

Abstract
Cystic fibrosis (CF) is a major genetic disease in Caucasians affecting 1 in 2500 newborns. Hepatobiliary pathology is a major cause of morbidity and mortality in CF second only to pulmonary disease. SULT1E1 activity is significantly elevated, generally 20–30-fold, in hepatocytes of mouse models of CF. SULT1E1 is responsible for the inactivation of β-estradiol (E2) at physiological concentrations via conjugation with sulfonate. The increase in SULT1E1 activity results in the alteration of E2-regulated protein expression in CF mouse liver. To investigate the mechanism by which the absence of CFTR in human cholangiocytes induces SULT1E1 expression in hepatocytes, a membrane-separated human MMNK-1 cholangiocyte and human HepG2 hepatocyte co-culture system was developed. The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in bile duct cholangiocytes but not hepatocytes, whereas SULT1E1 is expressed in hepatocytes but not cholangiocytes. CFTR expression in MMNK-1 cells was inhibited with siRNA by >90% as determined by immunoblot and immunohistochemical analysis. Control and CFTR-siRNA-MMNK-1 cells were co-cultured with HepG2 cells in a Transwell membrane-separated system. After 8 h of co-culture, HepG2 cells were removed from exposure to MMNK-1 cells and placed in fresh medium. After 24–48 h, expression of SULT1E1 and selected E2-regulated proteins was analyzed in the HepG2 cells. Results demonstrated that SULT1E1 message and activity were selectively induced in HepG2 cells co-cultured with CFTR-deficient MMNK-1 cells. The expression of E2-regulated proteins (IGF-1, GST-P1 and carbonic anhydrase II) was also altered in response to decreased E2 levels. Thus, the loss of CFTR activity in cholangiocytes stimulates the expression of SULT1E1 in hepatocytes by a paracrine mechanism. SULT1E1 expression in HepG2 cells is inducible by sterol mediated liver-X-receptor (LXR) activation although not by progestins that induce SULT1E1 in the endometrium. SULT1E1 induction in the human cholangiocyte/hepatocyte co-culture system is consistent with and supports the results observed in CF mice. The changes in hepatocyte gene expression affect liver biochemistry and may facilitate the development of CF liver disease.
Keywords: Cystic fibrosis, Liver disease, Cystic fibrosis transmembrane conductance, regulator, Sulfotransferase, SULT1E1, β-Estradiol, Hepatocytes, Cholangiocytes, Oxysterols, Liver-X-receptor
1. Background
Cystic fibrosis (CF) is a major lethal genetic disease in Caucasians that affects 1 in 2500 newborns. CF is an autosomal recessive disorder caused by disruption of the expression and function of the gene encoding the CF transmembrane receptor (CFTR) protein [1]. CFTR plays a crucial role in chloride (Cl−) secretion; absent or diminished CFTR activity confers a phenotypic deficit in Cl− secretion resulting in the clinical manifestations of CF. CFTR is a cAMP-regulated chloride channel located on the apical plasma membrane of most absorptive and secretory cells including cholangiocytes. Its dysfunction impairs Cl− secretion and therefore Cl−/HCO3- exchange resulting in the inability to maintain a normal secretory response across cellular membranes [2, 3].
In humans, the major clinical problems of CF involve a loss of pulmonary function due to changes in mucus structure and associated bacterial infections [4]. Over the last three decades, advances in the pulmonary and nutritional care of CF patients have resulted in an improved life expectancy and quality of life. Consequently, other chronic and systemic manifestations complicating the clinical course of 7CF have emerged as significant medical issues including pancreatic insufficiency, liver disease (LD) and cirrhosis, and gastrointestinal problems [4,5]. CFLD affects approximately 15–20% of CF patients and is the second leading cause of mortality and morbidity. The symptoms of CFLD generally occur during adolescence and gradually increase in severity with age, and the occurrence of CFLD appears to be limited to this group of CF patients [6]. CFLD can be life-threatening and result in the necessity for liver transplantation. The pathogenesis of many of these complications of CF is not understood. The underlying causes for the occurrence of severe CFLD, particularly early in life, and its limitation to approximately 20% of the CF population are not known. This pattern of CFLD incidence suggests that one or more genetic modifiers may be involved in the regulation of disease severity.
Retention of bile acids and damage to the biliary system have been proposed as the primary insult in CFLD [7]. Although steatosis is common in CF liver, the major pathological insult is considered to be focal biliary cirrhosis that slowly develops with age to multilobular cirrhosis requiring liver transplantation [6]. The Cl− transport activity of the CFTR is critical to the maintenance of normal hepatobiliary transport and bile flow. In normal liver CFTR is present in the apical membrane of intrahepatic bile duct epithelial cells or cholangiocytes as well as the gallbladder [2,3]. Loss of CFTR activity results in a viscous bile with an altered bile acid composition.
2. Sulfotransferases
Rozmahel et al. have reported that the severity of intestinal disease in CFTR(−/−) homozygous recessive mice cosegregates with gene loci that contain the genes for hydroxysteroid and estrogen sulfotransferases (SULTs) in mice [8] and humans [9]. Therefore, these SULT genes may serve as candidate modifiers of CF severity, although the mechanism for modulation of this severity is not known. Table 1 shows the names and chromosomal localization of the genes for the human SULT isoforms. The isoforms generally associated with estrogen and hydroxysteroid sulfation are SULTs 1E1, 2A1 and 2B1b [10–12]. SULTs 2A1 and 2B1b structural genes are located at the same chromosomal loci whereas the gene for SULT1E1 is on a separate chromosome.
