

Abstract
The stilbene derivative, cis-3, 4’, 5-trimethoxy-3’-aminostilbene (stilbene 5c), is a potentially potent antitumor agent that acts via binding to the colchicine-binding site in tubulin. The current studies were designed to investigate the effectiveness of stilbene 5c against the HCT-116 human colon cancer cell line and B16/F10 melanoma cells as well as human endothelial cell formation and tumor perfusion. Stilbene 5c produced a time-dependent decrease in cell viability in both cell lines and the capacity of the cells to proliferate was not restored upon removal of the drug. Treatment with stilbene 5c also promoted both senescence and autophagy in both cell lines. TUNEL and annexin 5 staining indicated that apoptosis also occurs in stilbene 5c-treated HCT-116 cells, but not in B16/F10 melanoma cells. DAPI staining revealed morphological changes in the cell nuclei (binucleated and micronucleated cells) indicative of mitotic catastrophe in HCT-116 cells but not in the B16/F10 melanoma cells. p53-null HCT-116 cells demonstrated a similar growth arrest/cell death response to stilbene as p53-wild type HCT-116 cells. Stilbene 5c also completely inhibited human endothelial cell tube formation on Matrigel, consistent with potential anti-angiogenic actions. Using a new method developed for monitoring the pharmacodynamic effects of stilbene 5c in vivo, we found that a single injection of stilbene 5c reduced tumor perfusion by 65% at 4 hours, returning to baseline by 24 hours, while subsequent daily injections of stilbene 5c produced progressively larger reductions and smaller rebounds. This work indicates that stilbene 5c could potentially be effective against melanoma and colon cancer through the promotion of multiple modes of growth arrest and cell death coupled with anti-angiogenic and antivascular actions.
Keywords: microtubules, autophagy, senescence, angiogenic, vascular disrupting
1. Introduction
Colorectal cancer and melanoma represent two of the most common and life-threatening cancers worldwide. First-line treatment for colon cancer includes combination therapy with 5-flourouracil, leucovorin, and oxaliplatin or irinotecan with bevacizumab [1]; unfortunately, the later stages of this disease show high resistance to the current therapeutics. Melanoma is generally managed by surgical removal followed by adjuvant therapy, frequently involving the alkylating agent decarbazine [2]. Although positive responses have been observed in up to 25% of melanoma patients treated with decarbazine in phase II clinical trials, the responses were generally transient and only 1-2% of the patients demonstrated a long-term response [3]. More recent studies have shown response rates of 10-12% [4-6]. Furthermore, treatment with the majority of current chemotherapeutic agents is generally associated with severe side effects. Hence, the identification of more effective and/or less toxic anticancer agents would improve the management of these diseases.
Microtubule binding-drugs such as the vinca alkaloids and the taxanes are used in the treatment of a variety of malignancies. These agents can be subdivided according to their binding sites on microtubules [7]. In addition to the binding sites for the vinca alkaloids and taxanes, many agents have been identified that bind to the colchicine binding site of tubulin; however, none are yet in clinical use. Combretastain A4-phosphate (CA4P), a phosphorylated form of the natural product combretastatin, and ZD6126 have been tested in phase I human clinical trials and CA4P has also undergone Phase II testing [8-9]. In addition to or as a consequence of their effects on microtubules, these agents have also been shown to act as vascular disrupting agents (VDAs); however, colchicine-site inhibitors (CSIs) have shown serious side effects such as neurotoxicity and cardiovascular toxicity [8-10]. Furthermore, resistance to these agents in vitro can occur as a consequence of differences in tubulin structure at the binding site of the drug [11-12], although agents that interact with the colchicine binding site generally have not been substrates for the conventional drug efflux pumps [13-14].
Stilbene 5c (cis-3, 4', 5-trimethoxy-3'-aminostilbene) binds to the colchicine-binding site on tubulin, causing destabilization of the microtubules (Figure 1) [15]. This compound has shown effectiveness against leukemic and ovarian cancer cells at nanomolar concentrations [15-16]. Stilbene 5c also selectively decreases vascular perfusion and microvessel density in tumors, without affecting these parameters in normal mouse organs [17]. In the current work, we have investigated the antitumor effect of stilbene 5c in HCT116 human colon carcinoma cells and B16 F10 murine melanoma cells. Consistent with previous studies in other experimental cancer models [15-17], stilbene 5c appeared to act through multiple pathways including the promotion of apoptosis, autophagy, mitotic catastrophe and senescence. Stilbene 5c also completely inhibited human endothelial cell tube formation in vitro, consistent with potential anti-angiogenic actions, and was found to be as potent as the vascular disrupting agent combretastatin A4. Finally, we provide a direct analysis of the effect of stilbene 5c on the kinetics of suppression and recovery of vascular perfusion in vivo, an approach which is critical to the clinical development of this class of vascular-targeting drugs.
Figure 1.
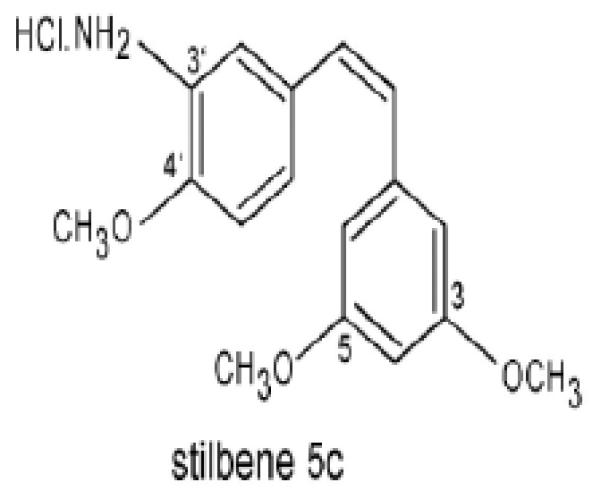
Chemical structure of Stilbene 5c (cis-3, 4', 5-trimethoxy-3'-aminostilbene)
2. Materials and methods
2.1 Cell culture
HCT-116 colon cancer cells were purchased from ATCC, maintained in 10% DMSO (Sigma Chemical, St. Louis, MO) with fetal bovine serum (FBS) (GIBCO Life Technologies, Gaithersburg, MD) and stored frozen in liquid nitrogen until ready for use. Cells were defrosted and cultured in a T75 flask (Cellstar) in RPMI 1640 medium with 5% fetal bovine serum, 5% bovine calf serum, 2 mM L-glutamine, and penicillin/streptomycin 0.5 mL/100 mL medium (10,000 units/mL penicillin and 10 mg/mL streptomycin (GIBCO Life Technologies, Gaithersburg, MD) and incubated at 37°C, 5% CO2, in a humidified environment. B16F10 murine melanoma cells were kindly given from Dr. Kimber White’s laboratory at Virginia Commonwealth University and kept frozen under liquid nitrogen in 10% DMSO (Sigma Chemical, St. Louis, MO) with Fetal Bovine Serum (FBS) until use. Cells were then thawed off and cultured in a T75 flask (Cellstar) in RPMI 1640 medium with 10% fetal bovine serum, 5% bovine calf serum, 2 mM L-glutamine, and penicillin/streptomycin 0.5 mL/100 mL medium (10,000 units/mL penicillin and 10 mg/mL streptomycin (GIBCO Life Technologies, Gaithersburg, MD) and incubated at 37°C, 5% CO2, in a humidified environment. Stilbene 5c was made up as a stock solution in DMSO (Sigma Chemical, St. Louis, MO).
2.2 Clonogenic Survival assay
The ability of cells to form colonies was evaluated by plating 200 cells in 6-well plates for control, 1% DMSO, 10 nM and 30 nM of stilbene 5c, and 2,000 cells for 100 nM, 300 nM and 600 nM of stilbene 5c. Cells were permitted to adhere overnight, and were then treated with the indicated concentrations for 2 days; drug was removed and fresh media added every other day. At day 9, cells were washed one time with 1X PBS before fixation with 100% of methanol and staining with crystal violet dye (1%) for 10 minutes. Colonies were counted visually in each well.
2.3 MTT dye assay for cell viability
In 96-well plates, 200 μL of medium containing 6,000 cells was added to each well and cells were allowed to attach overnight prior to drug treatment. After 72 hours of drug exposure, media was removed, MTT (2 mg/mL PBS) added and incubated at 37°C for 3 hours. 100 μL of DMSO was added for 5 minutes to each well to dissolve the formazan blue dye. Absorbance was read at 490 nm (KC Junior software, EL800 Universal Microplate Reader).
