

Abstract
Silymarin, an extract of the seeds of milk thistle (Silybum marianum), is used as an herbal remedy, particularly for hepatoprotection. The main chemical constituents in silymarin are seven flavonolignans. Recent studies explored the non-selective methylation of one flavonolignan, silybin B, and then tested those analogues for cytotoxicity and inhibition of both cytochrome P450 (CYP) 2C9 activity in human liver microsomes and hepatitis C virus infection in a human hepatoma (Huh7.5.1) cell line. In general, enhanced bioactivity was observed with the analogues. To further probe the biological consequences of methylation of the seven major flavonolignans, a series of 7-O-methylflavonolignans were generated. Optimization of the reaction conditions permitted selective methylation at the phenol in the 7-position in the presence of each metabolite’s 4–5 other phenolic and/or alcoholic positions without the use of protecting groups. These 7-O-methylated analogues, in parallel with the corresponding parent compounds, were evaluated for cytotoxicity against Huh7.5.1 cells; in all cases the monomethylated analogues were more cytotoxic than the parent compounds. Moreover, parent compounds that were relatively non-toxic and inactive or weak inhibitors of hepatitis C virus infection had enhanced cytotoxicity and anti-HCV activity upon 7-O-methylation. Also, the compounds were tested for inhibition of major drug metabolizing enzymes (CYP2C9, CYP3A4/5, UDP-glucuronsyltransferases) in pooled human liver or intestinal microsomes. Methylation of flavonolignans differentially modified inhibitory potency, with compounds demonstrating both increased and decreased potency depending upon the compound tested and the enzyme system investigated. In total, these data indicated that monomethylation modulates the cytotoxic, antiviral, and drug interaction potential of silymarin flavonolignans.
Keywords: Silymarin, Milk thistle, Silybum marianum, Flavonolignans, Methylation, Hepatitis C virus, Cytochrome P450, UDP-glucuronosyl-transferase
1. Introduction
The historical uses of milk thistle [Silybum marianum (L.) Gaertn. (Asteraceae)], particularly in the areas of cancer chemoprevention and hepatoprotection, have been reviewed extensively.1–8 From a chemistry perspective, silymarin is a crude extract of the seeds and affords a mixture of flavonolignans, which are diastereomeric and/or constitutional isomers of each other; a separate review explains the nomenclature of the various constituents.9 With respect to the purified compounds, there are seven key flavonolignans, which are termed silybin A (1), silybin B (3), isosilybin A (5), isosilybin B (7), silychristin (9), isosilychristin (11), and silydianin (13; Scheme 1).
Scheme 1.

Structures and conditions for the synthesis of 7-O-methylated analogues of the seven major flavonolignans from silymarin.
Recently, members of our team reported enhanced bioactivity of methylated analogues of silybin B (3), particularly with respect to assays for cytotoxic and antiviral activities.10 Therein, a non-selective methylation method using dimethyl sulfate afforded a series of five analogues of silybin B: mono-, di-, two different tri-, and tetra-methylated analogues. Since the objective of the research at that stage was to explore the biological activity of a few methylated analogues, the selectivity of the reaction was not critical. In fact, it was beneficial that those reaction conditions were non-selective, as this produced multiple analogues for testing.
The goal of the present study was to expand upon that initial SAR by selectively synthesizing 7-O-methyl analogues of the seven major flavonolignans (Scheme 1). However, rather than relying upon protecting groups, which require two additional synthetic steps for installation and removal, it was hypothesized that the inherent reactivity of the molecule could be harnessed to guide the selectivity. By judicious choice of reaction conditions, a single position, the C-7-phenol, was methylated selectively in the presence of multiple centers with similar reactivity, including 2 or 3 other phenolic positions, a similarly acidic secondary alcohol, and a primary alcohol. This chemoselectivity was comparable for all seven flavonolignans, which enabled a site-specific analysis of the importance of the methylated phenol for each compound’s activity in a suite of bioassays that probed cytotoxicity, anti-hepatitis C virus (HCV) activity, and inhibition of major drug metabolizing enzymes.
