

Abstract
Chemical exposures are important risks for development of hepatocellular carcinoma (HCC). One such chemical, diethylnitrosamine (DENA), is present in food products as well as in industrial and research settings. Further examination of tumors induced by DENA may yield clues to human risk. HCC from seven rhesus macaques exposed to DENA were selected from a tissue archive to examine for evidence of Wnt/β-catenin signaling events, which are frequently associated with HCC. DENA exposure durations ranged from 8 to 207 months, and total accumulated dose ranged from 0.7 to 4.08 mg. Unexposed colony breeder macaques served as controls. Previously unrecognized HCC metastases were discovered in lungs of three macaques. Overexpression of β-catenin and glutamine synthetase was detected by immunohistochemistry in six confirmed primary HCC and all metastatic HCC, which implicated Wnt/β-catenin activation. Concomitant β-catenin gene mutation was detected in one primary HCC; similar findings have been reported in human and rodent HCC. Neither β-catenin mutation nor β-catenin overexpression appeared to influence metastatic potential. Accumulation of intracellular proteins involved in Wnt/β-catenin signaling during HCC oncogenesis in rhesus macaques exposed to DENA appears to include other mechanisms, in addition to mutation of β-catenin gene.
Keywords: biological specimen banks, sequence analysis, DNA, carcinogens, mutagens, signal transduction pathway
Introduction
Hepatocellular carcinoma (HCC) is the most common hepatic malignancy and the third most common cause of cancer death in human beings (Parkin, Bray, and Devesa 2001; Roberts and Gores 2005). Chronic infection with hepatitis B and C viruses as well as the exposure to aflatoxin B1 are main risk factors believed to be responsible for approximately 80% of human HCC (Bosch, Ribes, and Borras 1999). Other factors that are linked with the development of HCC include heavy alcohol intake, use of oral contraceptives, genetic disorders, and exposures to chemical carcinogens (Calvisi et al. 2001). Pathogenesis of HCC can involve molecular alterations such as overexpression of growth factors or oncogenes, inactivation of tumor suppressor genes (e.g., p53 and Rb), and mutation activation of oncogenes (e.g., K-Ras) (Laurent-Puig and Zucman-Rossi 2006). Genetic modifications that activate Wnt/β-catenin signaling pathway have also been implicated in both human and rodent HCC (Laurent-Puig and Zucman-Rossi 2006; Thompson and Monga 2007).
β-catenin is a dual-function protein involved in cell adhesion and gene transcription (Katoh 2007; Daugherty and Gottardi 2007; Thompson and Monga 2007). During homeostasis, β-catenin complexes to E-cadherin at the plasma membrane and plays an important role in maintaining cell-cell adhesion. Free cytoplasmic β-catenin is phosphorylated at the N-terminal by axin/GSK-3β/APC complex and subsequently degraded by the ubiquitin-proteasome system. Therefore, the level of free cytoplasmic β-catenin remains low. When Wnt signaling is activated, GSK-3β activity is inhibited. This impairs degradation of β-catenin and causes the accumulation of β-catenin in the cytoplasm. Free cytoplasmic β-catenin then translocates into the nucleus and interacts with T cell factor/lymphoid enhancing factor transcription factors, resulting in up-regulation of its target genes (Giles, van Es, and Clevers 2003; Kikuchi, Kishida, and Yamamoto 2006). Nuclear accumulation of β-catenin has been reported in 17% to 40% of patients with HCC (Breuhahn, Longerich, and Schirmacher 2006), while 18% to 26% of HCC cases harbor mutations in the β-catenin gene CTNNB1 (Giles, van Es, and Clevers 2003; de La Coste et al. 1998; Miyoshi et al. 1998). The enhanced nuclear localization and activation of β-catenin has been linked with increased cell proliferation (Nhieu et al. 1999) and transition from hepatic adenoma to HCC (Ogawa et al. 1999).