2.4 Time course of drug toxicity and its impact on cell viability
Cells were cultured in 6-well plates, at a density of 50,000 cells per well for HCT-116 cells and 100,000 cells per well for B16/F10 melanoma cells, and allowed to attach overnight and then treated with drug. Viable cell number was determined based on trypan blue exclusion.
2.5 Assessment of mitotic catastrophe by DAPI staining
50,000 cells were seeded in 6-well plates and treated with stilbene 5c. Both adherent and non-adherent cells were collected by centrifugation, resuspended in PBS, and slides prepared using a cytospin (Shandon Cytospin 4, Thermal Electron Corp). Slides were fixed at room temperature with 4% formaldehyde in PBS for 10 minutes, rinsed in PBS and treated with acetic acid (1:2 in ethanol). Slides were washed with PBS for 5 minutes and 10 μL of Vectashield:Dapi (1:1000 dilution) added. Images were taken using an Olympus 1X 70 microscope and an Olympus SC 35 type camera. Three separate experiments were visualized and evaluated.
2.6 Assays for apoptosis
50,000 cells per well were plated and treated with stilbene 5c. Cells were harvested and collected on a cytospin slide, and fixed with formaldehyde and acetic acid/ethanol as described above. For the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, cells were blocked with BSA (1 mg/ml for 30 minutes), rinsed twice in PBS, and incubated with enzyme mixture (terminal transferase, 25mM CoCl2, fluorescein-12dUTP) for 1 hour. After washing with PBS, images were captured using an Olympus 1X 70 microscope and an Olympus SC 35 type camera.
For the PI/Annexin assay, adherent and non-adherent cells were harvested and 100 μL of binding buffer (BD Biosciences) was added to the pellets. 5 μL of Annexin-FITC (BD Biosciencec) and 5 μL of PI at 10 μg/mL (BD Biosciences) were added and cells were softly vortexed and incubated for 15 minutes in the dark at room temperature. Annexin V binding buffer 10X (BD Pharmingen) was diluted to 1X and 400 μL of binding buffer was added and samples were analyzed by flow cytometry at 530 nM.
2.7 Evaluation of autophagy by acridine orange staining
50,000 cells were seeded in 6-well plates, permitted to adhere overnight, and drug added. Cells were washed with PBS and acridine orange dye (1:10000 in PBS; prepared in the dark) was added for 15 minutes. Plates were washed, and photographs were taken with an Olympus 1X 70 microscope and an Olympus SC 35 camera. For flow cytometry, 10 μL of acridine orange solution (1:10000 final conc) was added to harvested cells which were then analyzed using excitation/emission wavelengths of 525 nM and 620 nM.
2.8 Transfection of HCT116 cells with RFP-LC3
HCT-116 cells were centrifuged and resuspended with a RFP-LC3 construct [18] using 100 μl of the Amaxa Nucleofector Kit V. The cell suspension, collected in a cuvette, was subjected to program D-032. Transfected cells were then mixed with 500 μL of warm medium and transferred to a Petri dish where Gentamycin (8ng/mL) was used to maintain the stable transfection.
2.9 Senescence detection by β-Galactosidase staining
Cells were plated in a density of 50,000 cells per well, allowed to adhere overnight, and then exposed to drug. Cells were then washed once with PBS, fixed with 2% formaldehyde/ 0.2% glutaraldehyde for 5 minutes, and a staining solution composed of 1 mg/mL 5-bromo-4-chloro-3-inolyl-β-galactosidase in dimethylformamide (20 mg/mL stock), 5 mM potassium ferricyanide, 150 mM NaCl, 40 mM citric acid/sodium phosphate, 2 mM MgCl2, at pH 6.0 was added (overnight at 37°C). The cells were then washed twice with PBS and images obtained using an Olympus 1X 70 microscope and an Olympus SC 35 type camera.
For quantification of β-galactosidase staining using flow cytometry, drug-treated cells were washed and incubated (37°C and 5% CO2 for 1 hour) with 100 nM of bafilomycin A1 in media to induce lysosomal alkalinization. After incubation, C12FDG was added to each well (final concentration of 33 μM) for an additional 1 hour. Cells were washed, collected by centrifugation, and resuspended in PBS. C12FDG is hydrolyzed by β-galactosidase and becomes fluorescent at wavelengths of 500–510 nM.
2. 10 Western blotting
100,000 cells were seeded in 60 mm dishes, allowed to adhere overnight, and drug added. Viable and non-viable cells were collected and mixed with 100 to 200 μL lysis buffer (1 M Tris-HCl, pH 6.8, 10% SDS) containing protease and phosphatase inhibitors (Sigma-Aldrich) and boiled for 5 minutes. Proteins were separated on 12% gels using SDS-PAGE, transferred onto nitrocellulose membrane and incubated with the primary antibody (overnight at 4°C). Primary antibodies used were anti-p62 (SQSTM1–Santa Cruz sc-28359), anti-caspase 3 (Cell Signaling 9665), anti-LC3 antibody (NB100-2220; Novus Biologicals, Littleton, CO), anti-ß actin (Santa Cruz sc-47778), and anti-PARP (Cell signaling 46D11). All primary antibodies presented were used at a 1:1,000 dilution. The following day, membranes were incubated with respective secondary antibodies for an hour. Secondary antibodies used were goat anti-mouse IgG (Amersham, GE Healthcare) and monkey anti-rabbit IgG (Amersham, GE Healthcare). Membranes were then washed three times and bands were detected using enhanced chemiluminescence detection reagents (Pierce, Rockford, IL).
2.11 Endothelial cell proliferation and vessel formation
HUVEC (Cascade Biologics) were grown in EBM2 medium (Clonetics) in a humidified 5% CO2 incubator at 37° C. To measure drug effects on cell proliferation, 10,000 HUVEC were placed in each well of a 96 well plate. After allowing for attachment overnight, stilbene 5c or combretastatin A4 were added, and the number of cells was determined after an additional 72 hours by staining with sulforhodamine B (Sigma). The spontaneous formation of capillary-like structures by HUVEC on a basement membrane matrix preparation, Matrigel (Becton Dickinson), was used to assess angiogenic potential. Fifteen-well angiogenesis slides (IBIDI, Munich, Germany) were coated with 10 μl growth-factor-reduced Matrigel, and HUVEC (10,000 cells/well) were seeded and incubated at 37°C for 30 min. Stilbene 5c and combretastatin A4 were added, and after 24 hours, tubules were fixed with 3% glutaraldehyde, stained with 0.1% toluidine blue, and photographed. Drug effects on tube formation were quantified manually by counting tubule branch points, defined as cell junctions containing at least three tubules.
2.12 Luciferase imaging of mouse xenograft perfusion
Nude mice were injected subcutaneously with PBS-washed UCI-101/luciferase tumor cells (5 × 106) to establish tumor xenografts. After tumors reached a size with the longest diameter greater than 10 mm, mice were treated with stilbene 5c at 50 mg/kg/day intraperitoneally for 3 days. All mice were anesthetized with an isoflurane and oxygen mix prior to imaging. Luciferase imaging was done at 4 and 24 hours after each daily administration of stilbene 5c. All images were collected by a Xenogen Live Imaging system every 2 min for 60 minutes total after injection of luciferin. The total number of photons in the tumor area of interest was calculated and plotted against time (min) during the collection period. Final imaging was done at 24 hr after the third stilbene 5c injection.
2. 13 Statistical analysis
Statistics were performed using one-way ANOVA followed by Bonferroni analysis. The significance of group values was determined based on a p-value of p<0.05.
3. Results
3.1 Sensitivity to stilbene 5c in HCT-116 colorectal cancer cells and B16/F10 melanoma cells
Previous reports have shown that stilbene 5c is effective in promoting cell death and growth arrest in both leukemic and ovarian cancer cells [15-16]. Figure 1A show that exposure of HCT-116 cells to stilbene 5c resulted in a concentration-dependent reduction in viable cell number, with a decline of approximately 75 to 80% at a drug concentrations of 100 nM . Figure 2A also shows a similar response pattern in a clonogenic survival assay, with an ~ 90% decline in colony formation at 100 nM. Similar studies performed in B16/F10 melanoma cells indicated that the melanoma cells are somewhat less sensitive to stilbene 5c, demonstrating 25% and 50% decreases in cell viability at 30 nM and 300 nM, respectively, with similar results in the clonogenic survival assay ( Figure 2B). Consequently 100 nM Stilbene 5c was used for all further studies with HCT-116 cells and 300nM Stilbene 5c was used for studies with the B16/F10 melanoma cells.
