

Abstract
Multiple Sclerosis (MS) is an incurable central nervous system (CNS) demyelinating disease affecting several million people worldwide. Due to the multifactorial and complex pathology of MS, FDA approved drugs often show limited efficacy inpatients. We earlier documented that both lovastatin (cholesterol lowering drug) and metformin (anti-diabetic drug) attenuate experimental autoimmune encephalomyelitis (EAE), a widely used model of MS via different mechanisms of action. Since combination therapy of two or more agents has advantage over monotherapy, we here assessed the therapeutic efficacy of metformin and lovastatin combination in EAE. We found that suboptimal doses of these drugs in combination had additive effect to attenuate established EAE in treated animals than their individual treatments. Histological, immunohistochemistry and western blotting analyses revealed that the observed demyelination and axonal loss as evident from reduced levels of myelin and neurofilament proteins in the spinal cords of EAE animals were attenuated by treatment with these drugs in combination. Accordingly, the observed infiltration of myelin reactive T cells (CD4 and CD8) and macrophages (CD68) as well as the increased expression of their signatory cytokines in the spinal cords of EAE animals were attenuated by this regimen as revealed by enzyme-linked immune-sorbent assay and real-time PCR analyses. In the periphery, this regimen biased the class of elicited anti-myelin basic protein immunoglobulins from IgG2a to IgG1 and IgG2b, suggesting a Th1 to Th2 shift which was further supported by the increased expression of their signatory cytokines in EAE animals. Taken together, these data imply that metformin and lovastatin combination attenuates T-cell autoimmunity and neurodegeneration in treated EAE animals thereby suggesting that the oral administration of these FDA approved drugs in combination has potential to limit MS pathogenesis.
Keywords: Multiple sclerosis, Experimental autoimmune encephalomyelitis, Lovastatin, Metformin, T-cell autoimmunity, Neuroprotection
Introduction
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the central nervous system (CNS) that affects 2.5 million people worldwide [1]. Experimental autoimmune encephalomyelitis (EAE) is the murine model of MS which can be induced by immunization of animals with myelin basic proteins [2]. The overall pathology of EAE/MS is the activation of myelin reactive CD4+ Th1 and Th17 cells including CD8+ T cells in the periphery and that cross the blood brain barrier resulting in inflammatory demyelination in the brain and spinal cord [3,4]. While Th2 and T regulatory (Treg) cells keep in check the generation of Th1 and Th17 cells, respecively, in the CNS of EAE/MS [5,6]. Different classes of immunomodulatory agents i.e., interferon (IFN)-β, glatiramer acetate (GA), mitoxantrone, natalizumab, and fingolimod are available for MS treatment [7-11]. However, these agents except fingolimod are partially effective as they specifically target the inflammatory phase, but not neurodegenerative phase of MS to limit long-term disability [12]. In addition, these current medications are associated with undesirable side-effects and potential toxicities [13-16]. Therefore, there is an urgent need to develop agent(s) that targets both inflammatory and neurodegenerative phases of MS with greater efficacy [17]. Secondly, the inherent complexity of MS pathogenesis is suggestive of combining existing or novel agents that may complement one another to limit clinical symptoms in patients [18].
Metformin (Metf) is an oral biguanide drug used to treat type-2 diabetes and its mechanism of protection is ascribed to the activation of AMP-activated protein kinase (AMPK), a sensor of cellular energy status [19,20]. Similar to the pharmacological AMPK activator, 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR), Metf also attenuates inflammatory response in the brain and endothelial cells [21,22]. In EAE, Metf attenuates the expansion of Th1 and Th17 cells, but enhances the differentiation and expansion of Th2 and Treg cells [23]. Likewise, Metf is reported to regulate Th17 cells generation in other autoimmune disease i.e., rheumatoid arthritis (RA) and type-2 diabetes patients [24,25]. In addition, Metf is reported to be safe and beneficial in diabetic patients [26,27].
Large body of evidence suggests that statin as cholesterol lowering drug has potential to treat T-cell mediated, organ specific autoimmune diseases or other inflammatory diseases [28,29]. Promising results were obtained in clinical trials of statin in MS and RA including inhibition of optic neuritis in MS patients [30-32]. In EAE, statin promotes the differentiation and expansion of Th2 cells and Treg cells, and inhibits the activation of antigen presenting cells [33,34] and the differentiation of Th1 and Th17 cells [35,36]. In addition, statin is reported to protect the breaching of blood brain barrier and inhibit the expression of intracellular adhesion molecules to limit cellular infiltration in the CNS of EAE animals [37,38]. These effects of statin were accompanied with neuroprotection and induction of myelin repair in EAE [39,40].
Statin-mediated immunomodulatory and neuroprotective effects in EAE are ascribed to the inhibition of Rho and Ras family GTPases [38,41,42]. The inhibition of Rho family GTPases is reported to upregulate peroxisome proliferator activated receptors (PPARs) activity in cells [43-45]. PPAR activity is reported to be regulated by AMPK in cells and that the agonists of PPARs are reported to attenuate EAE [46-49]. Based upon this knowledge, we hypothesized that Metf may complement to lovastatin (LOV) mediated attenuation of EAE by targeting PPARs, albeit with different mechanisms of action. Therefore, we here tested whether LOV and Metf in combination attenuate T-cell autoimmunity and neurodegeneration in EAE model.
Material and Methods
Chemicals and reagents
Unless otherwise stated, all chemicals were purchased from Sigma. LOV and Metf were purchased from EMD Millipore (Billercia, MA). TRIzol reagent was purchased from Invitrogen (Carlsbad, CA), and RNA easy cleaning kits were from Qiagen (Valencia, CA). Antibodies for myelin basic protein (MBP) (clone 1, 129-138) were purchased from Serotec (Raleigh, NC). Antibodies for phosphorylated-neurofilament-H (SMI-31), non-phosphorylated-neurofilament-H (SMI-32) and proteolipid protein (PLP) were purchased from Chemicon (Temecula, CA) and abcam (Cambridge, MA). Mouse anti-rat CD68 antibodies were purchased from Biosource (Camarillo, CA). Secondary antibodies i.e., goat anti-mouse IgG and anti-rabbit IgG conjugated with Texas Red (for CD68 and MBP) and FITC (for SMI-31) were purchased from Vector Laboratories, Inc. (Burlingame, CA). ECL-detecting reagents and nitrocellulose membranes were purchased from GE Healthcare Biosciences (Pittsburg, PA).