Diethylnitrosamine (DENA) is a potent carcinogen capable of inducing HCC experimentally in both rodents (Vesselinovitch and Mihailovich 1983; Nakae et al. 1992) and nonhuman primates (NHP) (Thorgeirsson et al. 1994). This has raised concern for human health risk since DENA is found in widely used products including tobacco, cheese, fish, and beer (World Health Organization 1978). Further investigation into possible mechanistic links between altered Wnt/β-catenin signaling and HCC within nonrodent species may provide additional insight into potential for human risk from DENA exposure. In a thirty-two-year chemical carcinogenesis study in NHP, multiple species were exposed to DENA using a variety of doses and routes of administration (Thorgeirsson et al. 1994). Archival tissues from this study served as specimens for investigating potential mechanisms underlying hepatic carcinogenesis. An additional objective was to determine whether the archive might serve as a source of biorepository specimens for comparative molecular studies. Tissues were selected and retrieved from a single species, rhesus macaques (Macaca mulatta), treated with DENA. Similar tissues from untreated macaque colony breeders served as negative controls. Immunohistochemical evaluations for expression of β-catenin as well as glutamine synthetase (GS), encoded by a β-catenin regulated gene GLUL, were carried out. In addition, DNA sequences generated from exon 3 of β-catenin, CTNNB1, corresponding to the N-terminal region containing the serine/threonine phosphorylation residues (Zucman-Rossi et al. 2007; Laurent-Puig and Zucman-Rossi 2006), were analyzed.
Materials and Methods
Tissues
Tissues used in this study were selected from a thirty-two-year chemical carcinogenesis study specimen archive of NHP (Thorgeirsson et al. 1994). In the original carcinogenesis study, several NHP species were exposed to a variety of compounds considered as putative carcinogens for human beings. In an attempt to determine the suitability of this rare collection for use in correlative studies of pathology and molecular analyses, formalin- or Zenker’s-fixed paraffin-embedded archival tissue samples were selected from DENA-treated rhesus macaques (Macaca mulatta) with HCC. Precise methods of fixation for each specimen were not indicated in archival records. Representative liver and lung specimens were selected from animals exposed to DENA either as newborn macaques or, in one case, at nine months of age. A level dose group included tissues from three macaques given a sustained DENA dose of 1 mg/Kg from birth throughout life span (Table 1). An escalating dose group consisted of tissues selected from four macaques given gradually increasing DENA dose, from 10 to 20 to 40 mg/Kg every fourteen days during the first six months of life, with dosing continued until a tumor was believed present (Thorgeirsson et al. 1994). Three untreated colony breeder macaques provided control tissues. Hematoxylin and eosin (H&E) staining on all the selected tissue sections was evaluated in an attempt to corroborate initial study diagnoses.
Table 1.
Hepatic and pulmonary histopathological and IHC findings for DENA-treated or untreated colony breeder rhesus macaques.
| IHC labeling results
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Treatment group | Animal ID | Dose schedule | Cumulative dose (g) | Duration on study (months) | Tissue examined | HCC diagnosis | β-catenin | GS |
| Level | 902L | 1 mg/Kg | 1.45 | 204 | Liver | + | + M, C, N | + |
| Lung | − | − | − | |||||
| 1102P | 1.86 | 164 | Liver | + | + M, C, N | + | ||
| Lung | − | − | − | |||||
| 1088P | 2.96 | 207 | Liver | + | + M, C, N | + | ||
| Lung | − | − | − | |||||
| Escalating | 716I | 10–20–40 mg/Kg | 0.7 | 8 | Liver | − | − | − |
| Lung | − | − | − | |||||
| 595G | 1.84 | 15 | Liver | + | + M, C | +Focal | ||
| Lung | + | + M, C | +Focal | |||||
| 1236T | 2.58 | 16 | Liver | + | + C | + | ||
| Lung | + | + M, C | + | |||||
| 4T | 4.08 | 26 | Liver | + | + C | +/− Rare | ||
| Lung | + | + M | +/− Rare | |||||
| Colony breeder | 303T | None | n/a | n/a | Liver | − | − | − |
| Lung | − | − | − | |||||
| 324G | n/a | n/a | Liver | − | n/d | n/d | ||
| 302T | n/a | n/a | Lung | − | − | − | ||
HCC = hepatocellular carcinoma; DENA = diethylnitrosamine; IHC = immunohistochemistry; GS = glutamine synthetase; M = membranous labeling; C = cytoplasmic labeling; N = nuclear labeling; focal = limited numbers of cells in a tissue region; rare = occasional isolated positive cells; + = positive; − = negative; n/a = not applicable; n/d = not determinable, tissue preservation not optimal for IHC.