Figure 2. Sensitivity of HCT-116 cells and B16/F10 cells to stilbene 5c.
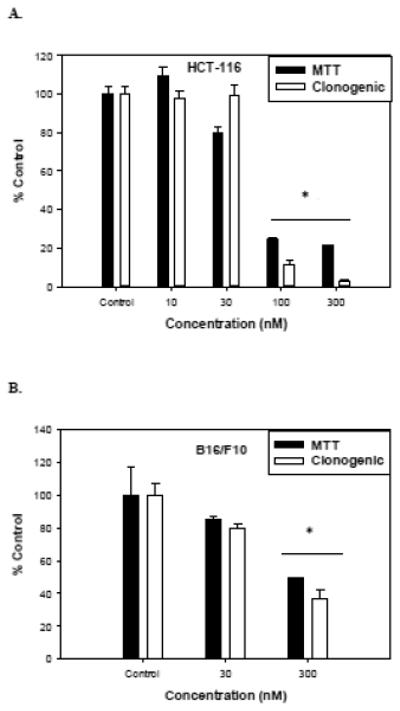
Dose response to stilbene 5c of HCT-116 cells (A) and B16/F10 cells (B) as measured by the MTT assay after 72 hours (closed bars) or clonogenic survival (open bars). Graphs represent pooled data from three replicate experiments. Error bars represent standard error (* p<0.05 compared to control).
In order to distinguish between cell death and growth arrest, the temporal effects of stilbene 5c on viability of HCT-116 and B16/F10 cells were monitored. Figure 3A indicates that subsequent to an initial decline in cell number, the HCT-116 tumor cell populations treated with stilbene 5c entered a state of growth arrest. To assess whether the drug-treated cells were able to recover the capacity to proliferate, drug was washed out on day 5 and fresh medium was added and replaced every other day for an additional 10 days. Our data indicates that the residual surviving tumor cell population had lost its capacity to recover, evidence of an apparently irreversible growth arrested state (not shown). Figure 3B indicates that B16/F10 melanoma cells treated with stilbene 5c at 300 nM also demonstrated growth arrest followed by a decline in viable cell number (with the exception that a short initial period of growth arrest preceding cell death; again, the growth arrest was prolonged and apparently irreversible despite removal of drug (not shown).
Figure 3. Effects of stilbene 5c on growth and survival of HCT-116 cells and B16/F10 melanoma cells.
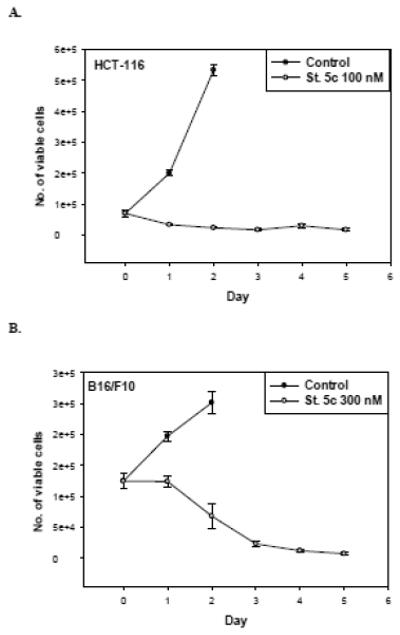
Viability of HCT-116 cells exposed to 100nM stilbene 5c (A) and B16/F10 cells exposed to 300nM stilbene 5c (B) was monitored over a period of 5 days by trypan blue exclusion. (✩) controls (○) Stilbene 5c. 50,000 HCT-116 cells and 100,000 B16F10 cells were plated in each plate of 6-well plates. Graphs represent pooled data from three replicate experiments. Error bars represent standard error (* p<0.05 compared to control).
3.2 Induction of senescence in HCT-116 colorectal cancer cells and B16/F10 melanoma cells
In view of the fact that residual surviving HCT-116 cells appeared to be incapable of proliferative recovery after drug treatment, it appeared likely that the cells had entered into a state of senescence arrest. Figure 4A indicates that the Stilbene 5c-treated cells demonstrated morphological markers of senescence such as granulations, flattening, and spreading as well as β-galactosidase staining, a hallmark of senescence. Quantification of the intensity of β-galactosidase staining by flow cytometry, indicated that between 25-30% of the cells had entered a state of senescence (Figure 4C). In addition, flow cytometry indicated that stilbene 5c promoted the enlargement of HCT-116 colon carcinoma cells (not shown), which would be consistent with senescence [19] and/or autophagy (see section 3.3). Stilbene 5c also induced morphological changes in B16/F10 melanoma cells as well as β-galactosidase staining consistent with the promotion of senescence (Figure 4B). Flow cytometry analysis (Figure 4C) indicated that approximately 30% of the melanoma cell population was senescent after treatment with stilbene 5c.
Figure 4. Senescence in surviving HCT116 colon carcinoma cells and B16/F10 melanoma cells treated with stilbene 5c.
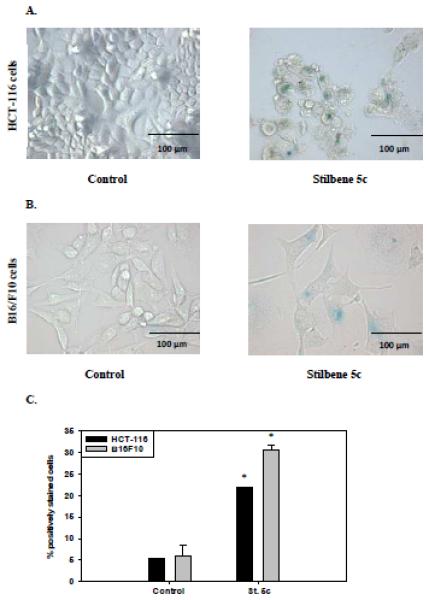
HCT-116 cells exposed to 100nM stilbene 5c (A) and B16/F10 cells exposed to 300nM stilbene 5c (B) were incubated with β-galactosidase staining buffer and X-gal. Images shown are representative of three replicate studies. C. Data presented in the graph were generated based on the percentage of cells that were positively stained with β-galactosidase in both treated and non-treated conditions as evaluated by flow cytometry at 72 hours_(* p<0.05 compared to control).
3.3 Promotion of autophagy by stilbene 5c in HCT-116 cells and B16/F10 melanoma cells
Autophagy is a regulated process in which misfolded proteins and damaged cellular components are broken down by lysosomal enzymes in double-membrane enclosures [20-22]. Recent studies have identified a linkage between autophagy and oncogene-induced, as well as chemotherapy-induced, senescence [23-25]. In addition, previous studies from our laboratory demonstrated that autophagy and senescence were induced in both HCT-116 cells and B16/F10 cells in response to another colchicine site-binding inhibitor, JG-03-14 [23]. Consequently, the induction of autophagy was evaluated in the HCT-116 and B16/F10 tumor cell lines.
During autophagy, acidified autophagosomes fuse with lysosomes to form acidic vacuoles, which can be visualized by staining with acridine orange dye. Figure 5A shows that control HCT-116 cells have a single orange vesicle located in the cytoplasm, which appears to be indicative of basal autophagy. HCT-116 cells treated with 100 nM stilbene 5c displayed an increased number of stained vacuoles (Figure 5A). In addition, cells exposed to stilbene 5c became enlarged in comparison with control cells, a morphological sign of autophagy (although, as indicated above, this could also reflect the senescence phenotype). Quantification of autophagy by flow cytometry indicated an increase from 5.3 ± 3.03% in controls to 25.07 ± 6.26% in cells exposed to stilbene 5c (Figure 5C), which closely parallels the extent of senescence induction .
Figure 5. Autophagy induction in HCT116 cells and B16/F10 melanoma cells by stilbene 5c.