Animals
Adult female Lewis rats weighing 250-300g were purchased from Charles River laboratory (Wilmington, MA) and housed in the animal care facility at the Medical University of South Carolina (MUSC) throughout the experiment and provided with food and water ad lib. All animal experimentations were conducted in accordance with accepted standards of humane care, as outlined in the ethical guidelines and approved by MUSC's Animal Ethics Committee.
EAE induction and evaluation
Procedures used for an induction of EAE are as described previously in our publications with slight modifications [39,50]. In brief, rats received a subcutaneous injection of 25 μg of guinea pig MBP in 0.1 ml of PBS emulsified with equal volume of CFA supplemented with 2 mg/ml of mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) in the hind limb footpads on days 0 and 7. Immediately and again 24 h later, rats received pertussis toxin (200 ng, intraperitoneally) in 0.1 ml PBS. Pertussis toxin was used as per the standardized protocol reported by us and other investigators for EAE induction. Similarly, healthy control group rats received subcutaneous injection of PBS and CFA emulsion in the hind limb footpads on days 0 and 7. Rats were examined for clinical scores by an experimentally blinded investigator daily. Clinical score assessed on a 0–5 scale: 0, no clinical disease; 1.0, piloerection; 2.0, loss in tail tonicity; 3.0, hind-leg paralysis; 4.0, paraplegia, and 5.0, moribund or dead. At several times during the study, rats were weighed. The clinical data of rats with clinical score >4.0 were not included for statistical analysis. On the peak clinical day of the disease (13th of 14th dpi) and at the conclusion of study (25th dpi), rats were euthanized, perfusion was done and lumbar spinal cords (SCs) were removed and snap frozen with liquid nitrogen and stored at -70oC till use. Alternatively, SCs of each rat was cut into 4 small pieces and fixed in 4% paraformaldehyde for histopathology or immunohistochemistry studies.
Drug treatments
Metf (150 mg/ml) was suspended in PBS and LOV (1 mg/kg) was suspended in ethanol/NaOH solution and administered alone or in combination by gavages, every day in 200 μl volumes. Treatment was started in rats with established EAE on 10th or 11th day of post immunization (dpi) and continued till the lessening of paralytic symptoms (on 25th dpi; recovery). EAE rats without drug treatment received PBS, once daily. Likewise, healthy control rats received vehicle or Metf dose, once daily.
Histology and immunohistochemistry studies
For single-labeling standard methodology was used. Briefly, slides were blocked by using ‘Image-iT® fixation and permeabilization kit' (Life technologies, Grand Island, NY) and incubated with appropriately diluted primary antibody (1:100) at 4°C overnight followed by washing and further incubation with secondary antibodies (1:500) for 1 h. For double-labeling, slides were incubated simultaneously or separately with both types of primary antibodies after blocking with a serum-PBS solution at 4°C overnight as described above. Secondary antibodies conjugated with Texas Red (MBP or CD68) or conjugated with FITC (SMI-31 or CNPase) were used. Slides were also incubated with Texas Red or FITC conjugated IgG antibodies without primary antibody as negative controls and an appropriate mouse IgG or rabbit polyclonal IgG as isotype controls. After thorough washings, slides were mounted with Fluoromount-G (Electron Microscopy Sciences) containing Hoechst. Slides were examined under fluorescence microscope (Olympus BX-60) and images were captured at magnifications ×400 with an Olympus digital camera (Optronics; Goleta, CA) using a dual-band pass filter. The contrast and brightness of images were processed using Adobe Photoshop CS 5 software.
RNA preparation, cDNA synthesis and real-time PCR analysis
Cells or tissues were carefully processed for RNA isolation using TRIzol reagent followed by cDNA synthesis and real-time PCR analysis using BIO-RAD CFX96 (BIO-RAD Laboratories). Primer sets used in the study were described previously [39,51]. IQTM SYBR Green Supermix was purchased from BIO-RAD (Hercules CA). Thermal cycling conditions were as follows: activation of iTaqTM DNA polymerase at 95°C for 10 min, followed by 40 cycles of amplification at 95°C for 30 s and 59-60°C for 30 s. The specificity and detection methods for data analysis are as described earlier [44]. In brief, realtime PCR specificity for each analysis was determined by melting curve analysis of the amplified product. The level of target gene transcripts was calculated relative to the expression of reference gene, β-actin in each sample. The detection of threshold was set above the mean baseline fluorescence determined by the first 20 cycles. A standard curve for each template was generated with a serial dilution of the template (cDNA).
Western blot analysis
Total cell lysates were prepared in ice-cold lysis buffer (RIPA buffer) and used for western blot analysis as described previously [52].
IgGisotype quantification
Anti-MBP-specific IgG isotypes were determined following solid phase ELISA as per the instructions of the manufacturer in serum samples of experimental animals. Briefly, the plates were coated with anti-MBP (2 μg/ml) in PBS overnight in a humidity controlled chamber. Coated plates were washed with PBS containing 0.05% Tween-20 and blocked with 1% BSA for 1 h at room temperature (RT). After washing, plates were incubated with serum samples diluted (1:100) with PBS for 2 h at RT. Bound antibody was detected by using anti rat IgG1, IgG2a and IgG2b secondary antibody: HRP enzyme system (Serotec, Raleigh, NC) employing Luminata Forte ELISA HRP substrate (EMD Millipore, Billerica, MA) and read in a luminometer.
Quantification of cytokines
Levels of various cytokines in serum and spinal cord samples were quantified by ELISA kits (eBiosciences Inc., San Diego,CA) as per instructions in the product manuals. Slices of the spinal cords (50 to 100 mg) of rats were sonicated in 0.5 mL of PBS and centrifuged at 12,000×g for 10 minutes at 4°C. The protein concentration in each sample supernatant was determined using DC Protein Assay (Bio-Rad). To quantify the levels of various cytokines in sample, recombinant cytokine proteins were used as standards. Levels of cytokines in each sample in triplicate were measured using an ELISA reader (UV-VIS spectrophotometer), and data were normalized with total protein concentration in each sample.
Statistical analysis
Data are given as mean ± S.E.M and analyzed by using Student's t test for two groups comparison or one-way multiple range analysis of variance (ANOVA) for multiple columns comparison followed by a Bonferroni post-test. P values were determined for three to four separate samples in each experiment using GraphPad Prism 5.0 software (Graph Pad Software Inc. San Diego, CA). P values <0.05 were considered significant.