2. Results and discussion
2.1. Chemistry
The synthesis of 7-O-methyl analogues of the natural flavonolignans was challenging due to the possibility of alkylation at more than one hydroxyl group (Scheme 1). Therefore, a variety of bases [Cs2CO3, K2CO3, and Diazabicycloundecene (DBU)], electrophilic methylating agents (dimethyl carbonate, methyl iodide), solvents (acetone, THF, acetonitrile), temperatures (rt to 75 °C under standard heating or up to 130 °C using microwave heating), and times (2 h–2 days) were examined, all with the goal of minimizing the formation of polymethylated side products. Selective monomethylation of the phenolic group at C-7 was possible for all seven major flavonolignans over a temperature range of 30–60 °C using methyl iodide in dry acetone in the presence of K2CO3. The 7-position was the most reactive for methylation, presumably due to both acidity, being an arylogous carboxylic acid, and steric accessibility. While the yields were moderate to low, ranging from approximately 20–40%, the reaction conditions were optimized for selective monomethylation rather than yield.
2.2. Purification
Published methods were utilized to isolate the individual flavonolignans as starting materials for the methylation reactions.11 To purify the reaction mixtures, a PFP column was used in the reverse phase with a range of CH3CN/H2O (0.1% formic acid) gradients until each compound was >98% pure by analytical HPLC (Fig. S1; Supplementary data).
2.3. Structure determination
Structures of the 7-O-methylated analogues were determined using a suite of NMR spectroscopy and mass spectrometry experiments (see Tables 1 and 2 and Supplementary data). Using the data for 7-O-methylsilybin A (2) as a representative example, two methoxy singlets were apparent at δH 3.78 (3H) and 3.77 (3H), due to the introduced methoxy group at C-7 and the natural one at C-3″, respectively. In contrast, the parent flavonolignan silybin A (1) displayed a sharp singlet at δH 10.86 for the C-7 phenolic proton signal. Further evidence for the methoxy group on C-7 in analogue 2 was observed in the meta-coupled protons at H-6 and H-8 (δH 6.12 and 6.10, respectively), which were shifted downfield slightly compared to 1 (Table 1). The 13C NMR data (Table 2) of each methylated analogue displayed 26 distinct signals, which was one more than the non-methylated starting materials, including resonances at δC 56.0 and 55.7 due to the two methoxy moieties at C-7 and C-3″, respectively; these assignments were supported by HSQC correlations. Moreover, data from the HMBC experiment demonstrated the connectivity of the methoxy moieties (Fig. 1), where correlations were observed between the protons at δH 3.78 to C-7 (δC 167.6) and δH 3.77 to C-3″ (δC 147.6), respectively, verifying that the introduced methoxy