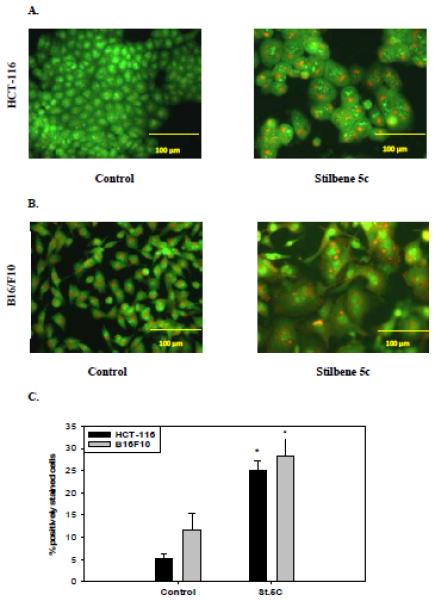
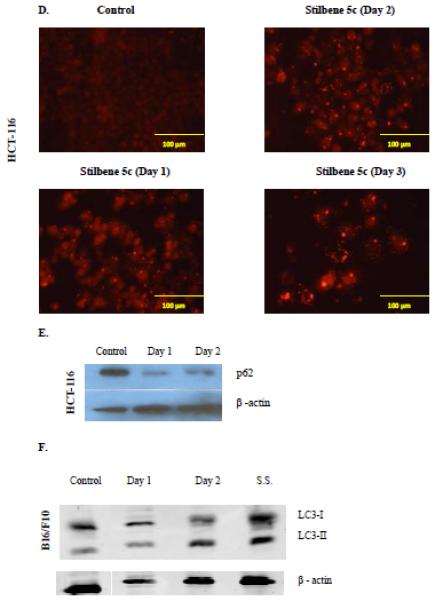
HCT-116 cells exposed to 100nM stilbene 5c (A) and B16/F10 cells exposed to 300nM stilbene 5c (B) were stained with acridine orange 72 hrs post treatment. All images were taken at 200X and data are representative of three replicate studies. C. The extent of autophagy was determined based on percentage of the cell population showing an increase in acridine orange staining by flow cytometry 72 hrs post treatment (* p<0.05 compared to control). D. Puncta formation in HCT116 cells transfected with an RFP-LC3 vector and treated with 100nM stilbene 5c as shown at 24 hrs, 48 hrs, and 72 hrs. E. Degradation of p62 in HCT-116 cells after exposure to stilbene 5c for 48 hrs. F. Induction of autophagy by western immunoblotting for the microtubule associated light chain protein (LC3-I) conversion to LC3-II in B16/F10 melanoma cells. B16F10 cells were seeded in a serum starved media for 48 hrs and Serum starved cells (S.S.) were used as a positive control.
Autophagy induction was confirmed by the redistribution of LC3-II, the lipidated form of the LC3 protein that binds to autophagosomal membranes during autophagy [26-27]. As shown in Figure 5D, whereas control cells transfected with RFP-LC3 show one small diffuse fluorescent punctua per cell (consistent with the acridine orange staining), red puncta were clearly increased in a time-dependent manner and redistributed in the treated cells.
Autophagic flux, which indicates that the process of autophagy has proceeded to completion, was also measured via p62 degradation, a protein sequestered in autophagosome and degraded by lysosomal hydrolases [28]. In HCT-116 colon cancer cells treated with 100 nM stilbene 5c, level of p62 clearly declined post treatment (Figure 5E).
Stilbene 5c was also shown to promote autophagy in B16/F10 melanoma cells. Figure 5B indicates that autophagic vacuole formation was clearly increased in B16/F10 melanoma cells upon treatment with 300 nM stilbene 5c, with approximately 30% of cells demonstrating autophagy by flow cytometry (Figure 5C), while Figure 5F indicates that the LC3 protein was converted to the lipidated form LC3-II (serum starvation, denoted as ss, was used as a positive control for autophagy). p62 was not detected in the B16/F10 cells.
3.4 Evaluation of apoptotic cell death induced by stilbene 5c in HCT-116 cells and B16/F10 melanoma cells
One highly desirable outcomes of therapeutic treatment of cancer is the promotion of apoptosis. The capacity of stilbene 5c to promote apoptosis in HCT-116 cells was evaluated by TUNEL and Annexin V-PI staining. Figures 6A and 6B demonstrate that the induction of apoptotic cell death in HCT-116 cells treated with stilbene 5c by the TUNEL assay. Figures 6C and 6D indicate the occurrence of both early and late apoptosis on days 3 and 5 (combined quadrants 2 and 4). The percentage of the cell population undergoing apoptosis at days 3 and 5 (approximately 20%) was similar to that determined by Annexin/PI staining (Figure 6B). The observation that apoptosis did not increase overtime is consistent with the fact that cell viability did not further decline between days 3 and 5 (Figure 3A). In addition, stilbene 5c was found to induce caspase 3 cleavage in HCT-116 cells (Figure 6H)1. The induction of apoptosis by stilbene 5c was also evaluated in B16/F10 melanoma. The TUNEL assay (Figure 6E), Annexin/PI staining (Figure 6F), lack of significant PARP cleavage (Figure 6G) and lack of caspase 3 cleavage 1(Figure 6H) all indicated that apoptosis does not appear to play a significant role in the response to stilbene 5c in B16/F10 melanoma cells.
Figure 6. Assessment of apoptosis in HCT116 colon cancer cells.
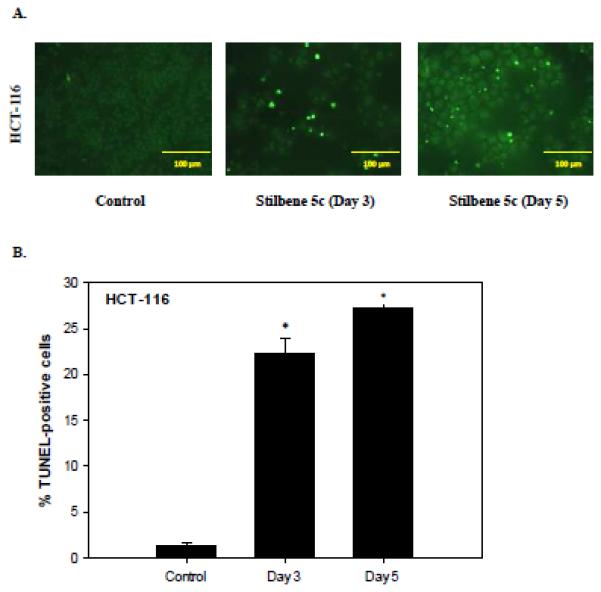
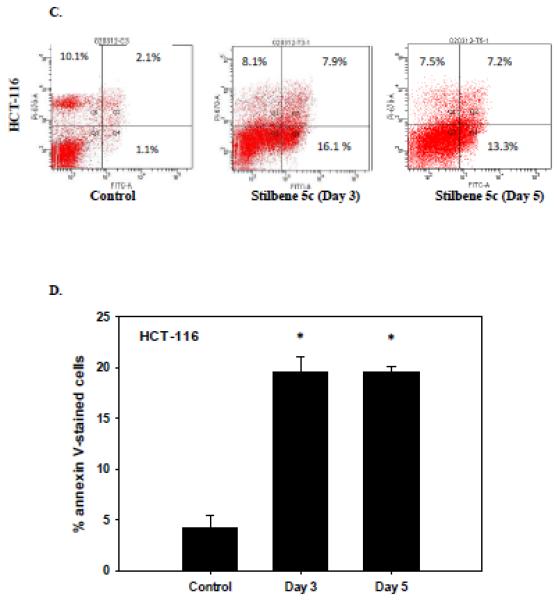
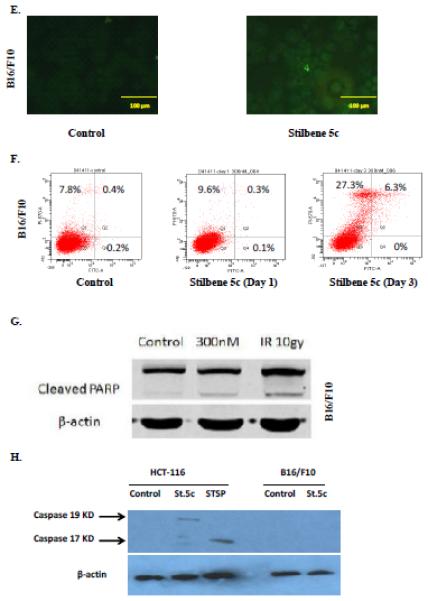
HCT-116 colon cancer cells were treated with 100nM stilbene 5c. A. TUNEL staining. B. Quantification of TUNEL staining (* p<0.05 compared to control). C. Staining with annexin V and PI . Q1- necrotic cells; Q2- late apoptotic cell population; Q3 – viable cells Q4- early apoptosis. Calculation of the percentage of apoptotic cells presented in Panel D is based on the combined values from Q2 and Q4 (* p<0.05 compared to control). Graphs represent pooled data from three replicate experiments. Error bars represent standard error (* p<0.05 compared to control). (E) Apoptosis evaluated in B16/F10 cells based on the TUNEL assay (F) PI/Annexin staining ( G) PARP cleavage and (H) Caspase 3 activation. For G and H, (IR): radiation (10 Gy) and staurosporine (1 μM) were used as positive control, respectively.
