

Abstract
The mechanistic target of rapamycin (mTOR), an evolutionally conserved serine and threonine kinase, plays a critical role in the promotion of cell growth and proliferation via integration of cellular and environmental cues. In adaptive immunity, the mTOR pathway orchestrates multiple physiological processes including the development and homeostasis of T cells under steady state, and their subsequent activation and differentiation upon antigen recognition. Associated with such fate decisions is the dynamic reprogramming of T cell metabolic pathways, as naïve, activated and memory cells are defined by distinct bioenergetic and biosynthetic activities. Emerging evidence indicates that mTOR signaling intersects with T cell metabolism at two major levels to constitute a critical control mechanism of T cell fate decisions. First, as a central environmental sensor, mTOR links immune signaling and the availability of nutrients, especially amino acids. Second, mTOR activates specific metabolic pathways in T cells such as aerobic glycolysis (also known as the “Warburg effect”) in a process dependent upon the induction of transcription factors MYC and HIF1α. Understanding how mTOR interplays with T cell metabolism to dictate T cell fates and functions will provide fundamental insights into the mechanism of immune responses and the development of novel therapeutics against immune-mediated diseases. In this review, we summarize the current advances on mTOR signaling and T cell metabolism in the control of development, homeostasis, activation and differentiation of T cells.
Keywords: mTOR, T cell metabolism, T cell quiescence, CD4+ T cell differentiation, CD8+ memory T cell
1. Introduction
The mechanistic target of rapamycin (mTOR; originally known as the mammalian target of rapamycin), an evolutionally conserved serine and threonine kinase, has been well defined as a critical environmental sensor. mTOR integrates a diverse array of cellular and environmental inputs, such as hormones, growth factors and nutrients, to orchestrate multiple cellular functions, including metabolism, growth, survival, and aging [1]. This kinase was originally identified as a crucial regulator in the control of cell proliferation via genetic screening of Saccharomyces cerevisiae. In mammalian cells, mTOR exists in two multi-protein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Fig. 1). mTORC1 contains the regulatory associated protein of mTOR (RAPTOR), mammalian lethal with Sec13 protein 8 (mLST8, also known as GbL), proline-rich AKT substrate 40 kDa (PRAS40) and DEP-domain-containing mTOR-interacting protein (DEPTOR). mTORC1 is sensitive to the immunosuppressant rapamycin and is involved in cell growth, proliferation and metabolism [1]. mTORC1 activity is tightly controlled by the upstream complex comprised of tuberous sclerosis complex 1 (TSC1) and TSC2, the mutation of which is observed in genetic hamartoma syndrome with overgrowth of multiple tissues [2]. The TSC1/TSC2 complex inhibits mTORC1 activation via suppression of the GTPase activity of RHEB (RAS homologue enriched in brain), while the TSC function itself is positively and negatively regulated by the distinct phosphorylation sites in TSC2 that are targeted by various signaling pathways, including PI3K-AKT and AMP activated kinase (AMPK). For downstream targets of mTORC1, S6K1 and 4E-BP1 are the two best characterized and they play an important role in mRNA translation to promote cell growth and proliferation. In contrast to mTORC1, the biological function of mTORC2 is less understood. mTORC2 consists of rapamycin-insensitive companion of mTOR (RICTOR), mammalian stress-activated protein kinase interacting protein (mSIN1), protein observed with Rictor-1 (PROTOR-1), mLST8, and DEPTOR (Fig. 1). Unlike mTORC1, mTORC2 is insensitive to acute rapamycin treatment but is activated by growth factors. It is involved in the regulation of cell survival, metabolism and cytoskeletal organization by activating AGC kinases, including AKT, SGK1 and PKCα [1].
Fig. 1.

Components of mTOR signaling. For mTORC1 activation, the TSC1–TSC2 complex integrates positive and negative signals from PI3K-AKT and LKB1-AMPK pathways, respectively. TSC-independent pathways may also feed into mTORC1 (e.g. AMPK-mediated direct phosphorylation of RAPTOR results in mTORC1 inhibition). Activation of mTORC2 is less studied, although the ribosome has been implicated in this process. Distinct downstream effector molecules mediate the functions of mTORC1 and mTORC2.
Among the plethora of functions regulated by mTOR, a pivotal role is on cellular metabolism [1,3,4]. Metabolism refers to the intracellular chemical reactions that convert nutrients and endogenous molecules into energy and biomass. T cells employ different metabolic programs to fulfill their bioenergetic and biosynthetic demands at different states (Fig. 2). Naïve T cells are in a quiescent state, and they utilize oxidative phosphorylation (OXPHOS) of glucose, amino acids and lipids in mitochondria to generate ATP for their survival [5–7]. IL-7 receptor signaling and autophagy are implicated in the shaping of cellular metabolism for the survival of naïve T cells [8,9]. In contrast, antigen-primed T cells switch from catabolism to anabolism to fulfill the increased metabolic and biosynthetic demands for extensive clonal expansion [5–7]. Activated T cells exhibit a marked increase in glycolysis but suppression of fatty acid β-oxidation. The reliance on glycolysis (even in the presence of high levels of oxygen) to generate ATP, which is far less efficient than OXPHOS, is an unusual metabolic aspect of proliferating T cells and cancer cells, a phenomenon known as the “Warburg effect”. Despite the low efficiency of energy generation, aerobic glycolysis provides the essential materials for the synthesis of nucleic acids and phospholipids. Moreover, activated T cells enhance nutrient availability by upregulation of amino acid transporters, the transferrin receptors and glucose transporters, to ensure a high-level metabolic supply for rapid proliferation [5–7]. Conversely, limitation of nutrient availability inhibits T cell functions and immune responses [10].