Table 1.
Human cytosolic SULTS.
| Isoform and names | Chromosomal loci |
|---|---|
| SULT1 or phenol SULT family | |
| SULT1A1: Phenol-sulfating PST, P-PST-1, thermostable-PST | 16p11.2–12.1 |
| SULT1A2: | 16p11.2–12.1 |
| SULT1A3: Monoamine-sulfating PST, M-PST, thermolabile-PST | 16p11.2–12.1 |
| SULT1B1: ST1B2, thyroid hormone-sulfating ST | 4q13.3 |
| SULT1C1: | 2q11.2 |
| SULT1C2: | 2q11.2 |
| SULT1C3: | 2q11.2 |
| SULT1E1: Estrogen-ST, EST | 4q13.3 |
| SULT2 or hydroxysteroid SULT family | |
| SULT2A1: Dehydroepiandrosterone-ST, DHEA-ST | 19q13.3 |
| SULT2B1a and 2B1b: | 19q13.3 |
| SULT4A1: Brain-selective SULT | 2p22.3 |
| SULT6A1: | 22q13.1 |
Sulfation involves the transfer of the sulfonate group of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to an acceptor compound to generate a charged hydrophilic product. Fig. 1 shows the sulfation of β-estradiol (E2) and the formation of the charged E2–3-sulfate. In the sulfation of drugs, xenobiotics and small endogenous compounds including steroids, this reaction is catalyzed by a family of cytosolic SULTs [13]. Sulfation has an important role in the synthesis and metabolism of estrogens and hydroxysteroids in humans. Conjugation of steroids with a sulfonate group is primarily an inactivation reaction because steroid sulfates do not bind and activate their appropriate receptors. Although several human SULT isoforms conjugate steroids, SULT1E1 is the major isoform for the inactivation of E2 and the regulation of its activity at physiological concentrations [10,14,15]. SULT1E1 is a member of the SULT1 or phenolic SULT gene family. The enzyme has a Km of 4 nM for the sulfation of E2 and demonstrates substrate inhibition with E2 concentrations above 20 nM [10]. SULT1E1 is expressed at high levels in human liver during the first trimester of development then the levels decrease to low adult levels shortly after birth [16]. SULT1E1 is also expressed in estrogen-responsive tissues including breast, endometrium, prostate and testis [17–20].
Fig. 1.
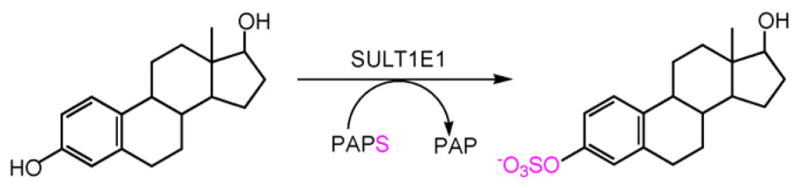
Formation of β-estradiol 3-sulfate by SULT1E1.
SULT2A1 is a member of the SULT2 or hydroxysteroid SULT gene family and is the most abundant SULT isoform expressed in liver [11]. SULT2A1 conjugates both 3α- and 3β-hydroxysteroids, estrogens and aliphatic hydroxyls in drugs and xenobiotics [11,21,22]. SULT2A1 is highly expressed in the fetal adrenal cortex and the reticular layer of the adult adrenal cortex where it is involved in the synthesis of dehydroepiandrosterone (DHEA) sulfate [11]. In contrast, SULT2B1b, the second member of the SULT2 family, is more selective for the sulfation of 3β-hydroxysteroids [12]; however, SULT2B1b is not expressed in human liver. SULT2B1b is expressed in other tissues including breast, prostate, lung and placenta [12,23,24].
3. CFTR-deficient mouse model
Transgenic mice with mutations in the CFTR have been widely used to investigate functional properties of these mutations and their role in the mechanism and pathology of CF. In humans, the most common CFTR mutation is the ΔF508 mutation that is responsible for approximately 60% of human CF and results in a severe form of the disease. The CFTR-ΔF508 protein is not properly trafficked in the cell and accumulates intracellularly preventing the Cl− transport function of the protein [25,26]. CFTR-knockout (KO) mice have also been generated that do not express the CFTR protein [25].
CFTR(−/−) mice do not demonstrate pulmonary disease although they display many of the manifestations of CF-associated GI and reproductive pathologies [25]. Homozygous CFTR(−/−) mice are generated by breeding heterozygous CFTR(+/−) mice. The CFTR(−/−) mice frequently die shortly after weaning due to meconium ilius unless maintained on a liquid diet. Many of the CFTR(−/−) mice do not gain weight or thrive on a liquid diet mimicking the growth deficits observed in human CF children. In contrast to humans, significant LD is not observed in young CFTR(−/−) mice although Durie et al. [27] report that many CFTR(−/−) mice over a year of age develop CFLD pathologies. Although there are differences in CF in humans and the CFTR(−/−) mouse models, these animals with disease-associated CFTR mutations can be analyzed through genotype/phenotype studies and provide a mechanistic framework with which to study CF disease mechanisms, genotype-based therapeutic approaches and pharmacologic interventions.