Immunohistochemistry (IHC) Analyses for β-Catenin and GS
IHC was performed as previously described (Custer et al. 2006) with minor modifications. Briefly, 6 μm-thick tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed by immersing tissues in target antigen retrieval buffer (TAR®, Dako, Carpinteria, CA) in a steamer (Black and Decker, Hunt Valley, MD) for 15 minutes, followed by 20 minutes cooling on the bench top. Primary antibodies included a rabbit polyclonal anti-β-catenin (Abcam, Cambridge, MA) used at a dilution of 1:100, and a mouse monoclonal anti-GS (BD Biosciences, Franklin Lakes, NJ) used at a dilution of 1:200. An antibody against a human hepatocyte-specific antigen, clone OCH1E5, (DakoCytomation, Carpenteria, CA) was used on a subset of specimens at 1:25 dilution as a means for analyzing metastatic carcinomas for evidence of liver origin. This antibody has been designated HepPar1 (Siddiqui et al. 2002). Biotinylated secondary antibodies, goat anti-rabbit IgG for anti-β-catenin and goat anti-mouse IgG for both anti-GS and the anti-hepatocyte marker, were diluted 1:500 (Dako, Carpinteria, CA). Reactions were developed using a peroxidase-based streptavidin detection method (Vector Labs, Burlingame, CA) and 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Invitrogen, Carlsbad, CA) as chromogen substrate. Positive IHC reaction controls included (1) positive labeling of bile ductular epithelial cells within liver sections for β-catenin antibody and (2) positive anti-GS labeling of human tonsil sections served as GS specific reaction controls. Additional positive control included use of a specimen of rhesus macaque liver fixed overnight in 10% neutral buffered formalin, which had been collected recently from a nonstudy colony animal (36821–6). This specimen was kindly provided by Dr. Matthew Starost, Diagnostic and Research Services Branch, NIH, Bethesda, MD. Negative reaction control included substitution of antibody diluent for the respective primary antibodies on tissue sections.
Genomic DNA Extraction from Paraffin-Embedded Tissues
For each tissue specimen, two 8 μm-thick paraffin-embedded tissue sections were placed in microcentrifuge tubes and paraffin was removed using xylenes. Genomic DNA was extracted using the DNeasy kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. In addition, lung specimens from macaques 595G, 4T, and 1236T, with HCC pulmonary metastases, were analyzed further. Cancer cells and adjacent normal lung tissues were separated from 10 μm-thick tissue sections using the LMD 6000 microdissection system (Leica Microsystems, Bannockburn, IL). DNA specimens from microdissected tissues were extracted using the PicoPure DNA Extraction Kit (Arcturus Bioscience, Mountain View, CA) according to the manufacturer’s instructions. The DNA concentrations were determined with the NanoDrop spectrophotometer (Thermo-Fisher Scientific, Waltham, MA).
β-Catenin DNA Sequence Analysis
The purified genomic DNA samples were used to amplify a 130 base pair (bp) fragment corresponding to the functionally important phosphorylation sites in exon 3 of the β-catenin gene. The sequences of primers used for polymerase chain reaction (PCR) were 105F: 5′-AAGCGGCTGTTAGTCACTGG-3′ (Forward) and 119R: 5′-GGACTTGGGAGGTATCCACA-3′ (Reverse). PCR amplification was carried out using Platinum PCR SuperMix (Invitrogen, Carlsbad, CA). Cycling conditions included an initial denaturation for 10 minutes at 95°C, thirty-five cycles of 95°C for 30 seconds, 55°C for 30 seconds and 72°C for 1 minute, followed by final extension for 10 minutes at 72°C. PCR products were separated by electrophoresis on 1.5% agarose gels and purified using MinElute Gel Extraction kit (Qiagen, Valencia, CA). The purified PCR products were subjected to direct sequencing using ABI Big Dye V1.1 sequencing kit (Applied Biosystems, Foster City, CA). The sequencing reactions were performed in both directions to ensure the accuracy of the sequences obtained. The primers used in the PCR reactions were used as sequencing primers. The published genome sequence of the β-catenin gene CTNNB1 in chromosome 2 of Macaca mulatta (NCBI Accession number NC 007859) was used as the template for sequence comparison in the search for mutations. The sequences were analyzed using SeqMan (DNASTAR, Madison, WI).
Results
Histopathology Findings in DENA-Treated Rhesus Macaques
Liver tissue sections from DENA-treated and breeder control rhesus macaques were obtained from the original chemical carcinogenesis study (Thorgeirsson et al. 1994) to analyze HCC for selected anatomic and molecular concordant features. The existence of HCC documented previously was corroborated in six of the seven selected DENA-treated animals (Table 1). HCC typically occurred as trabeculae of neoplastic hepatocytes, with some cancer foci arranged in compact solid nodules or as pseudoglandular structures admixed with foci of necrosis (Figure 1A–D). Evidence of the reported HCC in macaque 716I was not detected in either of the two available liver paraffin blocks; instead macaque 716I, which received lowest cumulative DENA dose (0.7g), had hepatocellular hyperplasia. Animals in escalating and level dose groups developing HCC accumulated DENA doses ranging from 1.45g to 4.08g (Table 1).