Fig. 2.

The interplay between mTOR and metabolism dictates T cell fate decisions. The specific signaling molecules and transcription factors that mediate the crosstalk with mTOR are shown in parentheses.
Emerging evidence indicates that mTOR signaling intersects with T cell metabolism at two major levels to constitute a critical control mechanism of T cell fate decisions (Fig. 2). First, as a central environmental sensor, mTOR links growth factor and immune signaling and the availability of nutrients, especially essential amino acids [1,11]. This ancient pathway of amino acid metabolism is acquired by the immune system as an important mechanism for immune regulation [12]. For example, pharmacological inhibition of amino acid metabolism attenuates mTORC1 activity, which contributes to the downregulation of T cell activation and the induction of T cell anergy [13,14]. Moreover, in response to Treg cell-mediated suppressive signals, dendritic cells upregulate the enzymes that consume multiple essential amino acids present in the immune microenvironment. Such nutrient starvation renders T cells to downregulate mTORC1 activity, and this induces the Treg-specific transcription factor FOXP3 [15]. Second, mTOR activation stimulates specific metabolic pathways, including glycolysis, lipid synthesis and mitochondrial activity [16,17]. In particular, in antigen-stimulated T cells, the glycolytic program is activated by mTOR via the downstream transcription factors HIF1α and MYC and their gene targets comprised of glucose transporters and metabolic enzymes (Fig. 3) [18,19]. Therefore, proper coordination of mTOR activity with T cell metabolism is essential to dictate T cell fate decisions.
Fig. 3.
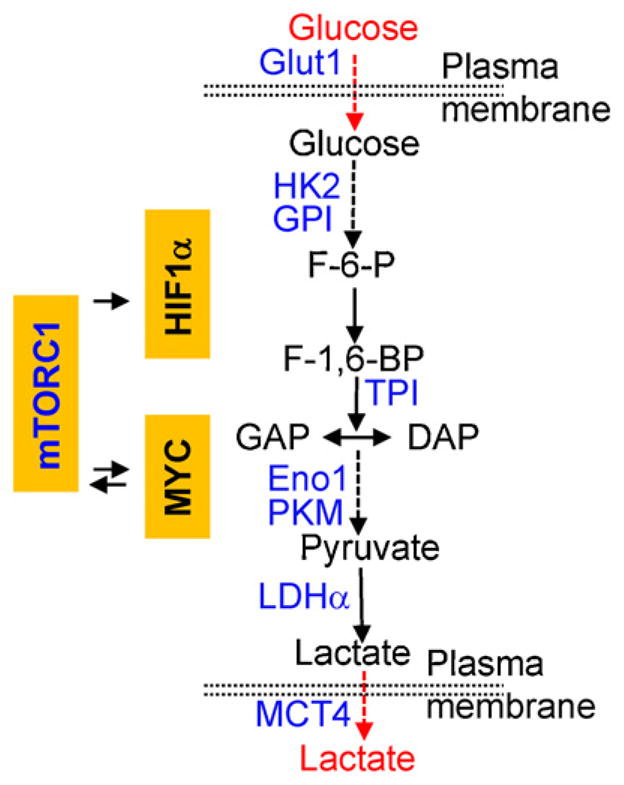
mTORC1 and metabolic control of glycolysis – the Warburg effects. mTORC1 activity induces the expression of MYC and HIF1α in T cells, which in turn mediate the expression of glycolytic enzymes and transporters (in blue), thereby promoting aerobic glycolysis as an important checkpoint for T cell activation and differentiation. MYC is also engaged in a reciprocal interaction with mTORC1, likely via the downstream target CD98 that is important for amino acid uptake and mTORC1 activation.
Here we discuss the recent advances in our understanding of how mTOR signaling and metabolic pathways, and in particular the interplay between them, orchestrate the development and homeostasis of T cells under steady state, and the activation and differentiation of T cells upon antigen recognition. Given the signal-dependent and context-specific regulation of mTOR and metabolic signaling, our discussion is mainly focused on the findings observed in T cells, especially those derived from the genetic systems.
2. Positive and negative components of mTOR regulate thymocyte development
The development of T cells in the thymus is a stepwise process involving the crosstalk of multiple signaling pathways. Starting as CD4−CD8−double-negative (DN) cells, T cell progenitors progress into CD4+CD8+ double-positive (DP) stage after β-selection, and then further mature into CD4+ and CD8+ single-positive cells following positive and negative selections [20]. Genetic dissection of positive and negative components of mTOR signaling has revealed their important roles in this highly ordered developmental process, although little direct evidence is available presently to link these functions to the metabolic control mechanism.
2.1. RICTOR/mTORC2
In the early stage of T cell development, NOTCH and pre-TCR provide critical signaling for thymocyte differentiation and proliferation [21]. Recent studies, via conditional deletion of RICTOR, have identified important functions of mTORC2 in thymocyte development [22,23]. Using Vav1-mediated hematopoietic deletion of RICTOR, Lee et al. reported that mTORC2 mediates NOTCH-driven proliferation and differentiation of pre-T cells. mTORC2 signals to both the canonical NF-κB and FOXO pathways, and the concerted actions of these two pathways mediate mTORC2 function in promoting the DN to DP transition. In contrast, RICTOR is dispensable for cell growth and glycolytic rate in thymocytes. Importantly, mTORC2 depletion diminishes NOTCH-driven T cell leukemia development [22]. In another independent study, Fang et al. reported that deficiency of RICTOR in the thymus impairs the proliferation of immature thymocytes and results in a partial block of thymic development. Notably, the effect of RICTOR deficiency is selective for the T cell lineage, as the development of B cells and myeloid cells is largely unaffected [23]. These findings provide important genetic evidence for the physiological functions of mTORC2 in thymocyte development. The function of mTORC1 in this process remains to be determined.