moiety was at the C-7 position in 2. For each of the analogues, a similar set of NMR experiments and data, coupled with comparisons to the data for the natural flavonolignans, were utilized to establish that the methoxy moiety was connected at the 7 position in all of the analogues. Finally, HRMS data supported the introduction of a single methyl moiety, where the [M–H]− ion peak for all of the analogues was fourteen mass units greater than that of the parent flavonolignans (see experimental).
Table 1.
1H NMR data of flavonolignans and 7-O-methylflavonolignans in DMSO-d6 (500 MHz, 30 °C)
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 5.08, d (11.5) | 5.14, d (11.5) | 5.08, d (11.5) | 5.14, d (11.5) | 5.11, d (10.9) | 5.16, d (10.9) | 5.11, d (11.5) | 5.16, d (11.5) | 5.02, d (11.4) | 5.05, d (11.4) | 5.17, d (11.9) | 5.22, d (11.4) | 4.86, dd (10.4) | 4.91, dd (10.4) |
| 3 | 4.63, dd (11.5, 6.3) | 4.69, dd (11.5, 6.3) | 4.61, dd (11.5, 6.3) | 4.67, dd (11.5, 6.3) | 4.60, dd (10.9, 6.3) | 4.66, dd (10.9, 6.3) | 4.61, dd (11.5, 5.9) | 4.67, dd (11.5, 6.3) | 4.52, dd (11.4, 6.4) | 4.58, dd (11.4, 6.3) | 4.64, dd (11.9, 6.5) | 4.67, dd (11.4, 6.9) | 4.44, dd (10.4, 5.4) | 4.48, dd (10.4, 5.4) |
| 6 | 5.91, d (1.7) | 6.12, d (1.7) | 5.91, d (2.3) | 6.12, d (2.3) | 5.92, d (2.3) | 6.13, d (2.3) | 5.92, d (2.0) | 6.13, d (2.3) | 5.91, d (2.0) | 6.12, d (2.3) | 5.93, d (1.9) | 6.13, d (2.0) | 5.91, d (2.5) | 6.12, d (1.9) |
| 8 | 5.86, d (1.7) | 6.10, d (1.7) | 5.87, d (2.3) | 6.10, d (2.3) | 5.89, d (2.3) | 6.12, d (2.3) | 5.89, d (2.0) | 6.12, d (2.3) | 5.86, d (2.0) | 6.09, d (2.3) | 5.88, d (1.9) | 6.11, d (2.0) | 5.88, d (2.5) | 6.11, d (1.9) |
| 2′ | 7.09, d (1.7) | 7.10, d (1.7) | 7.08, d (1.8) | 7.10, d (2.3) | 7.09, d (2.3) | 7.11, d (2.3) | 7.10, d (1.8) | 7.11, d (1.7) | 6.82, d (1. 5) | 6.84, d (1.2) | — | — | 3.44, dd (4.0, 2.0) | 3.45, dd (4.0, 2.0) |
| 5′ | 6.97, d (8.0) | 6.98, d (8.6) | 6.97, d (8.1) | 6.98, d (8.0) | 6.93, d (8.6) | 6.94, d (8.6) | 6.93, d (8.0) | 6.94, d (8.0) | — | — | 6.75, d (8.1) | 6.74, d (8.1) | 3.17, dd (8.9, 3.0) | 3.19, dd (6.9, 3.0) |
| 6′ | 7.00, dd (8.0, 1.7) | 7.01, dd (8.6, 1.7) | 7.02, dd (8.1, 1.8) | 7.02, dd (8.0, 2.3) | 6.98, dd (8.6, 2.3) | 6.99, dd (8.6, 2.3) | 6.98, dd (8.0, 1.8) | 7.00, dd (8.0, 1.7) | 6.87, br s | 6.88, br s | 6.96, d (8.1) | 6.97, d (8.1) | 5.92, d (8.9) | 6.01, d (6.9) |
| 2″ | 7.01, d (1.8) | 7.03, d (1.8) | 7.02, d (1.7) | 7.04, d (1.7) | 7.00, d (1.7) | 7.00, d (1.7) | 7.00, d (2.3) | 7.01, d (1.8) | 6.96, d (2.0) | 6.96, d (1.8) | 6.86, d (1.5) | 6.86, d (2.0) | 6.75, d (2.0) | 6.76, d (1.9) |
| 5″ | 6.80, d (8.6) | 6.80, d (8.6) | 6.79, d (8.6) | 6.80, d (8.0) | 6.80, d (8.6) | 6.80, d (8.0) | 6.80, d (8.6) | 6.80, d (8.2) | 6.76, d (8.4) | 6.76, d (8.1) | 6.70, d (7.9) | 6.69, d (8.5) | 6.63, d (8.5) | 6.64, d (8.4) |