Figure 1.

Hepatocellular carcinomas (HCC), rhesus macaques exposed to diethylnitrosamine (DENA), hematoxylin and eosin (H&E) stained tissue sections. (A) primary HCC, macaque 1102P liver. Hepatic architecture has been effaced by diffuse hepatocellular carcinoma accompanied by hemorrhage and necrotic foci. Bar = 500 μm. (B) Primary HCC exhibiting trabecular arrangement, macaque 1236T liver. Bar = 100 μm. (C) Primary HCC exhibiting pseudoglandular arrangement, macaque 1088P liver. Bar = 50 μm. (D) Primary HCC exhibiting solid pattern of growth with occasional trabecular arrangement, macaque 595G liver. Note focus of cancer invasion within hepatic blood vessel (arrow). Bar = 250 μm. (E) Pulmonary metastatic HCC exhibiting solid growth pattern with occasional trabecular arrangement, macaque 595G lung. Bar = 300 μm. (F) Metastatic HCC exhibiting trabecular arrangement observed in portions of the primary cancer illustrated in the corresponding primary HCC (D), macaque 595G lung. Bar = 50 μm.
In addition to primary HCC, previously undocumented evidence of metastatic HCC was observed in sections of lung from macaques 595G, 4T, and 1236T in the escalating dose group (Table 1). Large polygonal neoplastic epithelial cells similar to those in foci of primary HCC occurred as aggregates and invaded pulmonary parenchyma from vessels, implicating hematologic dissemination (Figure 1E–F). Hepatocyte-specific antigen was detected by IHC in these pulmonary metastatic lesion from macaque 1236T as further corroboration of the liver histogenesis of cancers in the lungs (data not shown). Pulmonary metastasis was not detected in macaques with HCC in the level dose group. No significant lesions were observed in livers and lungs of untreated macaques serving as colony breeders.
IHC Detection of β-Catenin and GS
Since aberrant β-catenin localization has been linked with HCC in human beings, IHC analysis of β-catenin was carried out to examine whether enhanced expression or abnormal cytolocalization of β-catenin occurred in macaques with HCC. In assessing subcellular distribution of β-catenin by IHC, general absence of membrane labeling in nonneoplastic hepatocytes in this series of macaque livers was tabulated as a negative finding (Table 1), even though some physiological presence at plasma membranes could be conceivable (Figure 2A). β-catenin was detected in each case of primary HCC with variable patterns of the antibody reaction within neoplastic hepatocytes; some lesions had predominately cytoplasmic expression of β-catenin, others exhibited both cytoplasmic and membranous labeling, while still other HCCs had a predominantly membranous pattern of β-catenin expression (Table 1 and Figure 2). In the level dose group, two out of three macaques exhibited nuclear localization of β-catenin, in addition to elevated cytoplasmic reactivity in HCC (Table 1 and Figure 2C). Membranous and cytoplasmic β-catenin reactivity was exemplified by one escalating dose group macaque with HCC (Figure 2E), while two other macaques in this group had prominent cytoplasmic β-catenin reactivity in primary HCC (Table 1). Additional β-catenin expression findings were observed in the three escalating dose group macaques with pulmonary metastatic HCC. In these, paired primary and metastatic neoplasms shared the immunoreactivity to β-catenin. Ability to localize β-catenin to cytoplasm and membrane in neoplastic hepatocytes differed for two macaques when evaluating primary versus metastatic foci, however (Table 1).
Figure 2.

β-catenin and glutamine synthetase (GS) immunoreactivity in macaques with and without hepatocellular carcinoma (HCC). Immunohistochemical signal presence of β-catenin and GS was developed using 3,3′-diaminobenzidine tetrahydrochloride (DAB) as chromogen, and tissues were counterstained with hematoxylin. (A) β-catenin labeled bile ductular membranes and cytoplasm in nonneoplastic livers, macaque 302T liver. Bar = 50 μm. Inset photomicrograph reveals robust labeling in bile ducts from a more recently collected colony specimen lacking HCC, using the same immunohistochemistry (IHC) protocol. Note faint, nonuniform labeling of β-catenin in nonneoplastic hepatocyte membranes from this specimen (arrow). Bar = 50 μm (B–D, serial sections of primary HCC, macaque 1088P liver). (B) IHC reaction control lacking primary antibody. Bar = 50 μm. (C) β-catenin immunoreactivity in neoplastic hepatocytes primarily localizes to cytoplasm and infrequently involves nuclei. Bar = 50 μm. (D) GS immunoreactivity in neoplastic hepatocytes primarily localizes to cytoplasm and occasionally involves nuclei. Bar = 50 μm. (E) β-catenin immunoreactivity in neoplastic hepatocytes primarily localizes to cytoplasmic membrane, revealing an alternate pattern of β-catenin expression in HCC, macaque 595G liver. Bar = 50 μm. (F) GS immunoreactivity in neoplastic hepatocytes comprising a pulmonary metastatic focus, macaque 595G lung. Bar = 50 μm.