2.2. PTEN
The tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a well-established negative regulator of the PI3-kinase pathway [24]. Loss of PTEN expression or activity is frequently associated with lymphomagenesis. T cell-specific deletion of PTEN results in CD4+ T cell lymphoma and an increased thymic cellularity, which is in part due to defective negative selection [25]. PTEN deficiency promotes the proliferation of developing thymic T cells, allowing them to bypass the requirement of IL-7R and pre-TCR signaling [26]. In line with the increased proliferation, PTEN-deficient thymocytes upregulate the expression of the MYC oncogene that could potentially evoke the metabolic machinery and cell cycle pathways [27,28]. Elevated activation of AKT and mTOR in PTEN-deficient thymocytes plays an important role in tumor development, as the lymphoma is blocked by rapamycin treatment or PDK1 deficiency [29,30]. Interestingly, PTEN deficiency does not overtly disrupt thymocyte homeostasis prior to the onset of malignancy, except for some modest disturbances, including a slight increase in cell size and modestly altered development of invariant natural killer T (iNKT) cells [31]. In addition to the alterations in the thymus, PTEN-deficient T cells are spontaneously active in the periphery and mediate the development of autoimmune disease [28]. The studies indicate that lack of PTEN in T cells results in lymphoma and autoimmunity that are mediated by T cells of distinct developmental stages.
2.3. LKB1
Tumor suppressor the liver kinase B1 (LKB1), a serine/threonine kinase, is inactivated in the Peutz-Jeghers familial cancer syndrome [32]. LKB1-AMPK plays an important role in the regulation of energy homeostasis and cell growth via the crosstalk with mTOR [32]. Recent studies highlight the functions of LKB1 in T cell development, survival, function and metabolism [33–36]. Deficiency of LKB1 in T cells profoundly affects thymocyte development, including a blockade of transition from DN3 to DN4 and disruption of positive selection by abrogating TCR signaling [33,35,36]. Defective thymocyte development is associated with increased cell death that is ascribed to the impaired AMPK activation and Bcl-XL expression [33,36]. LKB1 has also been implicated in the activation, survival and metabolism of peripheral T cells [36]. However, loss of AMPKα1, the predominant AMPK isoform in T cells, causes only slight disturbance of T cell homeostasis [36,37]. Therefore, further work is required to dissect the functions of LKB1 and AMPK and their crosstalk with mTOR signaling in T cell development and homeostasis.
3. Active control of mTOR activity enforces peripheral T cell homeostasis
Mature T cells in the extrathymic environment are long-lived cells in a quiescent state characterized by a small cell size, catabolic metabolism, and exit from the active cell cycle (G0). Engagement of TCRs by the self-peptide-MHC complex and the interaction between IL-7 and IL-7R keep naïve T cells alive for many weeks or months in the periphery [38]. In the steady state, quiescent T cells catabolically generate energy for their survival by oxidative phosphorylation of glucose, amino acids and lipids [5–7], but molecular pathways that establish quiescence in T cells remain poorly understood [39]. Although accumulating evidence indicates that mTOR functions as an integral sensor of cellular and environmental cues, mTOR deficiency unexpectedly does not alter homeostasis of peripheral T cells under the steady state [40]. In contrast, deficiency of TSC1 and the ensuing activation of mTORC1 disrupt naïve T cell homeostasis, associated with aberrant metabolism and defective survival, indicating that keeping mTORC1 in check is essential in the maintenance of T cell homeostasis [41–44].
TSC1 and TSC2 function as an integral complex in the stringent control of mTORC1 activity [2]. We and others have recently uncovered that TSC1 serves as a bona fide factor to enforce a quiescent program in naïve T cells to facilitate adaptive immune homeostasis and function [41–44]. TSC1 deficiency leads to the loss of quiescence manifested by a profound increase of cell size of naïve T cells, a hyperactive cellular response to acute TCR stimulation, and the spontaneous development of a unique ‘semi-activated’ (CD44+CD122−) population in vivo. Moreover, TSC1-deficient naïve T cells display upregulation of genes involved in multiple metabolic pathways, including OXPHOS and metabolism of nucleotides, sterols, amino acids and carbohydrates. Associated with the metabolic dysregulation, TSC1-deficient naïve T cells aberrantly enter active cell cycle but are prone to apoptosis, and importantly, are impaired in the generation of antigen-specific adaptive immunity [41]. These results suggest that TSC1 keeps the metabolic and cell cycle machineries in check and hence maintains the quiescence of naïve T cells. Mechanistically, TSC1-deficient T cells exhibit increased mTORC1 but diminished mTORC2 activities. Treatment of rapamycin in vivo partly restores the altered T cell homeostasis and survival, whereas loss of mTORC2 activity alone has no apparent effects on naïve T cell homeostasis [41]. These data illustrate that TSC1-dependent active control of mTORC1 is a key checkpoint to enforce quiescence of naïve T cells in the periphery.
In addition to TSC1, other negative regulators of mTOR are also involved in peripheral T cell homeostasis. As described above, PTEN deficiency predisposes the development of autoimmunity that can be exclusively mediated by peripheral T cells [28]. Loss of LKB1 results in the accumulation of activated T cells with excessive cytokine production in the periphery and an increased rate of glycolytic activity that is associated with elevated expression of glucose transporters and glycolytic enzymes. In addition, LKB1-deficient peripheral T cells show defective cell viability and proliferation upon TCR stimulation. The increased mTORC1 activity in LKB1-deficient T cells contributes to excessive cytokine production, but whether it leads to reduced cell survival or other defects is unclear [36]. Notably, although deletion of TSC1, PTEN or LKB1 each disrupts T cell homeostasis, the resulting phenotypes are rather different, indicative of the distinct downstream effector pathways mediated by these molecules. The precise mechanisms by which mTOR and metabolic pathways regulate the homeostasis of naïve T cells in the periphery await future investigation.