| 6″ | 6.86, dd (8.6, 1.8) | 6.86, dd (8.6, 1.8) | 6.86, dd (8.6, 1.7) | 6.86, dd (8.0, 1.7) | 6.86, dd (8.6, 1.7) | 6.85, dd (8.0, 1.7) | 6.86, dd (8.6, 2.3) | 6.85, dd (8.2, 1.8) | 6.80, dd (8.4, 2.0) | 6.81, dd (8.1, 1.8) | 6.76, dd (7.9, 1.5) | 6.76, dd (8.5, 2.0) | 6.55, dd (8.5, 2.0) | 6.56, dd (8.4, 1.9) |
| 7″ | 4.90, d (7.5) | 4.91, d (7.5) | 4.90, d (7.5) | 4.91, d (7.5) | 4.91, d (8.1) | 4.91, d (7.5) | 4.91, d (7.5) | 4.92, d (7.5) | 5.46, d (7.0) | 5.46, d (6.9) | 5.58, d (2.5) | 5.57, d (2.5) | 3.31, br s | 3.31, br s |
| 8″ | 4.17, ddd (7.5, 4.6, 2.3) | 4.18, ddd (7.5, 4.6, 2.3) | 4.16, ddd (7.5, 4.1, 2.3) | 4.17, ddd (7.5, 4.6, 2.3) | 4.16, ddd (8.1, 4.6, 2.3) | 4.17, ddd (7.5, 4.0, 2.3) | 4.17, ddd (7.5, 4.6, 2.3) | 4.17, ddd (7.5, 4.6, 1.7) | 3.47, ddd (12.6, 12.0, 7.0) | 3.47, ddd (12.6, 12.0, 6.9) | 3.67, m | 3.67, m | 2.72, br s | 2.73, br s |
| 9″a | 3.53, ddd (10.3, 5.2, 2.3) | 3.53, ddd (10.3, 5.2, 2.3) | 3.53, ddd (9.8, 4.6, 2.3) | 3.54, ddd (12.0, 5.2, 2.3) | 3.53, ddd (12.0, 4.6, 2.3) | 3.53, ddd (12.0, 4.6, 2.3) | 3.54, ddd (11.9, 5.2, 2.3) | 3.53, ddd (12.0, 5.2, 1.7) | 3.72, ddd (12.6, 10.9, 5.0) | 3.72, ddd (12.6, 10.9, 5.2) | 3.68, m | 3.68, m | 4.12, dd (8.0, 3.0) | 4.13, dd (8.0, 3.0) |
| 9″b | 3.33, m | 3.34, m | 3.33, m | 3.33, m | 3.33, m | 3.33, m | 3.33, m | 3.33, m | 3.63, ddd (12.6, 10.9, 5.6) | 3.63, ddd (12.6, 10.9, 5.7) | 3.45, m | 3.44, m | 3.78, d (8.0) | 3.78, d (8.0) |
| 7-OCH3 | — | 3.78, s | — | 3.78, s | — | 3.79, s | — | 3.79, s | — | 3.78, s | — | 3.80, s | — | 3.79, s |
| 3″-OCH3 | 3.77, s | 3.77, s | 3.77, s | 3.77, s | 3.78, s | 3.77, s | 3.77, s | 3.77, s | 3.76, s | 3.76, s | 3.70, s | 3.69, s | 3.73, s | 3.74, s |
| 3-OH | 5.83, d (6.3) | 5.90, d (6.3) | 5.83, d (6.3) | 5.89, d (6.3) | 5.83, d (6.3) | 5.91, d (6.3) | 5.84, d (5.9) | 5.91, d (6.3) | 5.77, d (6.4) | 5.81, d (6.3) | 5.76, d (6.5) | 5.81, d (6.9) | 6.02, d (6.3) | 6.04, d (6.3) |
| 5-OH | 11.90, s | 11.86, s | 11.90, s | 11.87, s | 11.90, s | 11.87, s | 11.90, s | 11.87, s | 11.91, s | 11.87, s | 11.93, s | 11.90, s | 11.81, s | 11.77, s |
| 7-OH | 10.86, s | — | 10.86, s | — | 10.85, s | — | 10.86 s | — | 10.84, s | — | 10.98, s | — | 10.91, s | — |
| 4″-OH | 9.18, s | 9.19, s | 9.17, s | 9.18, s | 9.15, s | 9.18, s | 9.16, s | 9.18, s | 9.03, s | 9.02, s | 9.00, s | 8.99, s | 8.79, s | 8.79, s |
| 9″-OH | 4.97, dd (5.7, 5.2) | 4.98, dd (5.7, 5.2) | 4.98, dd (5.7, 4.6) | 4.98, dd (5.7, 5.2) | 4.95, dd (5.7, 4.6) | 4.97, dd (5.7, 4.6) | 4.96, dd (5.5, 5.0) | 4.97, dd (4.6, 5.2) | 5.01, dd (5.6, 5.0) | 5.01, dd (5.7, 5.2) | 5.07, br dd (4.5, 2.0) | 5.06, dd (6.5, 4.5) | ||
| 3′-OH | — | — | — | — | — | — | — | — | — | — | — | — | 7.19, s | 7.17, s |
Chemical shifts in δ, coupling constants in Hz.
Table 2.
