10.1186/1471-2148-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes T, Liberles DA. The power-law distribution of gene family size is driven by the pseudogenisation rate’s heterogeneity between gene families. Gene. 2008;414:85–94. doi: 10.1016/j.gene.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 60.Sjöstrand J, et al. DLRS: gene tree evolution in light of a species tree. Bioinformatics. 2012;28:2994–2995. doi: 10.1093/bioinformatics/bts548. [DOI] [PubMed] [Google Scholar]
- 61.Than C, et al. Confounding factors in HGT detection: statistical error, coalescent effects, and multiple solutions. J Comput Biol. 2007;14:517–535. doi: 10.1089/cmb.2007.A010. [DOI] [PubMed] [Google Scholar]
- 62.Meng C, Kubatko LS. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: A model. Theoretical Population Biology. 2009;75:35–45. doi: 10.1016/j.tpb.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 63.Kubatko LS. Identifying hybridization events in the presence of coalescence via model selection. Systematic Biology. 2009;58:478–488. doi: 10.1093/sysbio/syp055. [DOI] [PubMed] [Google Scholar]
- 64.Jones G, et al. Statistical inference of allopolyploid species networks in the presence of incomplete lineage sorting. Systematic Biology. 2013;62:467–78. doi: 10.1093/sysbio/syt012. [DOI] [PubMed] [Google Scholar]
- 65.Marcussen T, et al. Inferring species networks from gene trees in high-polyploid north american and hawaiian violets (viola, violaceae) Systematic biology. 2012;61:107–126. doi: 10.1093/sysbio/syr096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hahn M. Bias in phylogenetic tree reconciliation methods: implications for vertebrate genome evolution. Genome Biology. 2007;8:R141. doi: 10.1186/gb-2007-8-7-r141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang J, Warnow T. Fast and accurate methods for phylogenomic analyses. BMC Bioinformatics. 2011;12:S4. doi: 10.1186/1471-2105-12-S9-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berglund-Sonnhammer AC, et al. Optimal gene trees from sequences and species trees using a soft interpretation of parsimony. Journal of Molecular Evolution. 2006;63:240–250. doi: 10.1007/s00239-005-0096-1. [DOI] [PubMed] [Google Scholar]
- 69.Durand D, et al. A hybrid micro-macroevolutionary approach to gene tree reconstruction. Journal of Computational Biology. 2006;13:320–335. doi: 10.1089/cmb.2006.13.320. [DOI] [PubMed] [Google Scholar]
- 70.Than C, Nakhleh L. SPR-based tree reconciliation: Non-binary trees and multiple solutions. Proceedings of the Sixth Asia Pacific Bioinformatics Conference. 2008:251–260. [Google Scholar]
- 71.Yu Y, et al. Algorithms for MDC-based multi-locus phylogeny inference. Lecture Notes in Bioinformatics; The 15th Annual International Conference on Research in Computational Molecular Biology (RECOMB), vol. 6577 of LNBI; 2011. pp. 531–545. [Google Scholar]
- 72.Yu Y, et al. Algorithms for MDC-based multi-locus phylogeny inference: Beyond rooted binary gene trees on single alleles. Journal of Computational Biology. 2011;18:1543–1559. doi: 10.1089/cmb.2011.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Than C, et al. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics. 2008;9:322. doi: 10.1186/1471-2105-9-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Than C, et al. Integrating sequence and topology for efficient and accurate detection of horizontal gene transfer. Proceedings of the Sixth RECOMB Comparative Genomics Satellite Workshop, vol. 5267 of LNBI; 2008. pp. 113–127. [Google Scholar]
- 75.Park H, et al. Bootstrap-based support of HGT inferred by maximum parsimony. BMC Evolutionary Biology. 2010;10:131. doi: 10.1186/1471-2148-10-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ané C, et al. Bayesian estimation of concordance among gene trees. Molecular Biology and Evolution. 2007;24:412–426. doi: 10.1093/molbev/msl170. [DOI] [PubMed] [Google Scholar]
- 77.Heled J, Drummond A. Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution. 2010;27:570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin G, et al. Inferring phylogenetic networks by the maximum parsimony criterion: a case study. Molecular Biology and Evolution. 2007;24:324–337. doi: 10.1093/molbev/msl163. [DOI] [PubMed] [Google Scholar]