4. mTOR and metabolic reprogramming drive T cell activation
T cell activation is a process in which cell proliferation is coordinated to the metabolic activity. Antigen-primed T cells are capable of undergoing rapid proliferation and doubling every 4–6 h at the peak of the response [45]. Concomitantly, T cells initiate a diverse array of transcriptional and translational responses to allow the uptake and utilization of extracellular nutrients. To ensure efficient proliferation, T cells metabolically switch from oxidative phosphorylation to anabolic metabolism including glycolysis (the Warburg effect) and glutaminolysis to meet the increased bioenergetic and biosynthetic demands [5–7,19]. Recent studies have identified specific signaling pathways and transcription factors involved in the coordination of cell proliferation and metabolism in activated T cells that dictate the outcome of adaptive immune responses.
4.1. mTOR in T cell activation and anergy
Early studies on mTOR signaling in T cell responses centered upon the anti-proliferative effect of the inhibitor rapamycin. Analysis of mTOR-deficient T cells has confirmed a role for mTOR in cell cycle progression, although T cell proliferation is delayed but not abolished [40]. Conversely, T cells deficient in TSC1 or TSC2 exhibit increased proliferation under selective TCR-stimulated conditions [46,47]. In addition to impairing T cell activation, inhibition of mTOR facilities the induction of T cell anergy. T cell anergy is usually induced by TCR (signal 1) alone, in the absence of co-stimulation (signal 2). Among the pathways strongly activated by signal 2 are AKT and mTOR. Blocking mTOR activity by rapamycin induces T cell anergy in vitro, even in the presence of both signals 1 and 2. In vivo, mTOR inhibition promotes T cell anergy under conditions that would normally induce active priming, indicating that mTOR plays a decisive role to distinguish T cell activation and anergy [13,14]. Consistent with this notion, T cells lacking TSC1 are refractory to the induction of T cell anergy [46]. Although rapamycin treatment inhibits the G1 phase of the cell cycle, blocking T cell proliferation alone does not induce anergy. Instead, blocking metabolic pathways necessary for mTOR activation promotes T cell anergy, suggesting that mTOR-dependent metabolic control is a key determinant of T cell activation and anergy [13,14].
4.2. Crosstalk between mTOR and MYC in T cell activation
MYC is an oncogenic transcription factor with a well-established role in the promotion of cell proliferation and metabolism that contributes to the transformation of cancer [48]. Recent evidence highlights that MYC acts as a pivotal regulator in the metabolic reprogramming during T cell activation [19]. Acute deletion of MYC markedly suppresses activation-induced glycolysis and glutaminolysis via abrogation of expression of rate-limiting enzymes in these processes. Disrupted T cell proliferation and growth in MYC-deficient T cells is partially rescued by the addition of nucleotides and polyamines [19], indicative of a particular importance of MYC-dependent glutaminolysis in cell proliferation. Furthermore, the elevated uptake of amino acids is an important event in proliferating cells, and mTOR couples the uptake of exogenous amino acids to the stimulation of cap-dependent translation in many cell types [11]. Interestingly, although general TCR-induced signaling pathways, including AKT and ERK, are largely intact in MYC-deficient T cells, mTOR activity is significantly suppressed, associated with the impaired expression of CD98, a surface molecule essential for the uptake of amino acids [19]. Given the important role of amino acids in mTOR activation, these results suggest that MYC-dependent CD98 expression and amino acid uptake establish a key crosstalk with mTOR during T cell activation. In a reciprocal manner, blocking mTOR activity with rapamycin diminishes TCR-induced MYC expression and glycolysis, thereby placing mTOR upstream of MYC as well (Fig. 3) [19]. The molecular details in the crosstalk between mTOR and MYC will be an important area to explore.
4.3. Other transcription factors in the metabolism of activated T cells
The estrogen-related receptor-alpha (ERRα), an orphan nuclear receptor, is a ligand-regulated transcription factor controlling multiple metabolic pathways. Aberrant expression of ERRα is associated with cancer development and metabolic disorders. A role for ERRα in the metabolic control of T cell growth and proliferation was recently identified [47]. ERRα is expressed at low levels in resting T cells, but is markedly induced by TCR and co-stimulation. Lack of ERRα impairs metabolic pathways including glycolysis and mitochondrial biogenesis, resulting in the impaired T cell growth and proliferation [47]. Further, the defective anabolic metabolism in ERRα-deficient T cells is associated with diminished effector T cell differentiation and autoimmune disease development [47]. Although the physiological ligand of ERRα remains unknown, ERRα broadly interacts with multiple signaling pathways to regulate T cell metabolism. ERRα-deficient T cells display altered mTOR and AMPK activities, suggesting that ERRα might regulate T cell metabolism through these pathways [47]. Additionally, both ERRα and MYC-deficient T cells exhibit similar defects in certain metabolic pathways, such as glycolysis and mitochondrial metabolism, suggesting the potential crosstalk between these transcription factors.