13C NMR data of flavonolignans and 7-O-methylated analogues in DMSO-d6 (125 MHz, 30 °C)
| Position | Type | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | CH | 82.6 | 82.7 | 82.5 | 82.7 | 82.5 | 82.7 | 82. 5 | 82.7 | 83.3 | 83.4 | 79.9 | 80.0 | 81.7 | 81.8 |
| 3 | CH | 71.4 | 71.5 | 71.4 | 71.5 | 71.5 | 71.6 | 71.5 | 71.6 | 71.7 | 71.8 | 71.5 | 71.7 | 70.8 | 71.0 |
| 4 | C | 197.8 | 198.5 | 197.7 | 198.5 | 197.8 | 198.4 | 197.8 | 198.4 | 197.8 | 198.4 | 198.2 | 198.7 | 196.5 | 197.6 |
| 4a | C | 100.4 | 101.4 | 100.4 | 101.4 | 100.5 | 101.4 | 100.5 | 101.4 | 100.5 | 101.4 | 100.6 | 101.5 | 100.3 | 101.2 |
| 5 | C | 163.3 | 163.0 | 163.3 | 163.0 | 163.3 | 163.0 | 163.3 | 163.0 | 163.3 | 163.0 | 163.4 | 163.1 | 163.4 | 163.0 |
| 6 | CH | 96.1 | 95.0 | 96.1 | 94.9 | 96.1 | 95.0 | 96.1 | 95.0 | 96.0 | 94.9 | 96.1 | 95.0 | 96.2 | 95.2 |
| 7 | C | 167.1 | 167.6 | 166.9 | 167.6 | 166.9 | 167.6 | 166.9 | 167.6 | 166.8 | 167.5 | 166.8 | 167.6 | 166.9 | 167.6 |
| 8 | CH | 95.1 | 93.9 | 95.0 | 93.9 | 95.1 | 93.9 | 95.1 | 93.9 | 95.0 | 93.8 | 95.1 | 93.9 | 95.0 | 93.8 |
| 8a | C | 162.5 | 162.4 | 162.4 | 162.4 | 162.5 | 162.4 | 162.5 | 162.4 | 162.6 | 162.5 | 162.6 | 162.5 | 162.0 | 162.0 |
| 1′ | C | 130.1 | 129.9 | 130.1 | 130.0 | 130.3 | 130.2 | 130.3 | 130.2 | 130.0 | 129.8 | 124.5 | 124.4 | 139.5 | 139.4 |
| 2′ | CH | 116.6 | 116.7 | 116.6 | 116.7 | 116.5 | 116.6 | 116.5 | 116.5 | 115.6 | 115.7 | 128.9 | 128.9 | 48.6 | 48.7 |
| 3′ | C | 143.3 | 143.3 | 143.2 | 143.2 | 142.9 | 142.9 | 142.9 | 142.9 | 146.4 | 146.4 | 141.6 | 141.6 | 96.7 | 96.7 |
| 4′ | C | 143.7 | 143.7 | 143.6 | 143.7 | 143.9 | 143.9 | 143.9 | 143.9 | 140.7 | 140.7 | 145.9 | 145.8 | 201.9 | 201.9 |
| 5′ | CH | 116.3 | 116.4 | 116.3 | 116.4 | 116.4 | 116.5 | 116.5 | 116.4 | 129.1 | 129.1 | 116.1 | 116.1 | 53.4 | 53.3 |
| 6′ | CH | 121.4 | 121.5 | 121.1 | 121.2 | 120.9 | 121.0 | 120.9 | 121.0 | 115.4 | 115.4 | 119.5 | 119.4 | 124.0 | 124.0 |
| 1″ | C | 127.5 | 127.5 | 127.5 | 127.5 | 127.5 | 127.5 | 127.5 | 127.5 | 132.4 | 132.4 | 132.8 | 132.8 | 133.0 | 133.0 |
| 2″ | CH | 111.6 | 111.6 | 111.6 | 111.6 | 111.7 | 111.7 | 111.7 | 111.7 | 110.4 | 110.9 | 110.2 | 110.3 | 112.4 | 112.4 |
| 3″ | C | 147.6 | 147.6 | 147.6 | 147.6 | 147.6 | 147.6 | 147.6 | 147.6 | 147.5 | 148.1 | 147.5 | 147.5 | 147.1 | 147.2 |
| 4″ | C | 147.0 | 147.0 | 147.0 | 147.0 | 147.0 | 147.0 | 147.0 | 147.0 | 147.1 | 147.7 | 146.3 | 146.3 | 145.1 | 145.1 |
| 5″ | CH | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.3 | 115.1 | 115.1 | 114.9 | 115.0 |
| 6″ | CH | 120.5 | 120.6 | 120.5 | 120.5 | 120.5 | 120.5 | 120.5 | 120.4 | 118.7 | 118.7 | 118.5 | 118.6 | 120.3 | 120.3 |
| 7″ | CH | 75.9 | 75.9 | 75.8 | 75.9 | 75.9 | 75.9 | 75.9 | 75.9 | 87.0 | 87.5 | 86.4 | 86.4 | 46.0 | 46.0 |
| 8″ | CH | 78.1 | 78.1 | 78.1 | 78.1 | 78.0 | 78.0 | 78.0 | 78.0 | 53.4 | 53.9 | 52.0 | 51.9 | 44.0 | 44.0 |
| 9″ | CH2 | 60.2 | 60.2 | 60.2 | 60.2 | 60.2 | 60.2 | 60.2 | 60.2 | 63.0 | 63.5 | 63.5 | 63.5 | 72.8 | 72.8 |
| 3″-OCH3 | CH3 | 55.7 | 55.7 | 55.7 | 55.7 | 55.7 | 55.7 | 55.7 | 55.7 | 55.6 | 55.6 | 55.6 | 55.6 | 55.4 | 55.4 |
| 7-OCH3 | CH3 | — | 56.0 | — | 56.0 | — | 56.0 | — | 56.0 | — | 55.9 | — | 56.0 | — | 56.0 |
Chemical shifts in δ. In compounds 9 and 10, position 5′ is quaternary; in compounds 11 and 12, position 2′ is quaternary.