Enhanced intracellular β-catenin level has the potential to up-regulate the expression of GS. Therefore, tissues were probed using an anti-GS antibody as a potential alternative indicator of increased β-catenin activity. Extensive GS immunoreactivity (Figure 2D) was similarly detected in serial sections from which β-catenin IHC (Figure 2C) was carried out, with the exception of macaques 595G and 4T (Table 1). IHC reactions for GS in liver and lung of macaque 595G were limited to only a few foci of neoplastic hepatocytes and were somewhat equivocal in macaque 4T. Overall, the expression of GS was limited to HCC; no anti-GS reactivity was detected in nonneoplastic liver or lung (Figure 2F). There was a general correlation between the presence of β-catenin and GS expression in HCC lesions by IHC.
β-Catenin Sequence Analysis
The finding of increased cytoplasmic and nuclear β-catenin in HCC from DENA-treated rhesus macaques led to an examination of the hypermutable region in exon 3 of the β-catenin gene for evidence of functionally significant mutations. Although genomic DNA from the formalin-fixed tissues was fragmented (data not shown), the 130bp target sequence spanning the phosphorylation region corresponding to nucleotides 56–186 of human β-catenin was amplified from all samples (Figure 3A). Sequences of the PCR products from HCC were compared to those of nontumor tissues from the same animals or to colony breeder control macaques. All sequences were also aligned to published genomic sequence of the macaque β-catenin gene CTNNB1. Sequence comparisons revealed one TCT → CCT mutation in animal 1102P, resulting in a serine to proline substitution at amino acid 33 (Figure 3B). No other mutations were detected in the remaining DENA-treated animals or breeder controls.
Figure 3.
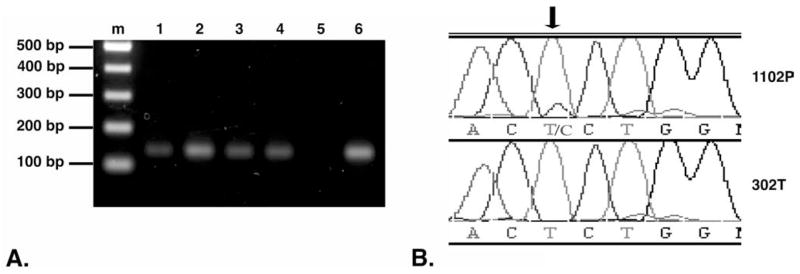
Detection of β-catenin mutation in hepatocellular carcinoma (HCC). (A) Polymerase chain reaction (PCR) amplification of β-catenin. PCR was carried out using genomic DNA isolated from paraffin-embedded tissues. PCR products were separated by electrophoresis on a 1.5% agarose gel. Lanes: (m) 100 bp DNA ladder, representative 130 bp amplified DNA product from unexposed and diethylnitrosamine (DENA)-exposed macaque livers: (1) 302T, (2) 4T, (3) 595G, (4) 1102P, (5) no DNA template negative control, and (6) genomic DNA isolated from FRhK4, a rhesus macaque kidney cell line (CRL-1688, American Type Culture Collection, Manassas, VA), used as a positive control. (B) Partial sequence chromatograms depicting nucleotide sequences of the β-catenin gene exon 3 in liver tissue of DENA-exposed macaque 1102P (top row), compared to sequence from colony breeder rhesus macaque 302T (bottom row). The T → C missense mutation (arrow) resulted in a Ser → Pro mutation at amino acid 33.
Due to the possibility that a rare mutation event may be difficult to detect amid a majority of DNA sequences derived from nonneoplastic cells lacking mutations, selected specimens from matched liver and lung metastatic HCC were microdissected to separate the cancers from surrounding non-neoplastic tissues. This approach yielded β-catenin sequence from enriched populations of primary and metastatic HCC cellularity yet did not reveal additional evidence of mutations (data not shown).