4. CFTR mice and SULT1E1 induction
To investigate the possible involvement of the steroid SULTs in CFLD their expression was investigated in the livers of the CFTR(−/−) mice. Falany et al. [28,29] have reported that SULT1E1 expression is increased up to 100-fold in the livers of both homozygous young adult CFTRΔF508 (−/−) mice and CFTR-KO(−/−) mice. The increase in SULT1E1 expression was not detected in the CFTRΔF508 (−/−) mice before weaning. Wildtype and heterozygous young adult CFTR(+/−) littermates showed no increase in liver SULT1E1 activity. The induction was selective for SULT1E1 activity since neither SULT1A nor SULT2 associated enzyme activities were increased [28]. Also, SULT1E1 activity was increased only in the livers of the CFTR(−/−) mice; SULT1E1 activity in the testes and endometrium of the CFTR(−/−) mice was not increased as compared to wildtype and CFTR(+/−) littermates (Table 2).
Table 2.
SULT1E1 activity in CFTRAF508 mouse tissues. Cytosol was prepared from different mouse tissues and assayed for SULT1E1 activity as described previously [14]. Results are expressed as pmol sulfated product/mg protein/min. ND means that there was no detectable activity.
| Tissue | ĘF508(+/+) Female | ĘF508(+/−) Female | ĘF508(−/−) Female | ĘF508(+/+) Male | ĘF508(+/−) Male | ĘF508(−/−) Male |
|---|---|---|---|---|---|---|
| Liver | 0.28 ± 0.10 | 0.15 ± 0.13 | 20.80 ± 10.4 | 0.24 ± 0.18 | 0.15 ± 0.15 | 13.05 ± 9.1 |
| Lung | 0.11 ± 0.03 | 0.14 ± 0.02 | 0.13 ± 0.04 | 0.11 ± 0.04 | 0.15 ± 0.03 | 0.12 ± 0.04 |
| Endometrium | 0.28 ± 005 | 0.29 ± 0.03 | 0.27 ± 0.05 | – | – | – |
| Testis | – | – | – | 26 ± 3 | 25 ± 7 | 27 ± 5 |
| Intestine | ND | ND | ND | ND | ND | ND |
| Colon | ND | ND | ND | ND | ND | ND |
| Kidney | ND | ND | ND | ND | ND | ND |
| Spleen | ND | ND | ND | ND | ND | ND |
Most of the CFTR(−/−) mice did not gain weight normally after weaning and the levels of hepatic SULT1E1 activity were negatively correlated with the loss in body weight although the liver to body weight ratios were not affected [28]. Histopathological examination of the CFTR(−/−) livers as well as determination of plasma levels of enzymes associated with liver damage showed no detectable pathology in the livers of these young adult mice.
To address the possibility that a unique SULT1E1 isoform may be expressed in these CFTR(−/−) livers, the SULT1E isoform induced in ΔF508(−/−) mice was cloned and sequenced for comparison to the published mouse testis SULT1E1 isoform [19]. The sequence of the CF liver isoform was identical to the sequence of the testis isoform indicating that the SULT1E1 isoform is present in CFTR mice. Subsequent examination of the mouse genome has not identified a unique related SULT1E1 gene indicating that a novel SULT1E1-related gene is not being induced.
5. Protein expression in livers of CFTR mice
The large increases in SULT1E1 activity in livers of CFTR(−/−) mice are expected to significantly decrease free E2 levels and thereby alter the expression of estrogen-regulated proteins. Li et al. [29] have reported that the expression of glutathione S-transferase P1 and carbonic anhydrase are down-regulated in the livers of CFTR(−/−) mice consistent with the loss of active E2. CYP2B9 expression was induced in livers of male CFTRΔF508(−/−) mice but not females and the levels of CYP2B9 expression correlated with the increases in SULT1E1 activity. These effects may be related in part to a decrease in the expression of estrogen receptor-α in CFTR(−/−) mice. Fig. 2 shows that the levels of immunoreactive SMAD3 are increased in female ΔF508(−/−) mice. SMAD3 is involved in TGF-β signal transduction pathways and acts as a transcription factor [30,31]. In mice, the pro-fibrotic properties of TGF-β are mediated by SMAD3 and are blocked in SMAD3-KO mice [31]. The association between SULT1E1 and SMAD3 expression has not been reported.
Fig. 2.

Immunoblot analysis of SMAD3 expression in liver cytosol from CFTRΔF508 mice. Cytosol (200 mg) prepared from livers of female CFTRΔF508 of each genotype (+/+, +/−, −/−) was resolved in a 12% SDS-polyacrylamide gel, electrotransferred to nitrocellulose membranes, immunoblotted with a rabbit anti-SMAD3 antibody (Calbiochem) and developed with the Pierce West Pico ECL substrate. Panel A shows the immunoreactivity of SMAD3 from two each female CFTRΔF508(+/+, +/−, −/−) mice. Panel B shows the levels of SMAD3 and SULT1E1 activity in liver cytosols of different genotype CFTRΔF508 mice.