3.5 Assessment of mitotic catastrophe by stilbene 5c
Several reports have shown that mitotic catastrophe might characterize a mitotic form of apoptosis [29-30]. Figure 7 demonstrates that HCT-116 cells treated with stilbene 5c have an abnormal enlarged morphology with multiple nuclei with the presence of DNA fragments, an indication of mitotic catastrophe induction. Studies in B16/F10 melanoma cells demonstrated only cells with single nuclei, with no markers of mitotic catastrophe (data not shown).
Figure 7. Mitotic catastrophe in HCT116 cells in response to stilbene 5c.
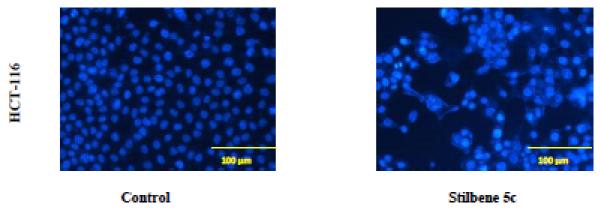
DAPI staining of HCT116 colon cancer cells exposed to 100 nM stilbene 5c for 72 hrs. Magnification 200X. Mitotic catastrophe is characterized by bi-nucleated and micro-nucleated cells. In control cells, nuclei are separated and well rounded; treated cell nuclei appear abnormal in size with more than one nucleus in the same proximity. Images are representative of three replicate studies.
3.6 Response to stilbene 5c in HCT-116 colon carcinoma cells lacking functional p53
The tumor suppressor gene p53 plays major roles in cell mechanisms that respond to genotoxic stress such as the induction of cell cycle arrest at G1/S phase and the initiation of apoptosis signaling pathways [31-32]. Cells lacking wild-type p53 have been shown to have the capability to evade apoptosis and are more likely to be resistant to anticancer therapy [33-35]. Thus, one of the more desirable properties of an antitumor agent is to be effective regardless of p53 status since more than 50% of tumors are p53 mutant. In order to evaluate sensitivity to stilbene 5c in cells lacking functional p53, HCT-116 (p53 −/−) cells were treated with 100 nM stilbene 5c and cell viability was evaluated in a time course study. The response to stilbene 5c in p53 −/− HTC-116 cells (Figure 8) was essentially identical to that in the p53 wild-type cells (Figure 2A), indicating that stilbene 5c does not require a wild-type p53 gene to suppress tumor cell growth.
Figure 8. Influence of Stilbene 5c on viability of HCT-116 (p53−/−) colon carcinoma cells.
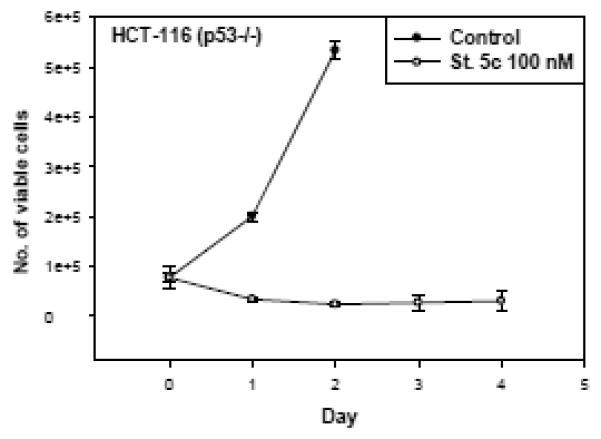
HCT-116 (p53−/−) colon cancer cells were treated with 100nM stilbene 5c and viable cell number was determined by trypan blue exclusion. Graphs represent pooled data from three replicate experiments.
3.7 Effects of pharmacological inhibition of stilbene 5c-induced autophagy in HCT-116 and B16/F10 cells
As shown above, stilbene 5c promotes autophagy in HCT-116 and B16/F10 cells. Autophagy has been shown to alternatively act as either a defensive mechanism in response to drug treatments or as a mode of cell death [36-37]. In order to assess the role of autophagy induced by stilbene 5c in both cell lines, we used the pharmacological autophagy inhibitors chloroquine for HCT-116 cells and bafilomycin A1 for B16/F10 cells, due to their relatively low toxicity in the respective cell lines [23]. Utilizing both the MTT dye assays and clonogenic survival, we observed that while treatment with both an autophagy inhibitor and stilbene 5c was more effective than treatment with stilbene 5c, the overall effect was essentially additive for the antiproliferative activity of each agent alone rather than reflecting sensitization through autophagy inhibition ( i.e. the autophagy induced by stilbene 5c did not appear to be cytoprotective for the HCT-116 cells or B16/F10 melanoma cells) (Figure 9).
Figure 9. Influence of autophagy inhibition on sensitivity to stilbene 5c in HCT-116 cells and B16/F10 melanoma cells.
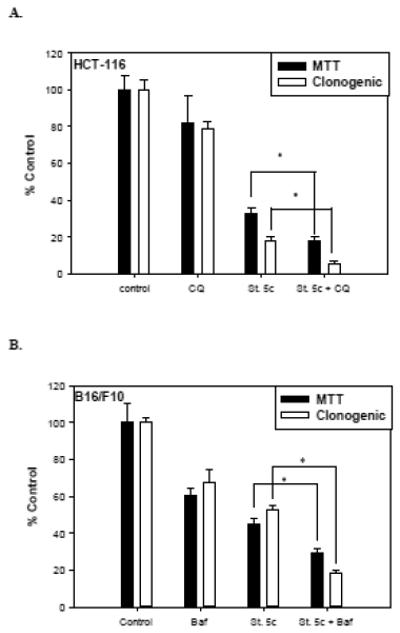
A. Viability and clonogenicity of HCT-116 cells upon exposure to 10 μM chloroquine, 100nM stilbene 5c, and chloroquine + stilbene 5c was measured by the MTT assay and clonogenic survival assay, respectively. B. Viability and clonogenicity of B16/F10 melanoma cells upon exposure to 10 nM bafilomycin, 300nM stilbene 5c, and bafilomycin + stilbene 5c was measured by MTT assay and clonogenic survival assay, respectively. In both assays, cells were pretreated with the autophagy inhibitor for 3 hours prior to stilbene 5c and then exposed to stilbene 5c for 72 hrs (* p<0.05 compared to St.5c alone).
3.8 Stilbene 5c possesses anti-angiogenic and anti-vascular properties
In addition to direct antitumor effects, agents that interfere with tubulin function have frequently been shown to also have anti-angiogenic and anti-vascular properties [38]. Figure 10A indicates that HUVEC human endothelial cells were highly sensitive to stilbene 5c, which inhibited their proliferation with an IC50 of 20 nM. Stilbene 5c also inhibited human endothelial cell tube formation on Matrigel, an in vitro correlate of anti-angiogenic actions (Figure 10B). Stilbene 5c caused a 74% inhibition at 10 nM and a 98% inhibition at 20 nM, while CoA4 only produced a 60% inhibition at 20 nM. For both endothelial cell tube formation and proliferation, stilbene 5c was found to be at least as potent as the vascular-disrupting agent combretastatin A4 (Figures 10A and 10B).
Figure 10. Stilbene 5c inhibits endothelial cell growth and tubule formation.