Similar as MYC, HIF1α is another well-documented transcription factor mediating the glycolytic activity of transformed cancer cells [48]. HIF1α is induced after T cell activation in an mTOR-dependent manner. However, specific deletion of HIF1α in T cells has no significant effects on glycolysis or T cell proliferation following immediate T cell activation [19]. Finally, the nuclear receptor Liver X receptor (LXR) is actively suppressed following T cell stimulation to promote cholesterol synthesis and clonal expansion [49]. Collectively, these results suggest that specific transcription factors are involved in the metabolic reprogramming of activated T cells, and these metabolic checkpoints serve as a key mechanism to facilitate activation-induced cell proliferation and growth. Given the pivotal role of mTOR in sensing cellular and environmental cues, it is important to further investigate how mTOR interplays with these metabolic pathways during T cell activation.
5. mTOR and metabolic pathways shape CD4+ T cell differentiation
Activated CD4+ T cells differentiate into a diverse range of lineages, including Th1, Th2 and Th17 effector cells and induced regulatory T (iTreg) cells, and this process is dictated by multiple cues from the immune microenvironment, especially the polarizing cytokines [50]. Distinct requirement of metabolic programs is associated with the differentiation of effector and regulatory T cell lineages. Generation of effector T cells is accompanied by strong upregulation of glycolysis, due to the transcriptional induction of glucose transporters and glycolytic enzymes [18,51]. In contrast, iTreg cells exhibit a low-degree of glycolytic activity but rely on lipid oxidation for their generation and function [18,51]. mTOR signaling and specific transcription factors have been identified to regulate the metabolic programs and fate decisions of CD4+ T cells.
5.1. Distinct roles of mTORC1 and mTORC2 in Th1 and Th2 differentiation
Delgoffe et al. reported the first genetic evidence on the role of mTOR in promoting the differentiation of effector T cells. T cells deficient in mTOR exhibit enhanced iTreg generation but defective differentiation into Th1, Th2 and Th17 effector cells [40]. Interestingly, the two different mTOR complexes exert disparate effects on effector T cell differentiation, as revealed by specific deletion of RHEB and RICTOR, which disrupt the activities of mTORC1 and mTORC2, respectively. RHEB deletion impairs Th1 and Th17 differentiation without a substantial effect on Th2 differentiation, and this is associated with the reduced development of autoimmune neuroinflammation [52]. Conversely, RICTOR-deficient T cells fail to become Th2 but retain the ability to develop into Th1 and Th17 cells. Mechanistically, RHEB and RICTOR have been shown to differentially regulate selective SOCS proteins and STAT signaling [52]. In an independent study, Lee et al. reported that RICTOR-deficient T cells display defective differentiation of both Th1 and Th2 cells. Impaired AKT and NF-κB activation downstream of mTORC2 contributes to defective Th1 and Th2 differentiation, respectively [53]. The role of mTOR in the metabolic activities of Th1 and Th2 cells has not been tested, although glycolysis is clearly required for T cells to differentiate into these lineages [18].
5.2. Inhibitory roles of mTOR for iTreg differentiation
Blocking mTOR activity by rapamycin has long been known to induce de novo FOXP3 expression and promote iTreg generation [54,55]. Deletion of mTOR results in spontaneous development of Foxp3+ T cells in TCR-activated cells [40], and this effect requires both mTORC1 and mTORC2 activities [52]. Conversely, increasing AKT activity, through either deletion of PTEN or enforced expression of constitutively active AKT, leads to mTOR-dependent inhibition of iTreg differentiation [56,57]. Moreover, several upstream receptors, including PD-L1 and S1PR1, inhibit iTreg differentiation by engaging the mTOR pathway [58–60]. Two downstream pathways have been shown to mediate the inhibitory effects of mTOR on iTreg differentiation. First, SMAD3, a key transcription factor transducing TGF-β signaling for iTreg cell generation, is constitutively phosphorylated in mTOR-deficient T cells [40]. Second, FOXO1 and FOXO3, which mediate FOXP3 gene expression, are inactivated by AKT-dependent phosphorylation [61–63], although whether this depends upon mTORC2 signaling requires additional experimental evidence.
5.3. The mTOR-HIF1α axis in Th17 differentiation
Shi et al. reported that the key glycolytic transcription factor HIF1α is selectively expressed in T cells undergoing Th17 differentiation [18]. Bioinformatic analysis of HIF1α-dependent gene expression profiles shows that the 10 pathways with the most significant enrichment are all associated with glycolytic or related metabolic pathways. Blocking glycolysis via HIF1α deletion or treatment with the pharmacological inhibitor 2-deoxyglucose (2-DG) impairs Th17 differentiation, and instead promotes iTreg cell generation and protects mice from autoimmune neuroinflammation [18]. In agreement with a role of mTOR in the induction of HIF1α during Th17 differentiation, rapamycin treatment suppresses glycolysis and Th17 differentiation [18]. Therefore, the mTOR-HIF1α axis and the downstream glycolytic program contribute to the lineage choices between Th17 and iTreg cells (Fig. 3). An independent study reveals that HIF1α promotes FOXP3 degradation and RORγt transcriptional activity to directly regulate iTreg and Th17 differentiation [64], and whether these activities interplay with the glycolytic program will be interesting to explore. Aside from a requirement in glycolysis, Th17 differentiation relies on amino acid metabolism [65], but is negatively controlled by LXR/SREBP-dependent lipid metabolism [66]. Whether mTOR regulates these additional metabolic processes in T cells remains to be established.
6. mTOR and metabolic pathways control CD8+ T cell memory
Engagement of antigen receptors on CD8+ T cells leads to the generation of effector T cells. While most terminally differentiated effector T cells die during the contraction phase, a small subset differentiates into long-lived memory T cells that facilitate subsequent responses to the re-challenge [67]. What determines the choice between effector and memory T cell differentiation is an important question, as it directly relates to the strategy of improving long-lasting protective T cell immunity. Emerging evidence indicates that mTOR and metabolic signals play an important role in this process, partly by coordinating cell metabolism and trafficking to dictate the final outcome of the adaptive immune response.