Results
Metf and LOV combination alleviates EAE clinical symptoms in treated rats
We earlier reported that 2 mg/kg dose of LOV attenuates EAE in MBP immunized rats [39,50]. Likewise, 100 mg/kg dose (orally; 37) of Metf attenuated EAE clinical symptoms in mice immunized with MOG or PLP peptides [23]. To test whether these agents in combination provide synergistic or additive effect to limit EAE, we treated EAE rats with the suboptimal dose of each drug in combination or alone. Drug treatment was started in established EAE (clinical score >3.0) rats to mimic MS clinical symptoms that are related to inflammatory demyelination. On clinical peak day (13th or 14th dpi), EAE clinical scores in rats treated with vehicle were higher (3.7 ± 0.28) as compared to those were treated with Metf (2.8 ± 0.16) or LOV (2.8 ± 0.1) alone. However, Metf and LOV combination demonstrated better suppression of EAE disease in rats (2.5 ± 0.15; P<0.05) as compared to their individual treatments (Figure 1A). On 25th dpi (Figure 1A), Metf and LOV combination significantly suppressed EAE disease in rats (0.36 ± 0.17; P<0.01) as compared to their vehicle treated counterparts (2.0 ± 0.5). These data provide evidence that Metf and LOV combination provides additive effect to attenuate EAE in animals.
Figure 1. Metf and LOV combination limits clinical impairments via inhibition of inflammation and demyelination in the SCs of EAE rats.
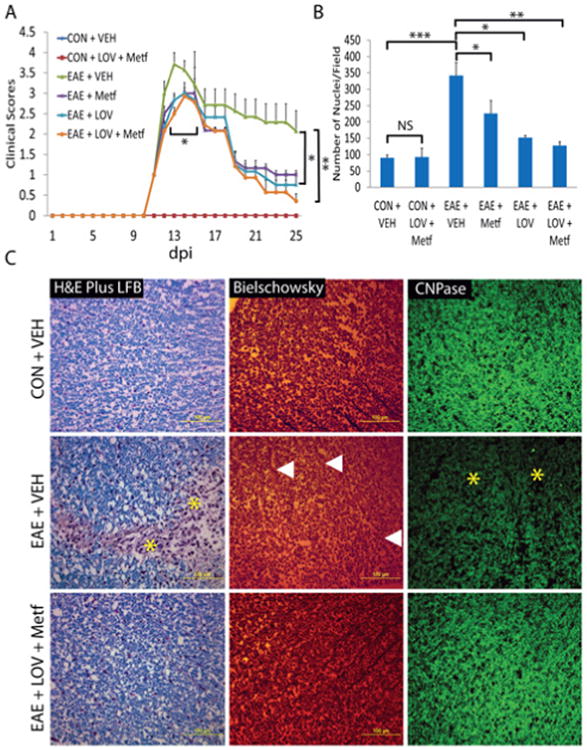
EAE was induced with guinea pig MBP (25 μg/rat) in female Lewis rats and treatment of LOV (1 mg/kg) and/or Metf (150 mg/kg) or vehicle were started orally by gavages in rats with established EAE or in healthy controls (CON). A: Shown is the composite mean ± SEM of clinical scores in rats (n=9)/group evaluated in three separate experiments. B: Shown is the composite mean ± SEM of three to four samples/group analyzed for cellular infiltration in the SCs of EAE rats on peak clinical day. C: Representative photographs of the cross-section of lateral funiculi of the SCs of rats (n=6)/group on peak clinical day, stained with LFB plus H&E (left panel) depicting cellular infiltration and demyelination, bielschowsky staining (middle panel) depicting axonal integrity and immunostaining with anti-CNPase (right panel) depicts demyelination. Asterisks depict cellular infiltration and demyelination in the white mater of the SC. Triangles depict neuronal axonal loss in the white matte of the SC. Differences were statistically significant as indicated: *p<0.05, **p<0.01 and ***p<0.001, and NS (not significant).
Metf and LOV combination reduces inflammation, demyelination and axonal loss in EAE
Since inflammatory demyelination and axonal loss are the pathological hallmark of EAE/MS [3,4], we examined the SCs of similarly treated EAE rats for cellular infiltration, demyelination and axonal loss on peak clinical day of EAE (13th or 14th dpi). As expected, cellular infiltration was increased in the lumbar region of the SCs of EAE rats treated with vehicle than controls and that was attenuated by treatment with Metf and LOV in combination or alone (Figure 1B). However, Metf and LOV combination effect was additive and greater than their individual treatments (Figure 1B). Consistent with cellular infiltration data, demyelination was greater in the SCs of EAE rats treated with vehicle than controls and that was rescued by treatment with Metf and LOV combination (Figure 1C; left panel). These data were further supported by the loss of CNPase, OL specific marker in the SCs of EAE rats treated with vehicle than controls and that was rescued by treatment with Metf and LOV combination (Figure 1C; right panel). Likewise, axonal loss was severe in the SCs of similarly treated EAE rats with vehicle than controls and that was attenuated by treatment with Metf and LOV combination (Figure 1C; middle panel). This Metf and LOV combination mediated attenuation of myelin and axonal loss in the SCs of EAE rats was greater than their individual treatments (not depicted). Together, these data provide evidence that Metf and LOV combination protects CNS integrity in EAE animals via attenuation of cellular infiltration, demyelination and axonal loss.
Metf and LOV combination limits T-cell autoimmunity in the peripheral and CNS compartments of EAE animals
Myelin reactive T cells are reported to play important role in the induction, maintenance and regulation of inflammatory demyelination in EAE/MS brain [53]. Therefore, we next determined as to what degree the protective effects of Metf and LOV combination in EAE rats are indeed due to their effects on the immune response in periphery or locally in the CNS. Levels of IFN-γ and IL-17A, signatory cytokines of Th1 and Th17 cells, respectively, were higher in the sera of EAE rats treated with vehicle than controls and that were attenuated significantly by treatment with Metf and/orLOV (Figures 2A and 2B). Since Th1 response mainlyprovokes IgG2a, while Th2 response provokes IgG1 in sera [54], we next assessed whether Metf and LOV influences the levels of MBP-specific immunoglobulin isotypes in treated EAE rats. As shown in the Figure 2E, level of anti-MBP immunoglobulin isotype IgG2a was elevated in the sera of EAE rats treated with vehicle than controls and that was reduced significantly by treatment with Metf and/or LOV. Conversely, levels of anti-MBP immunoglobulin isotype IgG1 and IgG2b were significantly elevated in the sera of EAE rats treated with the Metf and/or LOV than their vehicle treated counterparts (Figures 2C and 2D). Of note, the combinatorial effect of Metf and LOV on the regulation of anti-MBP immunoglobulin isotypes in the sera of EAE rats was additive, but not significant when compared to their individual treatments (Figures 2C-2E).
Figure 2. Metf and LOV combination inhibits inflammatory response in the blood of EAE rats.