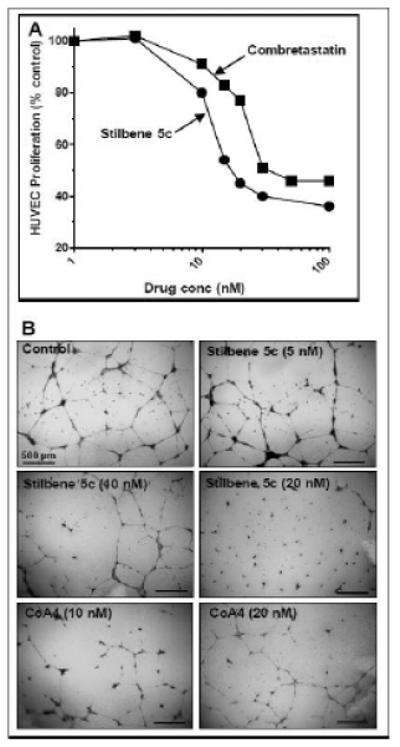
A. HUVEC were plated in 96 well plates and allowed to attach overnight. The indicated concentrations of stilbene 5c (✩) or combretastatin A4 (■) were added, and cell numbers determined after an additional 72 hours by staining with SRB. B. HUVEC were plated on growth-factor reduced Matrigel with the indicated concentrations of stilbene 5c or combretastatin A4. After 24 hours, the wells were fixed, stained and photographed, as described in Methods.
In a previous report, we found that stilbene 5c selectively suppressed ovarian cancer tumor perfusion in vivo, as measured by dynamic contrast-enhanced MRI (DCEMRI) [17]. While gadolinium-based DCE-MRI has been widely used to document the vascular-disrupting actions of the microtubule-binding drugs, including in phase I trials, due to its slow clearance it cannot be used to conduct longitudinal tumor perfusion imaging over time periods shorter than 24 hours in the same animal. We therefore utilized a method which would allow for the repeated measurement of tumor perfusion in the same mouse at short time intervals and/or after multiple drug treatments [39]. This method used luciferin as the imaging agent in mice with tumor xenografts derived from UCI-101/luciferase ovarian cancer cells. While luciferase-dependent imaging is most commonly used to quantify tumor burden, photon emission by luciferase in tumors is proportional not only to the tumor burden, but also directly correlated with tumor vascular perfusion in the early phase after luciferin injection; this is a consequence of the fact that the injected luciferin substrate needs to be delivered to the tumor through vessels to serve as a substrate for luciferase in the tumor to generate photons, which can be measured by the Xenogen Live Imaging system. This scenario is similar to an injection of gadolinium, which is delivered to the tumor to serve as the contrast to generate MRI signals.
A baseline analysis in untreated mice, injecting luciferin intraperitoneally at several time intervals, was first performed to evaluate the reproducibility of the imaging over a 72 hour period (Figure 11A). Photon emission was quantified by Xenogen Live Imaging system every two minutes for a total of 60 minutes. Photon emissions reached a peak at about 15-20 minutes and slowly decreased thereafter. After two hours, no trace of photon emission could be detected. Studies performed at various time intervals, (0, 4, 24, 48, and 72 hours) showed a high degree of consistency in the appearance of photon emission and tumor blood flow, with a coefficient of variation of 9.4% (Figure 11A).
Figure 11. Utilization of luciferase imaging for longitudinal study of tumor perfusion and the effect of stilbene 5c in mouse xenografts.
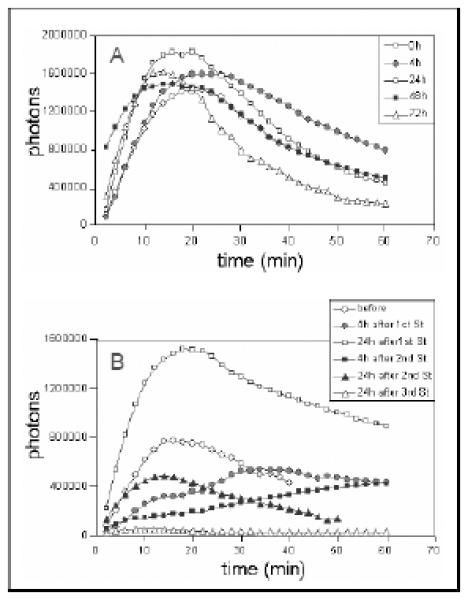
Mice with UCI-101/luciferase tumor xenografts were tested at multiple time points to assess tumor vascular perfusion. A. Luciferin was injected ip at 0, 4, 24, 48, 72 hr and images were collected by Xenogen Live Imaging system every 2 min for total 60 min after injection. In the representative mouse shown, the total number of photons in the tumor area of interest was calculated and plotted against time (min) during the collection period. B. Mice with UCI-101/luciferase tumor xenografts were treated with stilbene 5c at 50 mg/kg/day ip for 3 days. In the mouse shown here, luciferin was injected before, 4 hr after, and 24 hr after the first and second stilbene injections for imaging. Final imaging was done at 24 hr after the third stilbene injection. All images were collected by Xenogen Live Imaging system every 2 min for total 60 min after injection. The total number of photons in the tumor area was calculated and plotted against time (min) during the collection period.
Using this method, we evaluated tumor vascular perfusion after three daily injections of stilbene 5c. Imaging was performed at 4 hours after each stilbene administration so as to compare with our previous MRI findings [17]. We found that tumor perfusion was suppressed by 65% at 4 hours after intraperitoneal injection of stilbene 5c (50 mg/kg) (Figure 11B), data that was consistent with our MRI findings. Tumor perfusion partially rebounded at 24 hours, and along with the reestablishment of the tumor vasculature, this could have also been due to vasodilatation, which would increase tumor blood flow. When mice were given a second dose of stilbene 5c on day 2, there was a more pronounced suppression and a smaller rebound in tumor vascular perfusion, compared with the response to the first dose. A third dose of stilbene 5c nearly completely suppressed tumor perfusion (Figure 11B).
4. Discussion
Microtubules are dynamic proteins that play major functions in trafficking, signaling, division, and migration in eukaryotic and mammalian cells. Vinca alkaloids and colchicine-binding inhibitors destabilize microtubules leading to depolymerization while taxanes increase microtubule stability and polymerization [40-42]. Vinca alkaloids and taxanes were discovered more than 40 years ago and have been used effectively in the treatment of different solid and hematological malignancies [43-45]. While many colchicine-binding inhibitors have been developed and tested, none have been approved for any anticancer therapeutic indication, due largely to their neuro- and cardiotoxicities [7]. Previous studies of the colchicine binding site inhibitor, stilbene 5c, have shown that this drug disrupts tumor perfusion without affecting normal organ perfusion, leading to decreased delivery of blood and oxygen to tumor cells [17]. Stilbene 5c was also found to promote apoptosis in ovarian and leukemic cancer cells in vitro [15-16].
The current work evaluated the capacity of stilbene 5c to promote growth arrest and cell death in both HCT-116 colon carcinoma cells and B16/F10 melanoma. HCT-116 cells were found to be more sensitive to stilbene 5c than B16/F10 melanoma cells. One possible basis for this difference in sensitivity could be alterations in microtubule composition and dynamics upon treatment. For instance, a study by Arai et al showed that class II β-tubulin expression increases in B16/F10 melanoma cells when exposed to vincristine [46]. Overexpression of this protein likely makes melanoma cells less sensitive to anti-tubulin drugs [46].
Deficiencies in apoptotic signaling pathways can lead to the development of resistance to microtubule disrupting agents [47-48]. Because HCT116 colon cancer cells have a wild-type p53 protein and are apoptosis proficient, it was important to investigate whether apoptosis represents a primary mode of cell death in these cells. Our data clearly showed an increase in apoptosis after 72 hours of exposure to the drug. Unlike HCT-116 colon cancer cells, treatment of B16F10 melanoma cells with stilbene 5c did not generate markers of apoptosis, suggesting that apoptosis does not play a role in the toxicity of stilbene 5c in melanoma cells. Several studies have indicated that melanomas are generally resistant to apoptosis when exposed to chemotherapeutic agents via induction of different protective mechanisms [49-50]. Also, melanoma cells show minimal levels of apoptosis at the site of tumor lesions [51-52], and these characteristics may explain the high potential of metastasis in melanomas. These data suggest that B16F10 cells exposed to stilbene 5c are likely to undergo autophagy but not apoptosis.