Proteomic analysis of the effects of increased SULT1E1 activity was used to investigate changes in protein expression in both CFTRΔF508 and KO mice. Protein expression in a male CFTR-KO(−/−) mouse and a male ΔF508(−/−) mouse was compared to that of littermate controls. Cytosolic protein (100 μg) from each liver was focused in a Bio-Rad Criterion system using a pH 4–7 gradient. Focused proteins were subsequently resolved in 10–20% pre-cast SDS-PAGE gels. Gels were fixed and stained with Sypro Ruby, destained and imaged with a FX imager then analyzed using PDQuest software. Gels were analyzed for trends in increasing or decreasing intensities of protein spots in the CFTR-KO or ΔF508(−/−) mice compared to the appropriate control. In these experiments each gel possessed 375–400 detectable spots. Using a cut-off of a 2-fold change in spot intensity, PDQuest analysis identified approximately 40 spots in the KO gels and 25 spots in the ΔF508 gels as being significantly altered. Selected spots in each gel were isolated for identification by in-gel trypsin digestion and MALDI-TOF mass spectroscopy (MS). Table 3 shows the change in spot intensity between control and CFTR(−/−) mice as well as the identity of the selected spots. The spots identified in the CFTR(−/−) mice as being down-regulated by MALDI-TOF-MS included GST-P1 and CA that had been previously reported by Li et al. [29].
Table 3.
Proteomic analysis of cytosolic protein changes in male CFTR KO and AF508 mice. Cytosolic protein expression in a male KO(−/−) mouse (SULT1E1 13.74, pmol/min/mg) was compared to that of a littermate control (0.26 pmol/min/mg). In a separate experiment, cytosolic protein expression in a male AF508(−/−) mouse (16.1 pmol/min/mg) was compared to that of a littermate control (0.97 pmol/min/mg). Cytosol (100 μg) from each liver was focused in a Bio-Rad Criterion system using a 4–7 pH gradient and processed as describe above—duplicate gels were run for each sample and analyzed for trends in changing intensities of protein spots comparing the KO or AF508 to its control. In the ΔF508 mice, two control proteins whose intensities were not different were included.
| KO Mice
|
ΔF508 Mice
|
||||
|---|---|---|---|---|---|
| Spot | KO/Contrd@ | Identity | Spot | ΔF508/Contrd@ | Identity |
| 3401 | 8.15 | Apolipoprotein A IV | 0001 | 0.31 | Unknown |
| 5501 | 5.6 | Met Adenosyltrans. | 0012 | * | Major urinary protein |
| 706 | 0.24 | Albumin | 5005 | 2.0 | Adenosine kinase |
| 9201 | * | Glutathione S-transferase M1 | 4001 | * | Fatty acid binding protein# |
| 1102 | 0.02 | Major urinary protein | 3404 | ⊗ | Regucalcin |
| 4706 | 3.1 | Heat shock protein | 6001 | 6.1 | Fatty acid binding protein# |
| 2504 | 3.15 | ATP synthase | 4 | * | Apolipoprotein E |
| 2304 | 0.34 | Regucalcin | 3202 | 0.17 | ApolipoproteinA1# |
| 9203 | * | Carbonic anhydrase | 3203 | 0.38 | Apolipoprotein A1# |
| 10 | * | Glutathione S-transferase P1 | 3713 | Control | Heat shock protein |
| 1002 | 3.13 | Paralbumin | 3506 | Control | Actin |
No protein was detectable in the KO or ΔF508 but was detectable in the controls.
A protein modification occurred that affected gel migration.
These proteins are related isoforms.
6. Human cystic fibrosis and liver disease
Although the major site of CF pathology in humans is in the pulmonary system, most patients with severe forms of CF are diabetic and display absorptive problems in the GI tract. The CFTR is also important to the maintenance of normal hepatobiliary transport and bile flow. In normal liver, the CFTR is present in the apical membrane of intrahepatic bile duct epithelial cells or cholangiocytes as well as the gallbladder, but is not expressed in hepatocytes [2,3,32]. Disruption of CFTR function in cholangiocytes leads to the production of hyperviscous bile of abnormal composition and ultimately predisposes to hepatobiliary cell damage. It has been suggested that unchecked hepatobiliary damage leads to the emergence of disrupted cholesterol homeostasis and energy metabolism, portal hypertension and the focal biliary fibrosis that herald the onset of CFLD [33,34]. Little is known about the mechanisms of liver parenchymal damage in CF. Liver damage is not simply attributable to cholestasis since treatment with ursodeoxycholate can alleviate many problems with bile flow; however, ursodeoxycholate treatment has no effect on the development or course of CFLD [6].
Approximately 20% of patients with severe CF develop LD that can progress to a degree that liver transplantation is required. This progression in CFLD is not observed in the transgenic CFTR(−/−) mice and represents a fundamental difference from humans in this CF model. Therefore, elucidation of the biochemical and pathological changes in human liver that are associated with the loss of CFTR activity is needed. The biochemical changes in CF liver associated with the development of CFLD may involve the release of proinflammatory cytokines, growth factors, lipid peroxides/reactive oxygen species (ROS) and the activation of hepatic stellate cells [3,5,7]. Therefore, the pathogenesis of CFLD may in part involve the disruption of paracrine regulatory mechanisms operating between hepatocytes and cholangiocytes. There is precedent for hepatocyte–cholangiocyte cross-talk in the stringent control of bile production, processing and flow [35–37].
7. SULT1E1 expression in CF liver
The induction of SULT1E1 expression in CFTR(−/−) mouse liver results in alterations in the expression of E2-regulated proteins [29]. In contrast, the understanding of the pathophysiological and histopathological changes in human CF liver is severely limited due to the lack of availability of human CF liver tissue, obtained either during the course of the disease or at death, for scientific research. This has resulted in part from the increased lifespan of CF patients and the ability to diagnose CF genetically that has resulted in a decrease in autopsies. Liver biopsies that represent the best way to diagnose CFLD provide little tissue for research.