The induction of EAE in rats and treatment with Metf and/or LOV or vehicle were as detailed in Figure 1 legend. Shown are the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the levels of IFN-γ (A), IL-17A (B), anti-MBP IgG1 (C), anti-MBP IgG2b (D) and anti-MBP IgG2a (E) in the sera of EAE rats on peak clinical day. Differences were statistical significance as described for Figure 1 legend.
Next to determine whether Metf and LOV combination protects CNS functions via regulation of the local inflammatory responses, we performed ELISA and real-time PCR based analyses for T cell markers and their signatory cytokines in the SCs of EAE rats. Consistent with the observed higher levels of IFN-γ and IL-17A in sera (Figures 2A and 2B), levels of these cytokines were also elevated in the SCs of EAE rats treated with vehicle as compared to controls and that were reversed by treatment with Metf and/or LOV (Figures 3E and 3F). Likewise, levels of the mRNA transcripts for Th1 (IFN-γ) and Th2 (IL-4 and IL-10) cytokines including Th17 cell transcription factor, ROR- γt were elevated in the SCs of EAE rats treated with vehicle than controls and that were reversed by treatment with Metf and/or LOV (Figures 3A-3D). The observed effect of Metf and LOV combination was additive to attenuate IFN-γ (mRNA and protein) and IL-17A (protein) or induce IL-4 (mRNA) than their individual treatment in EAE rats (Figures 3A-3F). In addition, the activation of macrophages/microglia (CD68+ve) was greater in the SCs of EAE rats treated with vehicle than controls and that was attenuated by treatment with Metf and/or LOV (Figure 4). Furthermore, levels of IL-23 and TNF-α mRNA transcripts were elevated as the marker of antigen presenting cells activation in the SCs of EAE rats treated with vehicle than controls and that were attenuated by treatment with Metf and/or LOV (Figures 5A and 5B). The observed combinatorial effect of Metf and LOV on the attenuation of IL-23 and TNF-α expression in the SCs of EAE rats was additive, but not significant when compared with their individual treatments (Figures 5A and 5B). Consistent with cellular infiltration data (Figure 1B), levels of the CD4 and CD8 mRNA transcripts were significantly elevated indicating of the increased infiltration of CD4 and CD8 T cells in the SCs of EAE rats treated with vehicle than controls and that were reversed by treatment with Metf and/or LOV (Figures 5C and 5D). The observed combinatorial effect of Metf and LOV on the attenuation of CD4 and CD8 expression in the SCs of EAE rats was additive and that was greater than their individual treatments (Figures 5C and 5D). Together, these data suggest that Metf and LOV combination limits T-cell autoimmunity in the peripheral and CNS compartments of EAE animals via regulation of Th1, Th2 and Th17 cells immune responses.
Figure 3. Metf and LOV combination inhibits inflammatory response in the SCs of EAE rats.

The induction of EAE in rats and treatment with Metf and/or LOV or vehicle were as detailed in Figure 1 legend. Shown are the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the levels of IFN-γ (A), IL-4 (B), ROR-γt (C) and IL-10 (D) mRNA transcripts in the SCs of EAE rats on peak clinical day. Shown are the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the levels of IFN-γ (A), IL-17A (B) protein in the SCs of EAE rats on peak clinical day. Differences were statistical significance as described for Figure 1 legend.
Figure 4. Metf and LOV combination limits the expression of proinflammatory cytokines and T cells markers in the SCs of EAE rats.

The induction of EAE in rats and treatment with Metf and/or LOV or vehicle were as detailed in Figure 1 legend. Shown are the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the levels of TNF-α (A), IL-23 (B), CD4 (C) and CD8 (D) mRNA transcripts in the SCs of EAE rats on peak clinical day. Differences were statistical significance as described for Figure 1 legend.
Figure 5. Metf and LOV combination reduces CD68+ve in the SCs of EAE rats.

The induction of EAE in rats and treatment with Metf and/or LOV or vehicle were as detailed in Figure 1 legend. Representative photographs depicts CD68+ve cells in the lateral funiculi of the SCs of rats (n=6)/group on peak clinical day. Arrowhead depicts CD68+ve cells in the white matter of the SC.
Metf and LOV combination treatment limits neurodegeneration in EAE rats
To assess the effect of Metf and LOV combination on CNS repair, we next determined the expression of trophic factors in the SCs of ameliorating EAE rats on 25th dpi. Levels of CNTF (ciliary neurotrophic factor) mRNA transcripts and protein were significantly reduced in the SCs of EAE rats treated with vehicle as compared to controls and that were reversed by treatment with Metf and/or LOV (Figures 6A and 6E). Likewise, levels of brain derived neurotrophic factor (BDNF), insulin like growth factor-1 (IGF-1) and leukemia inflammatory factor (LIF) mRNA transcripts were found to be reduced significantly in the SCs of EAE treated with vehicle as compared to controls and that were reversed by treatment with Metf and/or LOV (Figures 6B-6D). This effect of Metf and LOV combination on the expression of trophic factors in the SCs of EAE rats was additive, however not significant as compared to their individual treatments (Figures 6A-6E). Further, to determine that these trophic factors contribute to neuroprotection in EAE animals, we analyzed the SCs of similarly treated EAE rats using anti-myelin and neurofilament proteins antibodies by western blotting and immunohistochemistry. As shown in Figure 6F, levels myelin (MBP and PLP) and neurofilament (SMI-31 and SMI-32) proteins were reduced in the SCs of EAE rats treated with vehicle compared with controls and that were protected by treatment with Metf and/or LOV.
Figure 6. Metf and LOV combination enhances promyelinating milieu in the SCs of EAE rats.

The induction of EAE in rats and treatment with Metf and/or LOV or vehicle were as detailed in Figure 1 legend. Shown are the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the levels of CNTF (A), BDNF (B), IGF-1(C) and LIF (D) mRNA transcripts in the SCs of EAE rats on 25th dpi. Shown is the composite mean ± SEM of three to four samples/group analyzed in triplicate to determine the level of CNTF (A) protein in the SCs of EAE rats on 25th dpi. Representative western blot (IB) depicts levels of different proteins in the SCs of EAE rats on 25th dpi. Differences were statistical significance as described for Figure 1 legend.
Consistent with these data, intensities of the neuronal axon-specific, phosphorylated-neurofilament heavy chain protein (SMI-31) and that of MBP were reduced in the SCs of EAE rats treated with vehicle as compared to controls and that were reversed by treatment with Metf and/or LOV (Figure 7). These effects of Metf and LOV combination on neuroprotection in EAE rats were greater than their individual treatments (Figures 6F and 7). Taken together, these data provide evidence that Metf and LOV combination influences the expression of trophic factors to limit neurodegeneration in EAE animals.