Several antitumor agents, including microtubule disrupting drugs, can trigger mitotic spindle check points [53]. It has also been shown that anti-microtubule agents induce mitotic arrest-associated cell death [54-55]. For instance, combretastatin A4, a colchicine-binding inhibitor, was found to promote cell death related to mitotic catastrophe [56]. The finding that HCT-116 cells may fail to complete the process of cytokinesis with the resultant generation of multiple micronuclei, a hallmark of mitotic catastrophe, is consistent with the previous findings of the cytotoxic effect of stilbene 5c in ovarian cancer cells [16]. These observations may indicate that cell death in HCT-116 colon cancer cells is not only due to apoptosis, but also occurs through mitotic catastrophe. B16/F10 melanoma cells did not show evidence of mitotic arrest/catastrophe upon treatment with stilbene 5c, supporting the possibility of differences in microtubule composition and dynamics between these cell lines.
Induction of autophagy in HCT-116 cells was evident based on acridine orange staining of autophagic vesicles as well as of the redistribution and puncta formation of an RFP/LC3 construct transfected into the cells. In addition, stilbene 5C promoted the degradation of p62, an indication of the induction of autophagic flux. Stilbene 5c also increased autophagic vacuole formation in B16F10 melanoma cells and promoted the conversion of LC3-I to LC3-II. However, we have previously reported that p62 is not detectable in the B16/F10 cells [23].
All of these observations point toward the capability of stilbene 5c to promote autophagy. However, although inhibition of autophagy in both cell lines increased the impact of Stilbene 5c on cell survival and viability, the impact of the combination treatment was essentially additive, which tends to exclude the possibility that autophagy might be playing a cytoprotective function in these experimental models.
Several studies have shown that induction of autophagy is associated with promotion of senescence in cancer cells [24-25]. In the current work, an essentially identical percentage of HCT-116 cells were shown to undergo both autophagy and senescence. This finding is consistent with a close association between these responses but is not direct evidence of a direct linkage. Our studies further demonstrated that subsequent to an initial phase of cell death, the residual surviving cells failed to recover after replenishment with drug-free medium. Previous studies investigated the effects of stilbene 5c on tubulin polymerization and these results indicated that stilbene 5c binds tubulin more potently than colchicine [15]. Thus, the apparent irreversible permanent arrest of treated cells could be due to unrepaired damage to microtubule function.
We have further confirmed the capacity of stilbene 5c to function as a vascular disrupting agent using the human endothelial cell tube formation Matrigel assay. In our previous studies, we found that stilbene 5c inhibited ovarian, pancreatic, breast and hepatocellular carcinoma cell proliferation with IC50s ranging from 30 to 80 nM while the current work shows that concentrations in the range of 100nM to 300nM are required for effectiveness against colon carcinoma and melanoma cells. However, endothelial cells are more sensitive to growth inhibition by stilbene 5c than were any of the carcinoma cells lines tested to date. This is consistent with our observations that treatment of HUVEC with stilbene 5c causes G2 cell cycle arrest, an 8-fold increase in the accumulation of cells with a sub G0/1 DNA content, the disruption of cellular microtubules, and the induction of apoptosis [17]. However, growth inhibition only partially accounts for the anti-vascular actions of the microtubule-binding drugs, which have also been shown to potently inhibit endothelial cell functions at even lower concentrations [38].
Alternative methods to DCE-MRI are needed to determine how fast tumor vascular perfusion can be suppressed by stilbenes, and how soon the re-establishment of tumor perfusion occurs. This information would be important in determining the optimal scheduling of stilbene 5c and related compounds, as studies suggest these drugs are most effective when administered more frequently and at shorter intervals [57-58]. We used luciferase imaging to quantify tumor blood flow, based on the assumption that delivery of the luciferin substrate for photon generation was dependent on tumor blood flow [39]. Since the luciferin was administered in excess and was therefore not a limiting factor for the luciferase reaction, a decrease in photons in the tumor was presumed to be a function of a suppression of tumor blood flow. In our experiments, photon detection was performed every two minutes, and the time to the peak of the curve and the area under the curve were used as indicators of tumor blood flow. A high degree of consistency was evident in repeated measurements of the same control mouse over 72 hours. Photon production, monitored after stilbene 5c administration, demonstrated a dramatic decrease and delay in achievement of the photon peak after 4 hours. The photon curve at 24 hours after stilbene 5c treatment demonstrated partial recovery compared to the 4 hour time point. A second treatment with stilbene 5c further suppressed the curve to an even lower level, and nearly all photon production was completely obliterated after the third dose even at 24 hours after the 3rd treatment.
In summary, stilbene 5c was shown to induce multiple growth arrest and cell death responses in two tumor cell lines, as well as to influence endothelial cell function at relatively low concentrations; we have also confirmed that this compound may work as a vascular disrupting agent by reducing tumor blood flow. Interestingly, we found that apoptosis is not absolutely necessary for the activity of stilbene 5c and both autophagy and senescence can contribute to its antitumor actions. These findings build upon our previous work that supports the potential utility of developing stilbene 5c as a promising agent for cancer therapy.
Footnotes
Footnote. The antibody uses detects both the 19kd and 17kd cleaved forms of caspase 3. In the HCT-116 cells, primarily the 19kd form was detected with stilbene treatment and the 17kd form with staurosporine. Neither form was detected with stilbene treatment in the B16/F10 melanoma cells.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
reference
- 1.Ohtsu A. Chemotherapy for metastatic gastric cancer: Past, present, and future. J Gastroenterol. 2008;43:256–64. doi: 10.1007/s00535-008-2177-6. [DOI] [PubMed] [Google Scholar]
- 2.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 3.Eigentler TK, Caroli UM, Radny P, Garbe C. Palliative therapy of disseminated malignant melanoma: A systematic review of 41 randomised clinical trials. Lancet Oncol. 2003;4:748–59. doi: 10.1016/s1470-2045(03)01280-4. [DOI] [PubMed] [Google Scholar]
- 4.Avril MF, Aamdal S, Grob JJ, Hauschild A, Mohr P, Bonerandi JJ, et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: A phase iii study. J Clin Oncol. 2004;22:1118–25. doi: 10.1200/JCO.2004.04.165. [DOI] [PubMed] [Google Scholar]
- 5.Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The oblimersen melanoma study group. J Clin Oncol. 2006;24:4738–45. doi: 10.1200/JCO.2006.06.0483. [DOI] [PubMed] [Google Scholar]
- 6.Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, et al. Dacarbazine (dtic) versus vaccination with autologous peptide-pulsed dendritic cells (dc) in first-line treatment of patients with metastatic melanoma: A randomized phase iii trial of the dc study group of the decog. Ann Oncol. 2006;17:563–70. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]
- 7.Jiang N, Wang X, Yang Y, Dai W. Advances in mitotic inhibitors for cancer treatment. Mini Rev Med Chem. 2006;6:885–95. doi: 10.2174/138955706777934955. [DOI] [PubMed] [Google Scholar]
- 8.Rustin GJ, Galbraith SM, Anderson H, Stratford M, Folkes LK, Sena L, et al. Phase i clinical trial of weekly combretastatin a4 phosphate: Clinical and pharmacokinetic results. J Clin Oncol. 2003;21:2815–22. doi: 10.1200/JCO.2003.05.185. [DOI] [PubMed] [Google Scholar]
- 9.Beerepoot LV, Radema SA, Witteveen EO, Thomas T, Wheeler C, Kempin S, et al. Phase i clinical evaluation of weekly administration of the novel vascular-targeting agent, zd6126, in patients with solid tumors. J Clin Oncol. 2006;24:1491–8. doi: 10.1200/JCO.2005.02.7458. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, et al. Phase i trial of the antivascular agent combretastatin a4 phosphate on a 5-day schedule to patients with cancer: Magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol. 2003;21:4428–38. doi: 10.1200/JCO.2003.12.986. [DOI] [PubMed] [Google Scholar]