To examine whether SULT1E1 is increased in the liver of CF patients, a set of 19 paraffin blocks of liver tissue from children diagnosed with CF prior to 1993 was obtained from The Children’s Hospital of Alabama and immunostained for SULT1E1. Three of the 19 specimens showed increased SULT1E1 immunostaining although the presence of CFLD in these patients was not recorded. Fig. 3 shows the increase in SULT1E1 immunostaining in two young CF patients, including one with obvious steatosis. The number of samples showing increased immunostaining for SULT1E1 is consistent with the anticipated percentage of CF patients with CFLD in this small population.
Fig. 3.

Immunohistochemical localization of SULT1E1 in human pediatric CF liver. Paraffin sections of liver from children with CF and age-matched controls were treated with a microwave antigen retrieval technique and incubated with rabbit anti-SULT1E1 IgG. After labeling with the biotin–streptavidin complex and counter-stained with hemotoxylin, visualization was carried out with DAB. Stained sections were photographed at 600× magnification. The upper panels are from CF patients and the bottom panels from controls. The upper-left panel (9 mo F) had steatosis and the highest staining, the upper-right panel was from a 4 yo F. The lower panels were from 9 mo M (left) and 4 yo F (right).
8. Cholangiocyte–hepatocyte co-culture model
The lack of tissue specimens from patients with CFLD and the anticipated interactions of cholangiocytes and hepatocytes during the development of CFLD have led to the use of model systems to study CFLD. To investigate interactions between cholangiocytes and hepatocytes in CF, a co-culture model has been developed [32]. Human MMNK-1 cholangiocytes were cultured with human HepG2 hepatocytes in a membrane-separated Transwell system. MMNK-1 cells are an immortalized human cell line that expresses CFTR. CFTR expression has been inhibited >85% using siRNA to replicate the loss of CFTR function in CF [32]. For these studies, MMNK-1 cells are plated in 60 mm dishes and allowed to grow overnight. HepG2 cells are placed in the Transwell chamber and allowed to attach overnight. The Transwell chamber is then placed in the 60 mm plate and the cells are cultured together for 8 h. The HepG2 cells are then removed and RNA is immediately prepared. For protein expression studies, the Transwell chamber is moved to fresh medium and incubated for 24–48 h to permit translation of synthesized message. Fig. 4 shows the selective expression of SULT1E1 compared to SULTs 1A1 and 2A1. The ability of CFTR repression in MMNK-1 cells to induce SULT1E1 expression in HepG2 cells even when cells are separated by a 0.4 μm membrane suggests involvement of a paracrine factor. Conditioned medium experiments also indicate that a permeable factor secreted by MMNK-1 cells is involved in the induction of SULT1E1 [32].
Fig. 4.

Quantitative RT-PCR analysis of SULT1E1 expression in HepG2 cells co-cultured with MMNK-1 cells. HepG2 cells were co-cultured with CFTR-siRNA or control-siRNA MMNK-1 cells for increasing times then total RNA was extracted and utilized to synthesize cDNA. Quantitative RT-PCR was performed with TaqMan® Gene Expression Assays for human SULTs 1E1, 2A1 and 1A1. Ribosomal 18S RNA was chosen as the endogenous control for total RNA normalization and mRNA expression levels were calculated using the Ct method, where the calibrators were the samples from normal HepG2 cells without co-culture with MNNK-1. Relative expression of SULT isoforms was presented as fold-change between co-culture with CFTR-siRNA and co-culture with control-siRNA. Each point represents the mean of four separate determinations.
9. Expression profiling of MMNK-1 cells
Information with respect to the transcriptional regulation of human SULT1E1 is limited. Falany and Falany [38] have demonstrated that progesterone regulates SULT1E1 expression in endometrial cancer cells. SULT1E1 expression is also greatly increased in normal human endometrial biopsies during the secretory phase of the menstrual cycle when progesterone levels are high [17]. Although low levels of progesterone receptor can be detected in human HepG2 cells, no effect on SULT1E1 message levels was observed following treatment of HepG2 cells with progesterone or medroxyprogesterone. It has recently been reported that SULT1E1 expression is regulated by liver-X-receptor (LXR) activation in mouse liver [39] and in cultured human breast cells [40].
To investigate the possible mechanisms by which inhibition of CFTR function in cholangiocytes may regulate SULT1E1 expression in hepatocytes, expression profiling of CFTR-siRNA-MMNK-1 cells and control-siRNA MMNK-1 cells was carried out. For expression profiling studies, total RNA was isolated from both control-siRNA and CFTR-siRNA-MMNK-1 cells and used to probe Affymatrix Human Gene 1.0 ST GeneChips. This array represents 28,869 genes with approximately 26 probes spread across the length of each gene to provide a more complete and more accurate picture of gene expression than 3′ based expression array designs. Table 4 shows the pathway analysis of cholesterol and sterol metabolism in the CFTR-siRNA-MMNK-1 cells. Expression of several genes associated with cholesterol/sterol biosynthesis or metabolism was significantly altered in the CFTR-siRNA-MMNK-1 cells. Included in the cholesterol synthesis pathway was the significant down-regulation of 7-dehydrocholesterol reductase and 24-dehydrocholesterol reductase (Table 5). Fig. 5 demonstrates that 7-dehydrocholesterol reductase and 24-dehydrocholesterol reductase, are responsible for the final steps in cholesterol synthesis. Both desmosterol and 7-dehydrocholesterol, substrates for 7-dehydrocholesterol reductase and 24-dehydrocholesterol reductase, respectively, are activators of the liver-X-receptor (LXR) [41,42]. Fig. 6 shows that desmosterol, 7-dehydrocholesterol, 24-hydroxycholesterol and the synthetic LXR agonist T0901317 are all capable of inducing SULT1E1 expression in HepG2 cells. Neither SULT1A1 or SULT2A1 expression was affected by LXR activation.