Figure 7. Metf and LOV combination restricts neurodegeneration in the SCs of EAE rats.
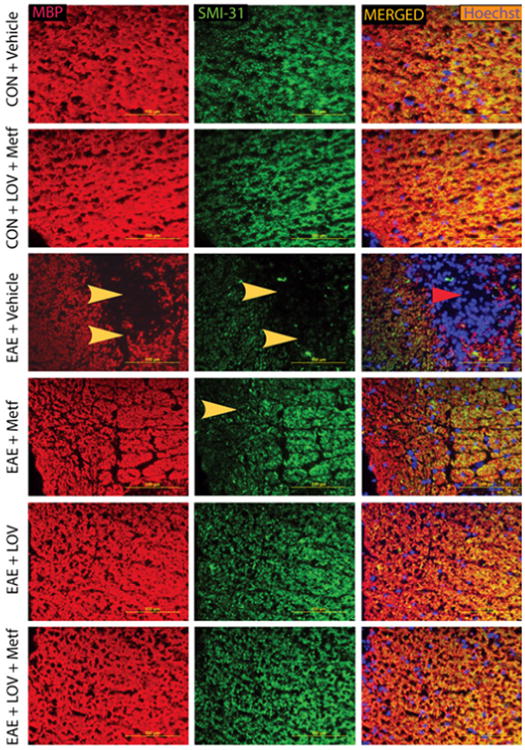
The induction of EAE in rats and treatment with Metf and/orLOV or vehicle were as detailed in Figure 1 legend. Representative photographs depicts MBP (left panel) and SMI-31 (middle panel) in the lateral funiculi of the SCs of rats (n=6)/group on 25th dpi. Arrowhead depicts loss of myelin and axonal protein in the white matter. Sections were counter stained with Hoechst dye (right panel).
Discussion
Single drug therapy often lacks its effectiveness in MS patients due to the complex pathology of disease. Clinical studies conducted recently using different immunomodulatory agents i.e., fingolimod, cladribine and teriflunomide demonstrated promising results in MS patients, but these medications were associated with serious side-effects [11,55,56]. We believe that combination of two or more agents using their suboptimal doses could minimize their potential side-effects and improve their beneficial effects to combat MS.
Major criteria of selecting agents for combination therapy for MS should be if: (a) agents act through different mechanisms of action, (b) each agent offers excellent safety profile and (c) agents not create potential toxicity when used in combination [57]. Metf and LOV meet these criteria as both characteristically attenuate clinical symptoms and provide neuroprotection in EAE animals [33,34,50]. In addition, both LOV and Metf [58] are reported to cross the BBB thereby these drugs can interfere with the neurodegeneration in MS brain. Our findings demonstrated that Metf and LOV combination treatment attenuates clinical symptoms in established EAE animals via inhibition of cellular infiltration and T-cell autoimmunity. Autoreactive Th1 and Th17 immune responses were reduced, while Th2 immune responses were enhanced in the peripheral and CNS compartments of EAE animals treated with Metf and LOV combination. In addition, demyelination and axonal loss were reduced, but the expression of trophic factors was enhanced suggesting that Metf and LOV combination limits neurodegeneration in EAE.
EAE model used in this study is widely used for preclinical testing of agents for MS. Several combinations of different agents including FDA approved MS drugs are tested in EAE model [18,57,59,60] as well as in MS patients [61]. Use of statins for MS is still under debate as mixed results are reported by combination of statins with IFN in different MS clinical studies [62-66]. The major cause of this observed inconsistency with the addition of statin to IFN therapy in MS is ascribed to their antagonizing mechanisms of action [67]. It suggests that statin should be tested in combination with immunomodulatory agents that does not interfere or negate its mechanism of action or vice versa. In this context, we and other earlier demonstrated that statin provides beneficial effects in combination with GA or other disease modifying in EAE [51,57,68,69]. In addition, we reported that AICAR in combination with LOV attenuates inflammation and provides neuroprotection in EAE animals [70]. The mechanism of action of both Metf and AICAR however is via AMPK activation, but Metf has greater cellular/tissue penetration than AICAR [19,20]. Therefore, Metf is the ideal candidate to be tested in combination with LOV in EAE. Atorvastatin and rapamycin combination has recently been documented to provide immunomodulatory synergy in EAE [71]. Rapamycin is known to block the activity of a serine/threonine protein kinase called mammalian target of rapamycin (mTOR) which is crucial for cell growth [72] and it is the downstream target of AMPK in cells [73]. Our findings demonstrate that LOV complements to the inhibitory effect of Metf in EAE animals that could be ascribed to their immunomodulatroy and neuroprotective activities as discussed below.
Immunomodulatory activities
The susceptibility of EAE development is thought to correlate with the expression of Th1 cytokines i.e., IL-23, IFN-γ and TNF-α, while that is controlled by Th2 cytokines i.e., IL-4 andIL-10 [3-6]. Activation of resident glial cells following infiltration of autoreactive Th1/Th17 cells and macrophages (CD68+ve) is responsible for CNS inflammation that eventually cause loss of myelin and axons in EAE animals. Statin is reported to shift the Th1 to Th2 biased phenotype responses in EAE [33] via regulation of T-bet and GATA3 transcriptional activities [34] and it inhibits Th17 cells generation and enhances the Treg cells expansion [35,36]. Likewise, Metf is reported to induce Th1 to Th2 biased phenotype responses and inhibits Th17 phenotype response in EAE [23]. Mechanistically, statin inhibits mevalonate pathway thereby reduces the biosynthesis of cholesterol and isoprenoids. The reduced level of cholesterol in statin treated cells is compensated by the uptake of extracellular cholesterol through LDL-receptors, while the reduced isoprenoids affects the secondary modification of Rho family GTPases in the cell membrane [74]. The statin-mediated depletion of isoprenoids is reported to shift the Th1 to Th2 bias in EAE animals via inhibition of the Ras and RhoA activities [41]. Metf is reported to attenuate EAE development via AMPK activation in T cells [23]. AMPK has also been implicated in different disease pathologies and the attenuation of cytokine-induced activation of endothelial cells via inhibition of the NF-κB activation [21,75]. In addition, we earlier documented that AICAR attenuates EAE via modulation of T-cell autoimmunity and the protection of blood-brain barrier [76,77].