6.1. Inhibitory effects of mTOR signaling on CD8+ T cell memory
Araki et al. reported that CD8+ memory T cell development is profoundly affected by rapamycin treatment during the course of acute infection with lymphocytic choriomeningitis virus (LCMV). Treatment of mice with rapamycin following LCMV infection enhances both the quantity and quality of CD8+ memory T cells [68]. Specifically, rapamycin treatment during the expansion phase increases the number of memory precursors, while the treatment during the contraction phase promotes the quality of CD8+ memory T cells. RNA interference targeting RAPTOR expression promotes CD8+ memory T cell differentiation, suggesting that mTORC1 is important in the regulation of memory T cell development [68]. Mechanistically, Rao et al. describes that mTOR exerts differential effects on the transcription factor T-bet and EOMES [69], which determine effector and memory T cell differentiation, respectively [70]. Blocking mTOR activity by rapamycin inhibits T-bet but promotes EOMES expression, thereby enhancing the generation of CD8+ memory T cells at the expense of effector cells [69]. In addition, inhibition of mTOR activity enhances homeostatic proliferation-induced CD8+ memory T cells, which is associated with increased tumor immunity [71]. More recently, transcription factor FOXO1 has been shown to be a key regulator that mediates the role of mTOR in the control of T-bet and EOMES expression [72]. Inactivation of FOXO1 leads to T-bet expression and subsequent effector T cell differentiation. However, inhibition of mTOR activity reverses FOXO1 activity to induce EOMES expression and subsequent memory development [72]. Collectively, mTOR acts as a central regulator by linking immune signals to downstream transcription factors to orchestrate effector and memory T cell differentiation. Targeting mTOR pathway may provide an effective strategy to improve vaccine- or infection-induced memory T cell responses.
6.2. Mitochondrial fatty acid oxidation in the promotion of memory responses
Modulation of mitochondrial metabolism is intricately involved in the development of CD8+ effector and memory T cells. During the expansion phase of CD8+ T cell responses, T cells upregulate glycolytic metabolism to fuel the bioenergetic and biosynthetic demands for cell growth and proliferation. In contrast to effector T cells, memory T cells switch the anabolic metabolism to oxidative phosphorylation for energy generation, indicative of reestablishment of a quiescent state [6]. Recent studies indicate that regulation of mitochondrial fatty acid oxidation (FAO) is an important mechanism underlying the differentiation of CD8+ memory T cells. Pearce et al. reported that TRAF6 (tumor necrosis factor receptor-associated factor 6) is an upstream regulator of FAO in CD8+ memory T cell development. TRAF6-deficient T cells elicit robust effector T cell responses, but are profoundly impaired to generate long-lived memory T cells [73]. Gene profiling analysis reveals that deficiency of TRAF6 leads to defective expression of genes involved in FAO. Moreover, AMPK activation is diminished in TRAF6-deficient T cells upon cytokine withdrawal, suggesting that AMPK likely mediates TRAF6-dependent FAO metabolism in the generation of memory T cells [73]. Consistent with this notion, activation of AMPK by metformin or inhibition of mTOR by rapamycin restores the generation of CD8+ memory T cells in TRAF6-deficient mice [73]. More recently, van der Windt et al. reported that CD8+ memory but not effector T cells possess substantial mitochondrial spare respiratory capacity (SRC) [74]. SRC is the extra capacity available in cells to produce energy in response to increased stress or work and as such is associated with cellular survival. IL-15, a cytokine critical for CD8+ memory T cells, regulates SRC and oxidative metabolism by promoting mitochondrial biogenesis and expression of carnitine palmitoyl transferase (CPT1a), a metabolic enzyme that controls the rate-limiting step to mitochondrial FAO [74]. Collectively, these studies highlight that regulation of FAO acts as a metabolic checkpoint in the differentiation of effector and memory T cells. How mTOR and AMPK signaling interplays with FAO and other mitochondrial metabolism awaits further investigation.
6.3. mTOR-mediated coordination of metabolism and trafficking
Chemokine receptors and trafficking molecules direct the systemic and local trafficking of T cells, which is an integral component of in vivo T cell responses. Homeostatic homing receptors, such as CCR7, CD62L and S1PR1, mediate the entry of naïve and memory T cells to peripheral lymph nodes [75,76]. In contrast, upregulation of pro-inflammatory chemokine receptors and adhesion molecules such as CXCR3 allows antigen-primed T cells to traffic to sites of inflammation to exert their effector function. mTOR differentially regulates the expression of homeostatic and pro-inflammatory homing receptors that further impacts the generation of effector and memory T cells. Inhibition of mTOR activity by rapamycin prevents downregulation of CCR7, CD62L and S1PR1 in antigen-primed CD8+ T cells, while increasing mTOR activity via PTEN deletion downregulates the expression of these receptors. Moreover, rapamycin facilitates the trafficking of T cells to secondary lymphoid tissues rather than to sites of inflammation [75], and this is associated with the increased CD8+ memory T cell generation [69]. At the molecular levels, mTOR regulates the expression of these receptors likely through its effect on the transcription factors FOXO1 and KLF2 [77,78], although detailed mechanisms are yet to be identified.