Table 4.
Pathway analysis of cholesterol and sterol metabolism in Human Gene 1.0 array data. Total RNA was isolated from control and CFTR-siRNA-MMNK-1 cells and used for expression profiling using the Human Gene 1.0 ST array. Both control and siRNA arrays were done in triplicate. Genes showing a significant difference in expression at p < 0.05 were used for further analysis. After statistical analysis, the resulting list (p < 0.05) was loaded into PathwayArchitect software (Agilent, CA, USA) for Gene Ontology (GO) analysis. Among 1775 probe sets matched to GO database of 90,204 total GO terms, 20 probe sets relate to cholesterol metabolism, e.g., with a p-value of 7.45E–16 when the two ratios of the same term (cholesterol metabolism) were compared.
| GO Term | # Genes in selection | Rows in selection | # Genes in database | Total rows in database | p-value |
|---|---|---|---|---|---|
| Cholesterol biosynthesis | 14 | 1775 | 33 | 90204 | 7.17 E–16 |
| Cholesterol homeostasis | 2 | 1775 | 7 | 90204 | 0.007 |
| Cholesterol metabolism | 20 | 1775 | 89 | 90204 | 7.45 E–16 |
| Cholesterol transport | 2 | 1775 | 10 | 90204 | 0.016 |
| Sterol biosynthesis | 14 | 1775 | 49 | 90204 | 1.79 E–14 |
| Sterol metabolism | 21 | 1775 | 100 | 90204 | 6.18 E–16 |
Table 5.
Selected genes associated with cholesterol metabolism with altered expression in CFTR-siRNA-MMNK-1 cells.
| Gene ST probe set | Mk cont Ave | Mk siRNA Ave | Gene symbol | mRNA Description | p-value | Regulation |
|---|---|---|---|---|---|---|
| 7945831 | 195 | 154 | OSBPL5 | Oxysterol binding protein-like 5 | 0.02 | Down |
| 7950067 | 2025 | 1435 | DHCR7 | 7-Dehydrocholesterol reductase | 0.03 | Down |
| 7913462 | 4326 | 3431 | DCHR24 | 24-Dehydrocholesterol reductase | 0.003 | Down |
Fig. 5.

Overview of cholesterol synthesis showing the roles of 24-dehydrocholesterol (desmosterol) and 7-dehydrocholesterol reductases in final steps to cholesterol formation.
Fig. 6.

Induction of SULT1E1 in HepG2 by LXR ligands. HepG2 cells were treated with TO901317, 24(S)-hydroxycholesterol, desmosterol and 7-dehydrocholesterol at varying concentrations for 48 h. Cytosol was prepared from the cells and SULT1E1 and SULT2A1 activities were determined using 20 nm [3H]-E2 or 4 μM [3H]-DHEA as substrate, respectively, with 25 μM PAPS as the sulfate donor as described previously [14]. SULT1A1 activity was assayed with 4 μM p-nitrophenol and [35S]-PAPS as described previously [43].
10. Summary
The gene loci encompassing the human and mouse steroid SULTs have mapped as possible genetic modifiers of CF intestinal disease. The mouse CFTRΔF508 and KO models of CF show selective large increases in liver SULT1E1 activity that alter the expression of estrogen-regulated genes. However, in mouse CF models early onset of CFLD does not occur, although CFLD may occur late in life. Approximately 20% of human CF patients have severe progressive LD that may ultimately require liver transplantation. The induction of SULT1E1 and potential effects of increased SULT1E1 expression in these patients has not been investigated. To determine whether SULT1E1 is induced in human CF liver, a co-culture model was utilized. Human MMNK-1 cholangiocytes with siRNA-inhibited CFTR expression are capable of inducing SULT1E1 expression in HepG2 hepatocytes that lack CFTR expression in a membrane-separated Transwell co-culture system. The induction appears to be selective for SULT1E1 since SULTs 1A1 and 2A1 as well as CYP3A4 are not induced. Expression profiling of CFTR-siRNA-MMNK-1 cells suggests that oxysterols in the cholesterol biosynthesis pathway may be involved in the paracrine regulation of SULT1E1. Desmosterol and 7-dehydrocholesterol, both immediate precursors in the synthesis of cholesterol, induce SULT1E1 expression in HepG2 cells via activation of LXR. Therefore, LXR activation resulting from changes in cholesterol synthesis in cholangiocytes during CF is a possible mechanism for the selective induction of SULT1E1 in hepatocytes. Results of this study indicate that the pathogenesis of CFLD may involve the disruption of paracrine regulatory mechanisms operating between hepatocytes and cholangiocytes.
Acknowledgments
Supported by a grant from Cystic Fibrosis Research, Inc. and NIH grant GM38953 to CNF.