Discussion
DENA is a potent hepatic carcinogen in rodents (Kim et al. 2008) and similarly DENA induces HCC in NHP in a dose-dependent manner (Thorgeirsson et al. 1994). HCC tissues from DENA-exposed NHP were examined for molecular evidence of aberrant β-catenin signaling pathway involvement. β-catenin expression, greater than that observed in control tissues, was detected by IHC in neoplastic hepatocytes of all six macaques with HCC evaluated. In animals with metastatic HCC, the β-catenin overexpression was concordant in the paired primary and pulmonary metastatic lesions. Furthermore, the level of GS expression, which is regulated by β-catenin, was highly associated with the presence of β-catenin in primary and metastatic HCC. Precise IHC localization of β-catenin within subcellular compartments varied among specimens. Some of the variation, that is, intensities of β-catenin labeling by IHC, could be due to technical issues such as unknown variation in postmortem interval, the type of fixative used, and length of fixation interval. The one DENA–exposed macaque (716I) developing benign hepatocellular hyperplasia, instead of carcinoma, lacked β-catenin and GS immunoreactivity in hepatocytes. This particular macaque received a cumulative DENA dose of 0.7g, suggesting some minimal threshold dose may be necessary for induction of β-catenin overexpression and hepatocellular carcinogenesis. An association of elevated GS expression and its correlation with β-catenin overexpression observed in this study, reported as evidence of Wnt/β-catenin signaling perturbation in human HCC (Audard et al. 2007; Zucman-Rossi et al. 2007), has not been documented previously in rhesus macaque HCC.
Mutations in the β-catenin gene have been mechanistically credited with activating Wnt signaling in HCC (Giles, van Es, and Clevers 2003). Mutations that impact β-catenin phosphorylation residues or the neighboring amino acids retard the degradation of β-catenin and result in its accumulation in cytoplasm and/or the nucleus. Despite uniformly positive β-catenin immunoreactivity among study macaques with HCC, this hallmark of Wnt/β-catenin activation was accompanied by limited evidence of β-catenin gene mutation. An S33P substitution was found in one primary HCC sample in this study. Ser33 is one of the phosphorylation targets that are crucial for the degradation of β-catenin. Mutation of this residue contributes to the impairment of the β-catenin degradation machinery and results in accumulation of intracellular β-catenin. This same mutation has been documented in human HCC (Zucman-Rossi et al. 2007; Nhieu et al. 1999; Miyoshi et al. 1998). The paucity of β-catenin mutations was validated in laser dissected primary and metastatic HCC tumors, limiting the likelihood of false negative sequence results due to DNA contamination from nonmalignant parenchymal and stromal tissues. Deletion mutations of this region of the β-catenin gene, reported in human HCC (Terris et al. 1999; Miyoshi et al. 1998), were not observed in this study. The one macaque with β-catenin mutation in HCC, which also exhibited nuclear accumulation of β-catenin, was not on study longest; it did not receive the greatest cumulative DENA dose; nor did it exhibit pulmonary metastasis. Since cumulative levels of DENA were similar in the two dose regimens, the finding of β-catenin in neoplastic hepatocyte nuclei in the level dose group only could not be attributed to DENA dosing. β-catenin accumulation in the nucleus, specifically in association with more aggressive foci of cancer within HCC (Calvisi, Ladu, et al. 2004), was not observed in this study. Although the number of HCC specimens in the current study of rhesus macaques was few, neither evidence of β-catenin mutation nor β-catenin overexpression appeared to influence metastatic potential.
The evidence for existence of β-catenin mutation in the presence of elevated cytoplasmic and nuclear localization of the protein is not categorical. In rodent studies, β-catenin gene mutations were found in seven of thirteen HCC (54%) in B6C3F1 mice (Ogawa et al. 1999), while DENA-induced HCC in c-Myc/TGF-β1 transgenic mice (Calvisi et al. 2001) and Cx32-wild type mice (Aydinlik et al. 2001) lacked β-catenin mutations. In a c-Myc/E2F1 transgenic model, 83% had nuclear accumulation of β-catenin, while only 42% of mice had β-catenin mutations (Calvisi, Factor, et al. 2004). Similarly, in a human study (Terris et al. 1999), among eighteen human HCC that had elevated cytoplasmic and nuclear β-catenin, ten (56%) harbored β-catenin sequence alterations. On the contrary, in a series of thirty-six human HCC specimens, 30% of which had increased cytoplasmic and nuclear localization of β-catenin, β-catenin gene mutation was found in just one patient (2.8%) (Kim et al. 2008). This latter finding is similar to that in the present study on rhesus macaques. The constellation of these studies across species documents variable occurrence of β-catenin mutation in association with β-catenin overexpression in HCC.