In addition to controlling the expression of homeostatic trafficking receptors, mTORC1 is critical for the expression of T-bet [69], which, among many of its downstream effects, directs the expression of pro-inflammatory chemokine receptors to coordinate T cell migration with effector functions. In particular, the T-bet target CXCR3 mediates the recruitment of activated CD8+ T cells to sites of inflammation and thereby promoting the effector function [79]. Consistent with such observations, CXCR3 promotes the formation of short-lived effector CD8+ T cells at the expanse of long-lived memory precursor CD8+ T cells [80–82]. Moreover, signaling via CXCR3 directly activates mTORC1 [83], indicative of the reciprocal interaction between mTORC1 and CXCR3. Therefore, mTORC1-dependent regulation of chemokine receptor expression and function is intricately involved in the decision between effector and memory cell generation. Whether this process is linked to the metabolic role of mTOR will be an interesting topic to explore.
7. Conclusions
Collectively, recent studies highlight that mTOR plays a pivotal role in adaptive immune responses by the coordination of cellular metabolism with T cell development, quiescence, activation and differentiation. The multiple negative regulators of mTOR actively control mTOR activity to ensure proper T cell development, survival and quiescence in the steady state. Upon antigen stimulation, elevation of mTOR activity by a diverse array of signals from immune microenvironment, including antigens, cytokines, inflammatory factors, nutrients and chemokines, reprograms cellular metabolism to orchestrate cell growth, proliferation and differentiation. The interplay between mTOR and cell metabolism during the dynamics of immune response likely serves as a central module to integrate upstream signals for the determination of the outcome of immune reactions. From this perspective, dissection of the crosstalk of mTOR with multiple upstream signals in the immune environment, in particular the metabolic cues such as availability of essential amino acids, is an important step to understand how T cells coordinate cellular metabolism with T cell growth, proliferation and differentiation.
Equally important is to identify the precise mechanisms whereby mTOR activates specific metabolic programs in T cells, and how these downstream pathways impinge upon T cell fate decisions. Although mTOR has been shown to regulate multiple metabolic pathways in cell lines, including glycolysis, lipid synthesis and mitochondrial activity [16,17], we are only beginning to appreciate the interplay between mTOR and the glycolytic program in T cells at the gene expression level [18,19]. Moreover, how the bioenergetic and biosynthetic pathways contribute to T cell fate decisions is unclear, and we have recently proposed three potential downstream mechanisms: signal crosstalk through metabolite-sensitive signaling targets, feedback control via mTORC1, and selective expansion and survival of metabolically fit populations [3]. The continued exploration of the crosstalk between mTOR and cell metabolism promises to provide new insight into the development of novel therapeutic strategies for effective vaccination as well as for the treatment of inflammatory and autoimmune diseases.
Acknowledgments
We acknowledge the large number of researchers who have contributed to this field whose work was not cited owing to space limitations. We thank Dr. Yanyan Wang for proof reading of the manuscript, and members of the Chi laboratory for helpful discussions. The authors’ research is supported by US National Institutes of Health (R01 NS064599 and R21 AI094089), National Multiple Sclerosis Society, Lupus Research Institute, and the American Lebanese Syrian Associated Charities (H.C.), and an Arthritis Foundation Postdoctoral Fellowship (K.Y.).
Footnotes
Competing interest statement
The authors declare no competing financial interests.
References
- 1.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews: Molecular Cell Biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mieulet V, Lamb RF. Tuberous sclerosis complex: linking cancer to metabolism. Trends in Molecular Medicine. 2010;16:329–35. doi: 10.1016/j.molmed.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nature Reviews: Immunology. 2012;12:325–38. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T Cell differentiation, function, and metabolism. Immunity. 2010;33:301–11. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–8. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Pearce EL. Metabolism in T cell activation and differentiation. Current Opinion in Immunology. 2010;22:314–20. doi: 10.1016/j.coi.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunological Reviews. 2010;236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs SR, Michalek RD, Rathmell JC. IL-7 is essential for homeostatic control of T cell metabolism in vivo. Journal of Immunology. 2010;184:3461–9. doi: 10.4049/jimmunol.0902593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pua HH, He YW. Mitophagy in the little lymphocytes: an essential role for autophagy in mitochondrial clearance in T lymphocytes. Autophagy. 2009;5:745–6. doi: 10.4161/auto.5.5.8702. [DOI] [PubMed] [Google Scholar]
- 10.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Lep-tin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 11.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunological Reviews. 2010;236:243–64. doi: 10.1111/j.1600-065X.2010.00915.x. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, et al. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. Journal of Immunology. 2007;178:2163–70. doi: 10.4049/jimmunol.178.4.2163. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. Journal of Immunology. 2009;183:6095–101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12055–60. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell. 2010;39:171–83. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 18.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1{alpha}-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. Journal of Experimental Medicine. 2011;208:1367–76. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–82. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpenter AC, Bosselut R. Decision checkpoints in the thymus. Nature Immunology. 2010;11:666–73. doi: 10.1038/ni.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annual Review of Immunology. 2005;23:945–74. doi: 10.1146/annurev.immunol.23.021704.115747. [DOI] [PubMed] [Google Scholar]
- 22.Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. Journal of Experimental Medicine. 2012;209:713–28. doi: 10.1084/jem.20111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang F, Wu Q, Ikenoue T, Guan KL, Liu Y, Zheng P. A critical role for rictor in T lymphopoiesis. Journal of Immunology. 2012;189:1850–7. doi: 10.4049/jimmunol.1201057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature Reviews: Molecular Cell Biology. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–34. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 26.Hagenbeek TJ, Naspetti M, Malergue F, Garcon F, Nunes JA, Cleutjens KB, et al. The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. Journal of Experimental Medicine. 2004;200:883–94. doi: 10.1084/jem.20040495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Karnell JL, Yin B, Zhang R, Zhang J, Li P, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. Journal of Clinical Investigation. 2010;120:2497–507. doi: 10.1172/JCI42382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. Journal of Experimental Medicine. 2009;206:2441–54. doi: 10.1084/jem.20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo W, Schubbert S, Chen JY, Valamehr B, Mosessian S, Shi H, et al. Suppression of leukemia development caused by PTEN loss. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1409–14. doi: 10.1073/pnas.1006937108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kishimoto H, Ohteki T, Yajima N, Kawahara K, Natsui M, Kawarasaki S, et al. The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood. 2007;109:3316–24. doi: 10.1182/blood-2006-07-038059. [DOI] [PubMed] [Google Scholar]