Footnotes
Lecture presented at the ‘18th International Symposium of the Journal of Steroid Biochemistry and Molecular Biology’, 18–21 September 2008, Seefeld, Tyrol, Austria.
References
- 1.Collins FS. Cystic fibrosis: molecular biology and therapeutic implications. Science. 1992;256:774–779. doi: 10.1126/science.1375392. [DOI] [PubMed] [Google Scholar]
- 2.Feranchak AP, Sokol RJ. Cholangiocyte biology and cystic fibrosis liver disease. Semin Liver Dis. 2001;21:471–488. doi: 10.1055/s-2001-19030. [DOI] [PubMed] [Google Scholar]
- 3.Feranchak AP. Hepatobiliary complications of cystic fibrosis. Curr Gastroenterol Rep. 2004;6:231–239. doi: 10.1007/s11894-004-0013-6. [DOI] [PubMed] [Google Scholar]
- 4.Jaffe A, Bush A. Cystic fibrosis: review of the decade. Monaldi Arch Chest Dis. 2001;56:240–247. [PubMed] [Google Scholar]
- 5.Modolell I, Alvarez A, Guarner L, De Gracia J, Malagelada JR. Gastrointestinal, liver, and pancreatic involvement in adult patients with cystic fibrosis. Pancreas. 2001;22:395–399. doi: 10.1097/00006676-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med. 2007;13:529–536. doi: 10.1097/MCP.0b013e3282f10a16. [DOI] [PubMed] [Google Scholar]
- 7.Freudenberg F, Broderick AL, Yu BB, Leonard MR, Glickman JN, Carey MC. Pathophysiological basis of liver disease in cystic fibrosis employing a {Delta}F508 mouse model. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1411–G1420. doi: 10.1152/ajpgi.00181.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui LC. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- 9.Zielenski J, Corey M, Rozmahel R, Markiewicz D, Aznarez I, Casals T, Larriba S, Mercier B, Cutting GC, Krebsova A, Macek M, Jr, Langfelder-Schwind E, Marshall BC, DeCelie-Germana J, Claustres M, Palacio A, Bal J, Nowakowska A, Ferec C, Estivill X, Durie P, Tsui L-C. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat Genet. 1999;22:128129. doi: 10.1038/9635. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Varlamova O, Vargas FM, Falany CN, Leyh TS. Sulfuryl transfer: the catalytic mechanism of human estrogen sulfotransferase. J Biol Chem. 1998;273:10888–10892. doi: 10.1074/jbc.273.18.10888. [DOI] [PubMed] [Google Scholar]
- 11.Falany CN, Comer KA, Dooley TP, Glatt H. Human dehydroepiandrosterone sulfotransferase. Purification, molecular cloning, and characterization. Ann N Y Acad Sci. 1995;774:59–72. doi: 10.1111/j.1749-6632.1995.tb17372.x. [DOI] [PubMed] [Google Scholar]
- 12.Falany CN, He D, Dumas N, Frost AR, Falany JL. Human cytosolic sulfotransferase 2B1: isoform expression, tissue specificity and subcellular localization. J Steroid Biochem Mol Biol. 2006;102:214–221. doi: 10.1016/j.jsbmb.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanchard RL, Freimuth RR, Buck J, Weinshilboum RM, Coughtrie MW. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics. 2004;14:199–211. doi: 10.1097/00008571-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Falany CN, Krasnykh V, Falany JL. Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. J Steroid Biochem Mol Biol. 1995;52:529–539. doi: 10.1016/0960-0760(95)00015-r. [DOI] [PubMed] [Google Scholar]
- 15.Kotov A, Falany JL, Wang J, Falany CN. Regulation of estrogen activity by sulfation in human Ishikawa endometrial adenocarcinoma cells. J Steroid Biochem Mol Biol. 1999;68:137–144. doi: 10.1016/s0960-0760(99)00022-9. [DOI] [PubMed] [Google Scholar]
- 16.Duanmu Z, Weckle A, Koukouritaki SB, Hines RN, Falany JL, Falany CN, Kocarek TA, Runge-Morris M. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and postnatal human liver. J Pharmacol Exp Ther. 2006;316:1310–1317. doi: 10.1124/jpet.105.093633. [DOI] [PubMed] [Google Scholar]
- 17.Falany JL, Azziz R, Falany CN. Identification and characterization of cytosolic sulfotransferases in normal human endometrium. Chem Biol Interact. 1998;109:329–339. doi: 10.1016/s0009-2797(97)00143-9. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T, Nakata T, Miki Y, Kaneko C, Moriya T, Ishida T, Akinaga S, Hirakawa H, Kimura M, Sasano H. Estrogen sulfotransferase and steroid sulfatase in human breast carcinoma. Cancer Res. 2003;63:2762–2770. [PubMed] [Google Scholar]
- 19.Song WC, Moore R, McLachlan JA, Negishi M. Molecular characterization of a testis-specific estrogen sulfotransferase and aberrant liver expression in obese and diabetogenic C57BL/KsJ-db/db mice. Endocrinology. 1995;136:2477–2484. doi: 10.1210/endo.136.6.7750469. [DOI] [PubMed] [Google Scholar]
- 20.Takase Y, Luu-The V, Poisson-Pare D, Labrie F, Pelletier G. Expression of sulfotransferase 1E1 in human prostate as studied by in situ hybridization and immunocytochemistry. Prostate. 2007;67:405–409. doi: 10.1002/pros.20525. [DOI] [PubMed] [Google Scholar]