Figure 1.
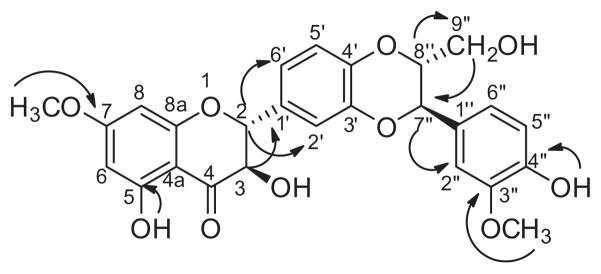
Key HMBC correlations of 7-O-methylsilybin A (2).
2.4. Biological studies
2.4.1. Cytotoxicity and cell viability
In comparing the cytotoxicity profiles of 7-O-methylated analogues versus parental counterparts (Table 3), addition of a single methyl group increased cytotoxicity against human hepatoma cells (Huh7.5.1). These findings were consistent with an earlier pilot study that examined a series of mono-, di-, tri- and tetra-methylated analogues of silybin B (3).10 Silychristin (9), isosilychristin (11), and silydianin (13) have been reported to be relatively non-toxic compared to the other silymarin flavonolignans.12 In the current study, marked increases in the cytotoxicity of silydianin (13), silybin A (1), and isosilybin A (5) were observed upon methylation (compounds 14, 2, and 6, with 13.8-, 8.4-, and 6.1-fold, respectively). However, the 7-O-methylation of isosilybin B (7) did not produce a pronounced increase in cytotoxicity (i.e., compound 8), which was interesting given that 7 has been shown to be the most cytotoxic of the parent flavonolignans, particularly in assays for prostate cancer chemoprevention.12–17
Table 3.
Cytotoxic activity of flavonolignans and 7-O-methylated analogues against human hepatoma (Huh7.5.1) cells
| Compound | IC50 values (μM) | Fold changea,b |
|---|---|---|
| Silybin A (1) | 80.0 | |
| 7-O-Methylsilybin A (2) | 9.5 | 8.4 |
| Silybin B (3) | 68.7 | |
| 7-O-Methylsilybin B (4) | 12.7 | 5.5 |
| Isosilybin A (5) | 80.0 | |
| 7-O-Methylisosilybin A (6) | 13.2 | 6.1 |
| Isosilybin B (7) | 27.0 | |
| 7-O-Methylisosilybin B (8) | 18.0 | 1.5 |
| Silychristin (9) | >130 | |
| 7-O-Methylsilychristin (10) | 66.4 | >2 |
| Isosilychristin (11) | >130 | |
| 7-O-Methylisosilychristin (12) | 27.6 | >4.7 |
| Silydianin (13) | >130 | |
| 7-O-Methylsilydianin (14) | 9.4 | 13.8 |
| Resveratrolc | 10.6 |
The fold change was calculated by dividing the IC50 value for the parent flavonolignan by the IC50 value for the corresponding 7-O-methylated analogue.
The average fold change for all analogues was approximately 6.
Typical average value as a positive control for the assay.