- 32.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature Reviews: Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Y, Li H, Liu H, Zheng C, Ji H, Liu X. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Research. 2010;20:99–108. doi: 10.1038/cr.2009.141. [DOI] [PubMed] [Google Scholar]
- 34.Cao Y, Li H, Liu H, Zhang M, Hua Z, Ji H, et al. LKB1 regulates TCR-mediated PLCgamma1 activation and thymocyte positive selection. EMBO Journal. 2011;30:2083–93. doi: 10.1038/emboj.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamas P, Macintyre A, Finlay D, Clarke R, Feijoo-Carnero C, Ashworth A, et al. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. European Journal of Immunology. 2010;40:242–53. doi: 10.1002/eji.200939677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maciver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase b1 is a central regulator of T cell development, activation, and metabolism. Journal of Immunology. 2011;187:4187–98. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. European Journal of Immunology. 2008;38:948–56. doi: 10.1002/eji.200738045. [DOI] [PubMed] [Google Scholar]
- 38.Sprent J, Normal Surh CD. T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nature Immunology. 2011;12:478–84. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends in Immunology. 2012;33:224–30. doi: 10.1016/j.it.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–44. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nature Immunology. 2011;12:888–97. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Q, Liu Y, Chen C, Ikenoue T, Qiao Y, Li CS, et al. The tuberous sclerosis complex-mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. Journal of Immunology. 2011;187:1106–12. doi: 10.4049/jimmunol.1003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brien TF, Gorentla BK, Xie D, Srivatsan S, McLeod IX, He YW, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. European Journal of Immunology. 2011;41:3361–70. doi: 10.1002/eji.201141411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Zhang H, Li L, Xiao Y, Rao E, Miao Z, et al. TSC1/2 signaling complex is essential for peripheral naive CD8+ T cell survival and homeostasis in mice. PLoS One. 2012;7:e30592. doi: 10.1371/journal.pone.0030592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, et al. Dynamic programming of CD8+ T lymphocyte responses. Nature Immunology. 2003;4:361–5. doi: 10.1038/ni912. [DOI] [PubMed] [Google Scholar]
- 46.Xie DL, Wu J, Lou YL, Zhong XP. Tumor suppressor TSC1 is critical for T-cell anergy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14152–7. doi: 10.1073/pnas.1119744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18348–53. doi: 10.1073/pnas.1108856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nature Reviews: Cancer. 2008;8:51–6. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 49.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–55. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. Journal of Immunology. 2011;186:3299–303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nature Immunology. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–53. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. American Journal of Transplantation. 2007;7:1722–32. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. Journal of Leukocyte Biology. 2008;83:1230–9. doi: 10.1189/jlb.1207851. [DOI] [PubMed] [Google Scholar]
- 56.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. Journal of Experimental Medicine. 2008;205:565–74. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7797–802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nature Immunology. 2009;10:769–77. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nature Immunology. 2010;11:1047–56. doi: 10.1038/ni.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. Journal of Experimental Medicine. 2009;206:3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nature Immunology. 2010;11:618–27. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 62.Harada Y, Elly C, Ying G, Paik JH, DePinho RA, Liu YC. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. Journal of Experimental Medicine. 2010;207:1381–91. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kerdiles YM, Stone EL, Beisner DL, McGargill MA, Ch’en IL, Stockmann C, et al. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–8. doi: 10.1126/science.1172638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. Journal of Clinical Investigation. 2011;121:658–70. doi: 10.1172/JCI42974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nature Reviews: Immunology. 2009;9:662–8. doi: 10.1038/nri2619. [DOI] [PubMed] [Google Scholar]
- 68.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–12. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature Immunology. 2005;6:1236–44. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 71.Rosenfeldt HM, Hobson JP, Maceyka M, Olivera A, Nava VE, Milstien S, et al. EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB Journal. 2001;15:2649–59. doi: 10.1096/fj.01-0523com. [DOI] [PubMed] [Google Scholar]
- 72.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–87. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–7. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nature Immunology. 2008;9:513–21. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nature Reviews: Immunology. 2011;11:109–17. doi: 10.1038/nri2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nature Immunology. 2009;10:176–84. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–71. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, et al. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. Journal of Immunology. 2006;177:5890–901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]
- 80.Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, et al. Inflammatory chemokine receptors regulate CD8(+) T cell contraction and memory generation following infection. Journal of Experimental Medicine. 2011;208:1621–34. doi: 10.1084/jem.20102110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, et al. Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short-lived effector cells leading to memory degeneration. Journal of Experimental Medicine. 2011;208:1605–20. doi: 10.1084/jem.20102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E118–27. doi: 10.1073/pnas.1101881108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwarz JB, Langwieser N, Langwieser NN, Bek MJ, Seidl S, Eckstein HH, et al. Novel role of the CXC chemokine receptor 3 in inflammatory response to arterial injury: involvement of mTORC1. Circulation Research. 2009;104:189–200. doi: 10.1161/CIRCRESAHA.108.182683. [DOI] [PubMed] [Google Scholar]