- 21.Meloche CA, Sharma V, Swedmark S, Andersson P, Falany CN. Sulfation of budesonide by human cytosolic sulfotransferase, dehydroepiandrosterone-sulfotransferase (DHEA-ST) Drug Metab Dispos. 2002;30:582–585. doi: 10.1124/dmd.30.5.582. [DOI] [PubMed] [Google Scholar]
- 22.Falany JL, Pilloff DE, Leyh TS, Falany CN. Sulfation of raloxifene and 4-hydroxytamoxifen by human cytosolic sulfotransferases. Drug Metab Dispos. 2006;34:361–368. doi: 10.1124/dmd.105.006551. [DOI] [PubMed] [Google Scholar]
- 23.He D, Frost AR, Falany CN. Identification and immunohistochemical localization of Sulfotransferase 2B1b (SULT2B1b) in human lung. Biochim Biophys Acta. 2005;1724:119–126. doi: 10.1016/j.bbagen.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Dumas NA, He D, Frost AR, Falany CN. Sulfotransferase 2B1b in human breast: differences in subcellular localization in African American and Caucasian women. J Steroid Biochem Mol Biol. 2008;111:171–177. doi: 10.1016/j.jsbmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholte BJ, Davidson DJ, Wilke M, De Jonge HR. Animal models of cystic fibrosis. J Cyst Fibros. 2004;3(Suppl 2):183–190. doi: 10.1016/j.jcf.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 26.Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164:1481–1493. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falany JL, Greer H, Kovacs T, Sorscher EJ, Falany CN. Elevation of hepatic sulphotransferase activities in mice with resistance to cystic fibrosis. Biochem J. 2002;364:115–120. doi: 10.1042/bj3640115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Falany CN. Elevated hepatic SULT1E1 activity in mouse models of cystic fibrosis alters the regulation of estrogen responsive proteins. J Cyst Fibros. 2007;6:23–30. doi: 10.1016/j.jcf.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 30.McKarns SC, Letterio JJ, Kaminski NE. Concentration-dependent bifunctional effect of TGF-beta 1 on immunoglobulin production: a role for Smad3 in IgA production in vitro. Int Immunopharmacol. 2003;3:1761–1774. doi: 10.1016/j.intimp.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He D, Wilborn TW, Falany JL, Li L, Falany C. Repression of CFTR Activity in Human MMNK-1 Cholangiocytes Induces Sulfotranserase1E1 Expression in Co-Cultured HepG2 Hepatocytes. Biochim Biophys Acta. 2008;1783:2391–2397. doi: 10.1016/j.bbamcr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart L. The role of abdominal ultrasound in the diagnosis, staging and management of cystic fibrosis liver disease. J R Soc Med. 2005;98(Suppl 45):17–27. [PMC free article] [PubMed] [Google Scholar]
- 34.White NM, Jiang D, Burgess JD, Bederman IR, Previs SF, Kelley TJ. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292:L476–486. doi: 10.1152/ajplung.00262.2006. [DOI] [PubMed] [Google Scholar]
- 35.Kanno N, LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2001;281:G612–G625. doi: 10.1152/ajpgi.2001.281.3.G612. [DOI] [PubMed] [Google Scholar]
- 36.Xia X, Francis H, Glaser S, Alpini G, LeSage G. Bile acid interactions with cholangiocytes. World J Gastroenterol. 2006;12:3553–3563. doi: 10.3748/wjg.v12.i22.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, Glaser S, Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falany JL, Falany CN. Regulation of estrogen sulfotransferase in human endometrial adenocarcinoma cells by progesterone. Endocrinology. 1996;137:1395–1401. doi: 10.1210/endo.137.4.8625916. [DOI] [PubMed] [Google Scholar]
- 39.Khor VK, Tong MH, Qian Y, Song WC. Gender-specific expression and mechanism of regulation of estrogen sulfotransferase in adipose tissues of the mouse. Endocrinology. 2008 doi: 10.1210/en.2008-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong H, Guo P, Zhai Y, Zhou J, Uppal H, Jarzynka MJ, Song WC, Cheng SY, Xie W. Estrogen deprivation and inhibition of breast cancer growth in vivo through activation of the orphan nuclear receptor liver X receptor. Mol Endocrinol. 2007;21:1781–1790. doi: 10.1210/me.2007-0187. [DOI] [PubMed] [Google Scholar]
- 41.Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, Hobbs HH. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem. 2006;281:27816–27826. doi: 10.1074/jbc.M603781200. [DOI] [PubMed] [Google Scholar]
- 42.Heverin M, Meaney S, Brafman A, Shafir M, Olin M, Shafaati M, von Bahr S, Larsson L, Lovgren-Sandblom A, Diczfalusy U, Parini P, Feinstein E, Bjorkhem I. Studies on the cholesterol-free mouse: strong activation of LXR-regulated hepatic genes when replacing cholesterol with desmosterol. Arterioscler Thromb Vasc Biol. 2007;27:2191–2197. doi: 10.1161/ATVBAHA.107.149823. [DOI] [PubMed] [Google Scholar]
- 43.Falany CN, Vazquez ME, Heroux JA, Roth JA. Purification and characterization of human liver phenol-sulfating phenol sulfotransferase. Arch Biochem Biophys. 1990;278:312–318. doi: 10.1016/0003-9861(90)90265-z. [DOI] [PubMed] [Google Scholar]