In the CNS, local antigen presentation is a critical event to initiate and perpetuate chronic inflammatory responses. While the CNS is devoid of professional antigen presenting cells (dendritic cells), however, MHC class II antigens and co-stimulatory CD80 and CD86 molecules are upregulated in microglia and macrophages in response to local cytokine production [33,78]. LOV has been reported to inhibit the expression of TNF-α and IL-1β in microglia and astrocytes including IFN-γ inducible transcription of MHC class II expression in microglia [79,80]. Likewise, AMPK is reported to inhibit IFN-γ induced expression of TNF-α and chemokines in astrocytes and microglia [81]. In addition, Metf-mediated induction of AMPK signaling is reported to attenuate inflammatory response in vascular cells [82]. In line with this, we earlier documented that AICAR attenuates LPS induced inflammatory response in glial cell cultures and in the rat brain [22]. These above described different mechanisms of action of Metf (as AMPK activator) and LOV (inhibitor Rho prenylation) may complement to restrict T-cell autoimmunity in the peripheral and CNS compartments of EAE animals.
Neuroprotective activities
Trophic factor secreted by brain glial cells play important role in many cellular events i.e., cell proliferation and differentiation [83]. We observed the increased levels of trophic factors i.e., CNTF, BDNF, LIF and IGF-1 in the SCs of EAE animals treated with Metf plus LOV combination suggesting that these drugs provide a promyelinating milieu in the CNS of EAE animals. In agreement with this, we earlier reported that LOV induces a promyelinating milieu in the CNS of ameliorating EAE animals [39]. Trophic factors i.e., IGF-1 and CNTF are reported to protect neurons and OL progenitor cells [84,85]. In addition, astrocytes, microglia and Th2 cells are known to secrete trophic factors i.e., BDNF and LIF [86,87]. Importantly, CNTF and BDNF induced AMPK activation was reported to regulate synaptic plasticity and cognitive functions [88,89]. In addition, we recently documented that Metf induced AMPK signaling protects OLs to restore CNS functions in EAE animals [90]. Statin-mediated PPAR-α activation protects OLs from cytokine toxicity, while PPAR-γ activation enhances their differentiation via inhibition of Rho family GTPase functions [44,91,92]. Accordingly, Metf and LOV combination treatment attenuated the loss of myelin and axonal proteins thereby clinical impairments in EAE animals (Figure 6F). The above described neuroprotective activities of Metf and LOV indicate that the combinatorial effect of these drugs may contribute to limit neurodegeneration in EAE animals.
In conclusion, our findings document that Metf and LOV combination treatment provides greater efficacy limiting clinical impairments in EAE animals. The overall contribution of the immunomodulatory and neuroprotective activities of these drugs in combination is complementing to limit EAE development. However, the precise mechanism behind this protection is not fully understood. As discussed above the beneficial mechanism of action of Metf and LOV are possibly ascribed to the regulation of PPARs activity both in the immune and CNS glial cells (Figure 8). Both statins and Metf are FDA approved oral medications for human patients other than MS. Based on the safety profile in humans and the observed efficacy of these drugs in combination in EAE warrants future clinical trials in MS patients.
Figure 8. The possible mechanism of action of Metf and LOV in immune and CNS cells in EAE.

LOV inhibits mevalonate pathway via inhibition of HMG-CoA reductase and lowers levels of cholesterol and isoprenoids. The depletion of isoprenoids affects Rho family GTPases functions that are reported to regulate PPARs activity in cells. Metformin (Metf) activates AMPK which in turn, regulates PPARs activity in cells. The observed targeting of PPARs by LOV and Metf in combination may inhibit Th1/Th17 cell expansion, but enhance Th2 cells generation that eventually inhibit CNS inflammation and demyelination in EAE.
Acknowledgments
We thank all members of the laboratory for their valuable comments and help during the course of this study at Children's Research Institute, Medical University of South Carolina; and Ms. Joyce Brian and Mrs. Chara William for their technical assistance.
This study was supported by grants from the VA-1BX001072, VA-BX001999 and NIH (NS-22576, NS-37766 and C06-RR018823).
Abbreviations
- MS
Multiple sclerosis
- EAE
Experimental autoimmune encephalomyelitis
- LOV
Lovastatin
- Metf
Metformin
- AICAR
5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside
- AMPK
AMP-activated protein kinase
- CNS
Central nervous system
- PPAR
Peroxisome proliferator activated receptor
- SC
Spinal cord
- CNTF
Ciliary neurotrophic factor
- BDNF
Brain derived neurotrophic factor
- IGF-1
Insulin like growth factor-1
- LIF
Leukemia inflammatory factor
References
- 1.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 2.Lublin FD. Role of myelin antigens in murine relapsing experimental allergic encephalomyelitis. J Clin Lab Immunol. 1984;13:179–182. [PubMed] [Google Scholar]
- 3.Hofstetter H, Gold R, Hartung HP. Th17 Cells in MS and Experimental Autoimmune Encephalomyelitis. Int MS J. 2009;16:12–18. [PubMed] [Google Scholar]
- 4.Ifergan I, Kébir H, Bernard M, Wosik K, Dodelet-Devillers A, et al. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. 2008;131:785–799. doi: 10.1093/brain/awm295. [DOI] [PubMed] [Google Scholar]
- 5.Mix E, Meyer-Rienecker H, Hartung HP, Zettl UK. Animal models of multiple sclerosis--potentials and limitations. Prog Neurobiol. 2010;92:386–404. doi: 10.1016/j.pneurobio.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsson T. Critical influences of the cytokine orchestration on the outcome of myelin antigen-specific T-cell autoimmunity in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Rev. 1995;144:245–268. doi: 10.1111/j.1600-065x.1995.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 7.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 8.Hartung HP, Gonsette R, König N, Kwiecinski H, Guseo A, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 9.Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- 10.Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 11.Doggrell SA. Oral fingolimod for relapsing-remitting multiple sclerosis Evaluation of: Kappos L, Radue E-M, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387-401; and Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362:402-15. Expert Opin Pharmacother. 2010;11:1777–1781. doi: 10.1517/14656566.2010.481671. [DOI] [PubMed] [Google Scholar]
- 12.Rizvi SA, Agius MA. Current approved options for treating patients with multiple sclerosis. Neurology. 2004;63:S8–14. doi: 10.1212/wnl.63.12_suppl_6.s8. [DOI] [PubMed] [Google Scholar]
- 13.Arbizu T, Alvarez-Cermeño JC, Decap G, Fernández O, Uría DF, et al. Interferon beta-1b treatment in patients with relapsing--remitting multiple sclerosis under a standardized protocol in Spain. Acta Neurol Scand. 2000;102:209–217. doi: 10.1034/j.1600-0404.2000.102004209.x. [DOI] [PubMed] [Google Scholar]