A precise role for β-catenin mutation in β-catenin overexpression and increased Wnt signaling events in HCC remains uncertain. Results obtained in neoplastic macaque hepatocytes included discovery of increased β-catenin and GS proteins in the absence of evident β-catenin mutation. Mechanisms other than β-catenin mutations have been suggested as an alternative explanation for such findings not only in HCC, but also in colon, ovarian, and breast cancers (Giles, van Es, and Clevers 2003). For example, a correlation between the mutation of axin gene and the abnormal expression pattern of β-catenin was reported in human HCC (Kim et al. 2008). Axin functions as a scaffold protein on which a complex including APC and GSK-3β is formed to phosphorylate β-catenin. This binding leads to subsequent degradation of β-catenin. Mutation of the axin gene causes the dysfunction of the GSK-3β complex and indirectly reduces the degradation of β-catenin. Likewise, mutations in APC or overexpression of the Frizzled Wnt receptor have also been linked with the activation of Wnt/β-catenin signaling events and ultimately to the formation of HCC (Giles, van Es, and Clevers 2003; Thompson and Monga 2007; Zucman-Rossi 2008). A study of liver cancer conducted in c-Myc/E2F1 transgenic mice documented the involvement of other members of the Wnt pathway in activation of β-catenin (Calvisi et al. 2005). Studies on the integrity or expression of genes other than β-catenin and GS were not included in the subject macaques.
The archival specimens collected for this investigation from the thirty-two-year study of risk assessment in NHP proved suitable for documenting the activation of Wnt/β-catenin pathway in NHP with HCC. The β-catenin overexpression in macaques with HCC was only rarely associated with β-catenin gene mutation. These findings are similar to those from other models and from patients and serve as a link to assessing risk in human exposures to DENA and the induction of HCC. The ability to carry out molecular correlates of disease pathology establishes the potential for this NHP specimen archive to serve as a resource in the search for mechanistic clues to other known and unknown human carcinogenic risks.
Acknowledgments
The authors appreciate pathology specimen reviews conducted by Schantel Hayes and Heather Shive and critical review of the manuscript by Kathryn Poindexter. The authors are grateful for scholarly discussions with Janelle Cortner on the project.
This research was supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute (NCI), Bethesda, MD. Heather S. Tillman was supported by an NCI Cancer Research Training Award Fellowship while a fourth-year veterinary medical student, University of Georgia, Athens. Dr. Tillman’s current address is Diagnostic Center for Population and Animal Health, Michigan State University, East Lansing, MI. L. Tiffany Reed was supported by an NCI Cancer Research Interns in Residence Training Award while a fourth-year veterinary medical student, University of Georgia, Athens. Dr. Reed’s current address is the School of Veterinary Medicine, Purdue University, West Lafayette, Indiana.
Abbreviations
- HCC
hepatocellular carcinoma
- DENA
diethylnitrosamine
- NHP
nonhuman primates
- GS
glutamine synthetase
- IHC
immunohistochemistry
- H&E
hematoxylin and eosin
- bp
nucleotide base pairs
- PCR
polymerase chain reaction
- Ser33
serine amino acid 33
- Pro
proline
- DAB
3,3′-diaminobenzidine tetrahydrochloride
References
- Audard V, Grimber G, Elie C, Radenen B, Audebourg A, Letourneur F, Soubrane O, Vacher-Lavenu MC, Perret C, Cavard C, Terris B. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J Pathol. 2007;212:345–52. doi: 10.1002/path.2169. [DOI] [PubMed] [Google Scholar]
- Aydinlik H, Nguyen TD, Moennikes O, Buchmann A, Schwarz M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of beta-catenin-mutated mouse liver tumors. Oncogene. 2001;20:7812–16. doi: 10.1038/sj.onc.1204982. [DOI] [PubMed] [Google Scholar]
- Bosch FX, Ribes J, Borras J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271–85. doi: 10.1055/s-2007-1007117. [DOI] [PubMed] [Google Scholar]
- Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Conner EA, Ladu S, Lemmer ER, Factor VM, Thorgeirsson SS. Activation of the canonical Wnt/b-catenin pathway confers growth advantages in c-Myc/E2F1 transgenic mouse model of liver cancer. J Hepatol. 2005;42:842–49. doi: 10.1016/j.jhep.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, V, Factor M, Ladu S, Conner EA, Thorgeirsson SS. Disruption of b-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterol. 2004;126:1374–86. doi: 10.1053/j.gastro.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, V, Factor M, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: Relationship to phenotype and tumor grade. Cancer Res. 2001;61:2085–91. [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Factor VM, Thorgeirsson SS. Activation of b-catenin provides proliferative and invasive advantages in c-myc/TGF-a hepatocarcinogensis promoted by phenobarbital. Carcinogenesis. 2004;25:901–8. doi: 10.1093/carcin/bgh083. [DOI] [PubMed] [Google Scholar]
- Custer MC, Risinger JI, Hoover S, Simpson RM, Patterson T, Barrett JC. Characterization of an antibody that can detect the Kai1/CD82 murine metastasis suppressor. Prostate. 2006;66:567–77. doi: 10.1002/pros.20386. [DOI] [PubMed] [Google Scholar]
- Daugherty RL, Gottardi CJ. Phospho-regulation of beta-catenin adhesion and signaling functions. Physiology (Bethesda) 2007;22:303–9. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–51. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;13:4042–45. doi: 10.1158/1078-0432.CCR-06-2316. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Kishida S, Yamamoto H. Regulation of Wnt signaling by protein-protein interaction and post-translational modifications. Exp Mol Med. 2006;38:1–10. doi: 10.1038/emm.2006.1. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sills RC, Houle CD. Overview of the molecular biology of hepatocellular neoplasms and hepatoblastomas of the mouse liver. Toxicol Pathol. 2005;33:175–80. doi: 10.1080/01926230590522130. [DOI] [PubMed] [Google Scholar]
- Kim YD, Park CH, Kim HS, Choi SK, Rew JS, Kim DY, Koh YS, Jeung KW, Lee KH, Lee JS, Juhng SW, Lee JH. Genetic alterations of Wnt signaling pathway-associated genes in hepatocellular carcinoma. J Gastroenterol Hepatol. 2008;23:110–18. doi: 10.1111/j.1440-1746.2007.05250.x. [DOI] [PubMed] [Google Scholar]
- Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–86. doi: 10.1038/sj.onc.1209547. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998;58:2524–27. [PubMed] [Google Scholar]
- Nakae D, Yoshiji H, Mizumoto Y, Horiguchi K, Shiraiwa K, Tamura K, Denda A, Konishi Y. High incidence of hepatocellular carcinomas induced by a choline deficient L-amino acid defined diet in rats. Cancer Res. 1992;52:5042–45. [PubMed] [Google Scholar]
- Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepato-cellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155:703–10. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Yamada Y, Kishibe K, Ishizaki K, Tokusashi Y. Beta-catenin mutations are frequent in hepatocellular carcinomas but absent in adenomas induced by diethylnitrosamine in B6C3F1 mice. Cancer Res. 1999;59:1830–33. [PubMed] [Google Scholar]
- Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer. 2001;37(Suppl 8):S4–66. doi: 10.1016/s0959-8049(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Roberts LR, Gores GJ. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin Liver Dis. 2005;25:212–25. doi: 10.1055/s-2005-871200. [DOI] [PubMed] [Google Scholar]
- Siddiqui MT, HosseinSaboorian M, Tunc Gokaslan S, Ashfaq R. Diagnostic utility of the HepPar1 antibody to differentiate hepatocellular carcinoma from metastatic carcinoma in fine-needle aspiration samples. Cancer Cytopathol. 2002;96:49–52. [PubMed] [Google Scholar]
- Terris B, Pineau P, Bregeaud L, Valla D, Belghiti J, Tiollais P, Degott C, Dejean A. Close correlation between beta-catenin gene alterations and nuclear accumulation of the protein in human hepatocellular carcinomas. Oncogene. 1999;18:6583–88. doi: 10.1038/sj.onc.1203051. [DOI] [PubMed] [Google Scholar]
- Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–1305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson UP, Dalgard DW, Reeves J, Adamson RH. Tumor incidence in a chemical carcinogenesis study of nonhuman primates. Regul Toxicol Pharmacol. 1994;19:130–51. doi: 10.1006/rtph.1994.1013. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD, Mihailovich N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983;43:4253–59. [PubMed] [Google Scholar]
- World Health Organization, International Agency for Research on Cancer (IARC) IARC Monogr Eval Carcinog Risk Chem Man. 1978;17:1–349. http://monographs.iarc.fr/ENG/Monographs/vol17/volume17.pdf. [Google Scholar]
- Zucman-Rossi J. Human and mouse hepatocellular adenoma and carcinoma display similar tumorigenesis pathway alterations. J Hepatol. 2008;48:884–86. doi: 10.1016/j.jhep.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P, Perret C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26:774–80. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]