- 79.Jin G, et al. Maximum likelihood of phylogenetic networks. Bioinformatics. 2006;22:2604–2611. doi: 10.1093/bioinformatics/btl452. [DOI] [PubMed] [Google Scholar]
- 80.Rokas A, et al. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- 81.Kubatko L, Degnan J. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Systematic Bioloyg. 2007;56:17–24. doi: 10.1080/10635150601146041. [DOI] [PubMed] [Google Scholar]
- 82.Wu CI. Inferences of species phylogeny in relation to segregation of ancient polymorphisms. Genetics. 1991;127:429–435. doi: 10.1093/genetics/127.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mossel E, Roch S. Incomplete lineage sorting: Consistent phylogeny estimation from multiple loci. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2010;7:166–171. doi: 10.1109/TCBB.2008.66. [DOI] [PubMed] [Google Scholar]
- 84.Liu L, Pearl DK. Species trees from gene trees: Reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Systematic Biology. 2007;56:504–514. doi: 10.1080/10635150701429982. [DOI] [PubMed] [Google Scholar]
- 85.Joly S, et al. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am Nat. 2009;174:E54–E70. doi: 10.1086/600082. [DOI] [PubMed] [Google Scholar]
- 86.Holland B, et al. Using supernetworks to distinguish hybridization from lineage-sorting. BMC Evol Biol. 2008;8:202. doi: 10.1186/1471-2148-8-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arenas M, et al. Characterization of reticulate networks based on the coalescent with recombination. Molecular Biology and Evolution. 2008;25:2517–2520. doi: 10.1093/molbev/msn219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lanier H, Knowles L. Is recombination a problem for species-tree analyses? Systematic Biology. 2012;61:691–701. doi: 10.1093/sysbio/syr128. [DOI] [PubMed] [Google Scholar]
- 89.Bapteste E, et al. Evolutionary analyses of non-genealogical bonds produced by introgressive descent. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18266–18272. doi: 10.1073/pnas.1206541109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu YC, et al. Evolution at the subgene level: domain rearrangements in the Drosophila phylogeny. Molecular Biology and Evolution. 2012;29:689–705. doi: 10.1093/molbev/msr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Freeling M, Thomas BC. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome research. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- 92.Slatkin M, Pollack JL. The concordance of gene trees and species trees at two linked loci. Genetics. 2006;172:1979–1984. doi: 10.1534/genetics.105.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hobolth A, et al. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS genetics. 2007;3:e7. doi: 10.1371/journal.pgen.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dutheil JY, et al. Ancestral population genomics: the coalescent hidden Markov model approach. Genetics. 2009;183:259–274. doi: 10.1534/genetics.109.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang H, et al. Sources of error inherent in species-tree estimation: impact of mutational and coalescent effects on accuracy and implications for choosing among different methods. Systematic Biology. 2010;59:573–583. doi: 10.1093/sysbio/syq047. [DOI] [PubMed] [Google Scholar]
- 96.Chung Y, Ané C. Comparing two Bayesian methods for gene tree / species tree reconstruction: A simulation with incomplete lineage sorting and horizontal gene transfer. Systematic Biology. 2011;60:261–275. doi: 10.1093/sysbio/syr003. [DOI] [PubMed] [Google Scholar]
- 97.Knowles L, et al. Full modeling versus summarizing gene-tree uncertainty: method choice and species-tree accuracy. Molecular Phylogenetics and Evolution. 2012;65:501–509. doi: 10.1016/j.ympev.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 98.Griffiths R, Marjoram P. Ancestral inference from samples of DNA sequences with recombination. Journal of Computational Biology. 1996;3:479–502. doi: 10.1089/cmb.1996.3.479. [DOI] [PubMed] [Google Scholar]
- 99.Bapteste E, et al. Networks: Expanding evolutionary thinking. Trends in Genetics. 2013;29:439–441. doi: 10.1016/j.tig.2013.05.007. [DOI] [PubMed] [Google Scholar]