2.4.2. Antiviral activity
In evaluating the anti-HCV profiles of these compounds (Fig. 2), some of the methylated flavonolignans displayed improved anti-HCV activity [i.e. 7-O-methylsilychristin (10), 7-O-methylisosilychristin (12)] relative to the corresponding parent compounds. In particular, silychristin (9), isosilychristin (11), and silydianin (13) did not show appreciable antiviral activity until they were methylated (i.e., 10, 12, and 14). On the other hand, for parent compounds that were active in blocking HCV activity, such as silybin A (1), silybin B (3), and isosilybin A (5), the activities of their methylated counterparts were either retained (2) or reduced (4 and 6). The most toxic parent compound, isosilybin B (7), had a slight enhancement of both antiviral activity and cytotoxicity (Table 3) by methylation (compound 8). Thus, it appeared that HCV antiviral activity tracked with cytotoxicity. That is, as compounds became more toxic, the more likely antiviral activity was detected. However, that is not to say that antiviral activity coincided with cytotoxicity, as we were always able to detect anti-HCV activity that was independent of cytotoxicity. We hypothesize that flavonolignans target shared cellular functions that confer both cytotoxic and antiviral activities, although this requires further experimentation to verify.
Figure 2.

Antiviral profile of parent and 7-O-methyl flavonolignans. Huh7.5.1 cells were infected with JFH-1 at a multiplicity of infection of 0.05. Virus inoculum was removed after 5 h and compounds were added. Cultures were incubated for 72 h before protein lysates were analyzed for HCV core protein expression by Western blot analysis. Detection of actin protein served as a protein loading control. Panel A depicts an example of a parent compound that lacked anti-HCV activity (9) and acquired anti-HCV activity upon methylation (10). Panel B depicts an example of a parent compound that had anti-HCV activity (3) and decreased anti-HCV activity upon methylation (4). The graphs below panels A and B present quantitation of HCV core pixel intensity following normalization to actin pixel intensity, expressed as percent inhibition relative to DMSO controls. Graphs below panel A depict parent compounds that lacked anti-HCV activity (9, 11, 13) and acquired anti-HCV activity upon methylation (10, 12, 14). Graphs below panel B depict parent compounds that had anti-HCV activity (1, 3, 5, 7), and retained (2), decreased (4, 6), or enhanced (8) anti-HCV activity upon methylation. Doses used were derived from toxicity data, such that two non-toxic concentrations and one near the IC50 value were evaluated in the antiviral assay. Concentrations for compounds 1, 3, and 5: 6.2, 20.7, 62.1 μM. Concentrations for compounds 2, 4, 6, and 14: 0.8, 2.7, 8.0 μM. Concentrations for compounds 7 and 8: 1.6, 5.4, 16.1 μM. Concentrations for compounds 9, 11, 12 and 13 12.4, 41.4, 124.2 μM. Concentrations for compound 10: 6.0, 20.1, 60.3 μM.
2.4.3. Inhibition of drug metabolizing enzymes
The propensity of methylation at the 7-O position to alter drug interaction liability was evaluated by testing all compounds as inhibitors of major drug metabolizing enzymes using the probe substrates (S)-warfarin (CYP2C9; Fig. 3), midazolam (CYP3A4/5; Fig. 4), and 4-methylumbelliferone (UGT; Fig. 5) and human liver or intestinal microsomes (HLM or HIM, respectively). Although CYP2C9 and CYP3A4/5 are expressed in both the human intestine and liver,18 intestinal CYP2C9 has not yet been shown to have clinical impact on drug disposition in vivo. As such, HLM were selected for incubations with (S)-warfarin, whereas HIM were selected for incubations with midazolam.
Figure 3.

Effects of selected flavonolignans and 7-O-methylated analogues on CYP2C9-mediated (S)-warfarin 7-hydroxylation in human liver microsomes (HLM). Incubation mixtures consisted of HLM (0.1 mg/mL), (S)-warfarin (4 μM), flavonolignan or 7-O-methylated analogue (10 or 100 μM; hatched and solid bars, respectively), and potassium phosphate buffer (0.1 M, pH 7.4). Reactions were initiated by the addition of NADPH (1 mM) and were terminated after 30 min with ice-cold MeOH containing 4-chlorowarfarin as internal standard. Activity in the presence of vehicle control (0.75% methanol, v/v) was 2.5 ± 0.1 pmol/min/mg microsomal protein. Bars and error bars denote means and SDs, respectively, of triplicate incubations. *p < 0.05, 10 versus 100 μM; #p < 0.05, flavonolignan versus 7-O-methylated analogue at 100 μM (two-way ANOVA with Bonferroni adjustment).