- 14.Dhib-Jalbut S. Glatiramer acetate (Copaxone) therapy for multiple sclerosis. Pharmacol Ther. 2003;98:245–255. doi: 10.1016/s0163-7258(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 15.Yousry TA, Major EO, Ryschkewitsch C, Fahle G, Fischer S, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354:924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 17.Al Jumah MA, Abumaree MH. The Immunomodulatory and Neuroprotective Effects of Mesenchymal Stem Cells (MSCs) in Experimental Autoimmune Encephalomyelitis (EAE): A Model of Multiple Sclerosis (MS) Int J Mol Sci. 2012;13:9298–9331. doi: 10.3390/ijms13079298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soos JM, Stüve O, Youssef S, Bravo M, Johnson HM, et al. Cutting edge: oral type I IFN-tau promotes a Th2 bias and enhances suppression of autoimmune encephalomyelitis by oral glatiramer acetate. J Immunol. 2002;169:2231–2235. doi: 10.4049/jimmunol.169.5.2231. [DOI] [PubMed] [Google Scholar]
- 19.Ota S, Horigome K, Ishii T, Nakai M, Hayashi K, et al. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem Biophys Res Commun. 2009;388:311–316. doi: 10.1016/j.bbrc.2009.07.164. [DOI] [PubMed] [Google Scholar]
- 20.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 21.Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- 22.Giri S, Nath N, Smith B, Viollet B, Singh AK, et al. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, et al. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumarac-Dumanovic M, Jeremic D, Pantovic A, Janjetovic K, Stamenkovic-Pejkovic D, et al. Therapeutic improvement of glucoregulation in newly diagnosed type 2 diabetes patients is associated with a reduction of IL-17 levels. Immunobiology. 2013;218:1113–1118. doi: 10.1016/j.imbio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Kang KY, Kim YK, Yi H, Kim J, Jung HR, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol. 2013;16:85–92. doi: 10.1016/j.intimp.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. No authors listed. [PubMed] [Google Scholar]
- 27.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 28.Mach F. Toward a role for statins in immunomodulation. Mol Interv. 2002;2:478–480. doi: 10.1124/mi.2.8.478. [DOI] [PubMed] [Google Scholar]
- 29.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 30.McCarey DW, McInnes IB, Madhok R, Hampson R, Scherbakov O, et al. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet. 2004;363:2015–2021. doi: 10.1016/S0140-6736(04)16449-0. [DOI] [PubMed] [Google Scholar]
- 31.Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 32.Tsakiri A, Kallenbach K, Fuglø D, Wanscher B, Larsson H, et al. Simvastatin improves final visual outcome in acute optic neuritis: a randomized study. Mult Scler. 2012;18:72–81. doi: 10.1177/1352458511415452. [DOI] [PubMed] [Google Scholar]
- 33.Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 34.Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004;172:1273–1286. doi: 10.4049/jimmunol.172.2.1273. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Markovic-Plese S. Statins' immunomodulatory potential against Th17 cell-mediated autoimmune response. Immunol Res. 2008;41:165–174. doi: 10.1007/s12026-008-8019-z. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Tao Y, Wang J, Garcia-Mata R, Markovic-Plese S. Simvastatin inhibits secretion of Th17-polarizing cytokines and antigen presentation by DCs in patients with relapsing remitting multiple sclerosis. Eur J Immunol. 2013;43:281–289. doi: 10.1002/eji.201242566. [DOI] [PubMed] [Google Scholar]
- 37.Prasad R, Giri S, Nath N, Singh AK, Singh I. Lovastatin inhibits endothelial-monocyte interaction via down regulation of adhesion molecules: involvement of PI3K-Akt-NF-kB pathway (Manuscript under preparation) 2004 [Google Scholar]
- 38.Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, et al. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003;17:905–907. doi: 10.1096/fj.02-1014fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, et al. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. FASEB J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- 40.Paintlia AS, Paintlia MK, Singh I, Skoff RB, Singh AK. Combination therapy of lovastatin and rolipram provides neuroprotection and promotes neurorepair in inflammatory demyelination model of multiple sclerosis. Glia. 2009;57:182–193. doi: 10.1002/glia.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunn SE, Youssef S, Goldstein MJ, Prod'homme T, Weber MS, et al. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–412. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paintlia AS, Paintlia MK, Singh AK, Singh I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol. 2008;73:1381–1393. doi: 10.1124/mol.107.044230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz-Velasco N, Dominguez A, Vega MA. Statins upregulate CD36 expression in human monocytes, an effect strengthened when combined with PPAR-gamma ligands Putative contribution of Rho GTPases in statin-induced CD36 expression. Biochem Pharmacol. 2004;67:303–313. doi: 10.1016/j.bcp.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Paintlia AS, Paintlia MK, Singh AK, Orak JK, Singh I. Activation of PPAR-γ and PTEN cascade participates in lovastatin-mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia. 2010;58:1669–1685. doi: 10.1002/glia.21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Cir Res. 2007;100:1442–1451. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 46.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng R, Pei Z, Zhang A, Zhou Y, Cai X, et al. AMPK activation enhances PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch Biochem Biophys. 2011;511:1–7. doi: 10.1016/j.abb.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Dunn SE, Bhat R, Straus DS, Sobel RA, Axtell R, et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med. 2010;207:1599–1608. doi: 10.1084/jem.20091663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206:2079–2089. doi: 10.1084/jem.20082771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, et al. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- 51.Paintlia AS, Paintlia MK, Singh I, Singh AK. Combined medication of lovastatin with rolipram suppresses severity of experimental autoimmune encephalomyelitis. Exp Neurol. 2008;214:168–180. doi: 10.1016/j.expneurol.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paintlia MK, Paintlia AS, Singh AK, Singh I. S-Nitrosoglutathione Induces Ciliary Neurotrophic Factor Expression in Astrocytes, Which Has Implications to Protect the Central Nervous System under Pathological Conditions. J Biol Chem. 2013;288:3831–3843. doi: 10.1074/jbc.M112.405654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hooper DC, Morimoto K, Bette M, Weihe E, Koprowski H, et al. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J Virol. 1998;72:3711–3719. doi: 10.1128/jvi.72.5.3711-3719.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 56.O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 57.Stüve O, Youssef S, Weber MS, Nessler S, von Büdingen HC, et al. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116:1037–1044. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lv WS, Wen JP, Li L, Sun RX, Wang J, et al. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012;1444:11–19. doi: 10.1016/j.brainres.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 59.Brod SA, Lindsey JW, Wolinsky JS. Combination therapy with glatiramer acetate (copolymer-1) and a type I interferon (IFN-alpha) does not improve experimental autoimmune encephalomyelitis. Ann Neurol. 2000;47:127–131. [PubMed] [Google Scholar]