- 11.Keates RA, Sarangi F, Ling V. Structural and functional alterations in microtubule protein from chinese hamster ovary cell mutants. Proc Natl Acad Sci U S A. 1981;78:5638–42. doi: 10.1073/pnas.78.9.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabral F, Sobel ME, Gottesman MM. Cho mutants resistant to colchicine, colcemid or griseofulvin have an altered beta-tubulin. Cell. 1980;20:29–36. doi: 10.1016/0092-8674(80)90231-7. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29:2943–71. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wesolowska O, Wisniewski J, Bielawska-Pohl A, Paprocka M, Duarte N, Ferreira MJ, et al. Stilbenes as multidrug resistance modulators and apoptosis inducers in human adenocarcinoma cells. Anticancer Res. 2010;30:4587–93. [PubMed] [Google Scholar]
- 15.Cao TM, Durrant D, Tripathi A, Liu J, Tsai S, Kellogg GE, et al. Stilbene derivatives that are colchicine-site microtubule inhibitors have antileukemic activity and minimal systemic toxicity. Am J Hematol. 2008;83:390–7. doi: 10.1002/ajh.21104. [DOI] [PubMed] [Google Scholar]
- 16.Durrant D, Richards JE, Walker WT, Baker KA, Simoni D, Lee RM. Mechanism of cell death induced by cis-3, 4', 5-trimethoxy-3'-aminostilbene in ovarian cancer. Gynecol Oncol. 2008;110:110–7. doi: 10.1016/j.ygyno.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 17.Durrant D, Corwin F, Simoni D, Zhao M, Rudek MA, Salloum FN, et al. Cis-3, 4', 5-trimethoxy-3'-aminostilbene disrupts tumor vascular perfusion without damaging normal organ perfusion. Cancer Chemother Pharmacol. 2009;63:191–200. doi: 10.1007/s00280-008-0726-6. [DOI] [PubMed] [Google Scholar]
- 18.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: From autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 19.Polewska J, Skwarska A, Augustin E, Konopa J. DNA-damaging imidazoacridinone c-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J Pharmacol Exp Ther. 2013;346:393–405. doi: 10.1124/jpet.113.203851. [DOI] [PubMed] [Google Scholar]
- 20.Eskelinen EL, Saftig P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–73. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biggers JW, Nguyen T, Di X, Gupton JT, Henderson SC, Emery SM, et al. Autophagy, cell death and sustained senescence arrest in b16/f10 melanoma cells and hct-116 colon carcinoma cells in response to the novel microtubule poison, jg-03-14. Cancer Chemother Pharmacol. 2013;71:441–55. doi: 10.1007/s00280-012-2024-6. [DOI] [PubMed] [Google Scholar]
- 24.Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, et al. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther. 2012;343:763–78. doi: 10.1124/jpet.112.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The atg16l complex specifies the site of lc3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskelinen EL. New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol. 2008;266:207–47. doi: 10.1016/S1937-6448(07)66005-5. [DOI] [PubMed] [Google Scholar]
- 28.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 29.Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–70. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- 30.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated g2 checkpoint abrogation. Cancer Res. 2007;67:339–45. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 31.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 32.O'Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, et al. Characterization of the p53 tumor suppressor pathway in cell lines of the national cancer institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997;57:4285–300. [PubMed] [Google Scholar]
- 33.McGill G, Fisher DE. Apoptosis in tumorigenesis and cancer therapy. Front Biosci. 1997;2:d353–79. doi: 10.2741/a197. [DOI] [PubMed] [Google Scholar]
- 34.Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S, et al. Bcl-2 family proteins: Regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem. 1996;60:23–32. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C23::AID-JCB5%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Rudin CM, Thompson CB. Apoptosis and disease: Regulation and clinical relevance of programmed cell death. Annu Rev Med. 1997;48:267–81. doi: 10.1146/annurev.med.48.1.267. [DOI] [PubMed] [Google Scholar]
- 36.Shintani T, Klionsky DJ. Autophagy in health and disease: A double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16:12–20. doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15:2594–601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao D, Richer E, Antich PP, Mason RP. Antivascular effects of combretastatin a4 phosphate in breast cancer xenograft assessed using dynamic bioluminescence imaging and confirmed by mri. FASEB J. 2008;22:2445–51. doi: 10.1096/fj.07-103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geney R, Chen J, Ojima I. Recent advances in the new generation taxane anticancer agents. Med Chem. 2005;1:125–39. doi: 10.2174/1573406053175292. [DOI] [PubMed] [Google Scholar]
- 41.Larkin JM, Kaye SB. Epothilones in the treatment of cancer. Expert Opin Investig Drugs. 2006;15:691–702. doi: 10.1517/13543784.15.6.691. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat. 2007;10:162–81. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Pajk B, Cufer T, Canney P, Ellis P, Cameron D, Blot E, et al. Anti-tumor activity of capecitabine and vinorelbine in patients with anthracycline- and taxane-pretreated metastatic breast cancer: Findings from the eortc 10001 randomized phase ii trial. Breast. 2008;17:180–5. doi: 10.1016/j.breast.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Norris B, Pritchard KI, James K, Myles J, Bennett K, Marlin S, et al. Phase iii comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National cancer institute of canada clinical trials group study ma8. J Clin Oncol. 2000;18:2385–94. doi: 10.1200/JCO.2000.18.12.2385. [DOI] [PubMed] [Google Scholar]
- 45.Dimitroulis J, Stathopoulos GP. Evolution of non-small cell lung cancer chemotherapy (review) Oncol Rep. 2005;13:923–30. [PubMed] [Google Scholar]
- 46.Arai K, Matsumoto Y, Nagashima Y, Yagasaki K. Regulation of class ii beta-tubulin expression by tumor suppressor p53 protein in mouse melanoma cells in response to vinca alkaloid. Mol Cancer Res. 2006;4:247–55. doi: 10.1158/1541-7786.MCR-05-0183. [DOI] [PubMed] [Google Scholar]
- 47.Murphy M, Hinman A, Levine AJ. Wild-type p53 negatively regulates the expression of a microtubule-associated protein. Genes Dev. 1996;10:2971–80. doi: 10.1101/gad.10.23.2971. [DOI] [PubMed] [Google Scholar]
- 48.Zhang CC, Yang JM, White E, Murphy M, Levine A, Hait WN. The role of map4 expression in the sensitivity to paclitaxel and resistance to vinca alkaloids in p53 mutant cells. Oncogene. 1998;16:1617–24. doi: 10.1038/sj.onc.1201658. [DOI] [PubMed] [Google Scholar]
- 49.Grossman D, Altieri DC. Drug resistance in melanoma: Mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 2001;20:3–11. doi: 10.1023/a:1013123532723. [DOI] [PubMed] [Google Scholar]
- 50.Satyamoorthy K, Bogenrieder T, Herlyn M. No longer a molecular black box--new clues to apoptosis and drug resistance in melanoma. Trends Mol Med. 2001;7:191–4. doi: 10.1016/s1471-4914(01)02013-5. [DOI] [PubMed] [Google Scholar]
- 51.Mooney EE, Ruis Peris JM, O'Neill A, Sweeney EC. Apoptotic and mitotic indices in malignant melanoma and basal cell carcinoma. J Clin Pathol. 1995;48:242–4. doi: 10.1136/jcp.48.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staunton MJ, Gaffney EF. Tumor type is a determinant of susceptibility to apoptosis. Am J Clin Pathol. 1995;103:300–7. doi: 10.1093/ajcp/103.3.300. [DOI] [PubMed] [Google Scholar]
- 53.Tao W. The mitotic checkpoint in cancer therapy. Cell Cycle. 2005;4:1495–9. doi: 10.4161/cc.4.11.2130. [DOI] [PubMed] [Google Scholar]
- 54.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in hela cells by low concentrations of paclitaxel (taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–25. [PubMed] [Google Scholar]
- 55.Milross CG, Mason KA, Hunter NR, Chung WK, Peters LJ, Milas L. Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. J Natl Cancer Inst. 1996;88:1308–14. doi: 10.1093/jnci/88.18.1308. [DOI] [PubMed] [Google Scholar]
- 56.Nabha SM, Mohammad RM, Dandashi MH, Coupaye-Gerard B, Aboukameel A, Pettit GR, et al. Combretastatin-a4 prodrug induces mitotic catastrophe in chronic lymphocytic leukemia cell line independent of caspase activation and poly(adp-ribose) polymerase cleavage. Clin Cancer Res. 2002;8:2735–41. [PubMed] [Google Scholar]
- 57.Kanthou C, Tozer GM. Microtubule depolymerizing vascular disrupting agents: Novel therapeutic agents for oncology and other pathologies. Int J Exp Pathol. 2009;90:284–94. doi: 10.1111/j.1365-2613.2009.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006;66:11520–39. doi: 10.1158/0008-5472.CAN-06-2848. [DOI] [PubMed] [Google Scholar]