- 60.Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol. 2005;165:83–91. doi: 10.1016/j.jneuroim.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 61.Ytterberg C, Johansson S, Andersson M, Olsson D, Link H, et al. Combination therapy with interferon-beta and glatiramer acetate in multiple sclerosis. Acta Neurol Scand. 2007;116:96–99. doi: 10.1111/j.1600-0404.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- 62.Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2008;71:1390–1395. doi: 10.1212/01.wnl.0000319698.40024.1c. [DOI] [PubMed] [Google Scholar]
- 63.Lanzillo R, Orefice G, Quarantelli M, Rinaldi C, Prinster A, et al. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: a longitudinal controlled trial of combination therapy. Mult Scler. 2010;16:450–454. doi: 10.1177/1352458509358909. [DOI] [PubMed] [Google Scholar]
- 64.Sorensen PS, Lycke J, Erälinna JP, Edland A, Wu X, et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10:691–701. doi: 10.1016/S1474-4422(11)70144-2. [DOI] [PubMed] [Google Scholar]
- 65.Rudick RA, Pace A, Rani MR, Hyde R, Panzara M, et al. Effect of statins on clinical and molecular responses to intramuscular interferon beta-1a. Neurology. 2009;72:1989–1993. doi: 10.1212/WNL.0b013e3181a92b96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamm CP, El-Koussy M, Humpert S, Findling O, von Bredow F, et al. Atorvastatin added to interferon beta for relapsing multiple sclerosis: a randomized controlled trial. J Neurol. 2012;259:2401–2413. doi: 10.1007/s00415-012-6513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feng X, Han D, Kilaru BK, Franek BS, Niewold TB, et al. Inhibition of interferon-beta responses in multiple sclerosis immune cells associated with high-dose statins. Arch Neurol. 2012;69:1303–1309. doi: 10.1001/archneurol.2012.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paintlia AS, Paintlia MK, Hollis BW, Singh AK, Singh I. Interference with RhoA-ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am J Pathol. 2012;181:993–1006. doi: 10.1016/j.ajpath.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luccarini I, Ballerini C, Biagioli T, Biamonte F, Bellucci A, et al. Combined treatment with atorvastatin and minocycline suppresses severity of EAE. Exp Neurol. 2008;211:214–226. doi: 10.1016/j.expneurol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 70.Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Z, Chen L, Niu X, Liu J, Ping M, et al. Immunomodulatory synergy by combining atorvastatin and rapamycin in the treatment of experimental autoimmune encephalomyelitis (EAE) J Neuroimmunol. 2012;250:9–17. doi: 10.1016/j.jneuroim.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 72.Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003;35:7S–14S. doi: 10.1016/s0041-1345(03)00211-2. [DOI] [PubMed] [Google Scholar]
- 73.Dong LX, Sun LL, Zhang X, Pan L, Lian LJ, et al. Negative regulation of mTOR activity by LKB1-AMPK signaling in non-small cell lung cancer cells. Acta Pharmacol Sin. 2013;34:314–318. doi: 10.1038/aps.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 75.Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, et al. AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3) Diabetologia. 2010;53:2406–2416. doi: 10.1007/s00125-010-1856-z. [DOI] [PubMed] [Google Scholar]
- 76.Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
- 77.Nath N, Giri S, Prasad R, Salem ML, Singh AK, et al. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 78.Cornet A, Bettelli E, Oukka M, Cambouris C, Avellana-Adalid V, et al. Role of astrocytes in antigen presentation and naive T-cell activation. J Neuroimmunol. 2000;106:69–77. doi: 10.1016/s0165-5728(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 79.Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwak B, Mulhaupt F, Veillard N, Pelli G, Mach F. The HMG-CoA reductase inhibitor simvastatin inhibits IFN-gamma induced MHC class II expression in human vascular endothelial cells. Swiss Med Wkly. 2001;131:41–46. doi: 10.4414/smw.2001.06144. [DOI] [PubMed] [Google Scholar]
- 81.Meares GP, Qin H, Liu Y, Holdbrooks AT, Benveniste EN. AMP-activated protein kinase restricts IFN-γ signaling. J Immunol. 2013;190:372–380. doi: 10.4049/jimmunol.1202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SA, Choi HC. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem Biophys Res Commun. 2012;425:866–872. doi: 10.1016/j.bbrc.2012.07.165. [DOI] [PubMed] [Google Scholar]
- 83.Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 84.Fernandez-Galaz MC, Morschl E, Chowen JA, Torres-Aleman I, Naftolin F, et al. Role of astroglia and insulin-like growth factor-I in gonadal hormone-dependent synaptic plasticity. Brain Res Bull. 1997;44:525–531. doi: 10.1016/s0361-9230(97)00238-4. [DOI] [PubMed] [Google Scholar]
- 85.Linker RA, Mäurer M, Gaupp S, Martini R, Holtmann B, et al. CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med. 2002;8:620–624. doi: 10.1038/nm0602-620. [DOI] [PubMed] [Google Scholar]
- 86.Vanderlocht J, Hendriks JJ, Venken K, Stinissen P, Hellings N. Effects of IFN-beta, leptin and simvastatin on LIF secretion by T lymphocytes of MS patients and healthy controls. J Neuroimmunol. 2006;177:189–200. doi: 10.1016/j.jneuroim.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 87.Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, et al. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005;102:19045–19050. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Escartin C, Pierre K, Colin A, Brouillet E, Delzescaux T, et al. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci. 2007;27:7094–7104. doi: 10.1523/JNEUROSCI.0174-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gomez-Pinilla F, Vaynman S, Ying Z. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J Neurosci. 2008;28:2278–2287. doi: 10.1111/j.1460-9568.2008.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paintlia AS, Paintlia MK, Mohan S, Singh AK, Singh I. AMP-Activated Protein Kinase Signaling Protects Oligodendrocytes that Restore Central Nervous System Functions in an Experimental Autoimmune Encephalomyelitis Model. Am J Pathol. 2013 doi: 10.1016/j.ajpath.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sim FJ, Lang JK, Ali TA, Roy NS, Vates GE, et al. Statin treatment of adult human glial progenitors induces PPAR gamma-mediated oligodendrocytic differentiation. Glia. 2008;56:954–962. doi: 10.1002/glia.20669. [DOI] [PubMed] [Google Scholar]
- 92.Paintlia AS, Paintlia MK, Singh A, Singh I. Modulation of Rho-ROCK Signaling Pathway Protects Oligodendrocytes Against Cytokine Toxicity via PPAR-a Dependent Mechanism. Glia. 2013 doi: 10.1002/glia.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]