Figure 4.

Effects of selected flavonolignans and 7-O-methylated analogues on CYP3A-mediated midazolam 1′-hydroxylation in human intestinal microsomes (HIM). Incubation mixtures consisted of HIM (0.05 mg/ml), midazolam (4 μM), flavonolignan or 7-O-methylated analogue (10 or 100 μM; hatched and solid bars, respectively), and potassium phosphate buffer (0.1 M, pH 7.4) supplemented with magnesium chloride (3.3 mM). Reactions were initiated by the addition of NADPH (1 mM) and were terminated after 4 min with ice-cold acetonitrile containing alprazolam as internal standard. Activity in the presence of vehicle control (0.1% DMSO, v/v) was 700 ± 100 pmol/min/mg microsomal protein. Bars and error bars denote means and SDs, respectively, of triplicate incubations. *p <0.05, 10 versus 100 μM; #p < 0.05, flavonolignan versus 7-O-methylated analogue at 100 μM (two-way ANOVA with Bonferroni adjustment).
Figure 5.

Effects of selected flavonolignans and 7-O-methylated analogues on UGT-mediated 4-methylumbelliferone (4-MU) glucuronidation in human liver microsomes (HLM). Incubation mixtures consisted of HLM (0.4 mg/mL), 4-MU (100 μM), flavonolignan or 7-O-methylated analogue (10 or 100 μM; hatched and solid bars, respectively), bovine serum albumin (BSA, 0.05%), and Tris–HCl buffer (0.1 M, pH 7.5) supplemented with magnesium chloride (5 mM). Reactions were initiated by the addition of UDPGA (4 mM) followed by fluorescence analysis of 4-MU depletion over a period of 10 min. Activity in the presence of vehicle control (0.2% MeOH, v/v) was 8.0 ± 0.4 nmol/min/mg microsomal protein. Bars and error bars denote means and SDs, respectively, of triplicate incubations. *p < 0.05, 10 versus 100 μM; #p < 0.05, flavonolignan versus 7-O-methylated analogue at 100 μM (two-way ANOVA with Bonferroni adjustment).
All flavonolignans and 7-O-methylated analogues showed concentration-dependent inhibition of CYP activity (Figs. 3 and 4), except for isosilybin A (5) against CYP2C9 activity (Fig. 3). The parent versus methylated analogue pairs at 100 μM differed in inhibition potency except for silybin B (3) versus 7-O-methylsilybin B (4) against CYP2C9 activity (Fig. 3), silydianin (13) versus 7-O-methylsilydianin (14) against CYP3A4/5 activity, and silychristin (9) versus 7-O-methylsilychristin (10) against CYP3A4/5 activity (Fig. 4). Amongst the analogue series, compounds with an ‘iso’ configuration inhibited the CYP enzymes (Figs. 3 and 4) to a greater extent than corresponding regioisomers. For example, 7-O-methylisosilybin A (6), 7-O-methylisosilybin B (8), and 7-O-methylisosilychristin (12) were more potent against CYP activities than 7-O-methylsilybin A (2), 7-O-methylsilybin B (4), and 7-O-methylsilychristin (10). Sulfaphenazole and ketoconazole, known potent inhibitors of CYP2C9 and CYP3A4/5 activity, respectively, inhibited corresponding activities by 67% and 81% (data not shown).
Compared to the CYPs, inhibition of UGTs was less extensive (Fig. 5). However, in general, the parent compounds were more potent than the analogues. For example, silybin A (1), silybin B (3), and silydianin (13) at 100 μM were more potent than the respective methylated analogues (i.e., 2, 4, and 14), suggesting that methylation reduced activity against UGT. One exception was 7-O-methylisosilychristin (12), which was more potent than isosilychristin (11). Diclofenac, a non-specific inhibitor of the UGTs, inhibited activity by 56% (data not shown).
Collectively, CYP2C9 activity was most sensitive to inhibition, by both parent flavonolignans and corresponding methylated analogues, followed by CYP3A4/5 and UGT activity (up to 91%, 72%, and 44% inhibition, respectively). 7-O-Methylation of these isolated flavonolignans highlights the importance of this metabolically labile site, providing mechanistic insight into the enzyme–ligand interaction.
3. Conclusion
In summary, a series of 7-O-methyl flavonolignan derivatives has been synthesized selectively, and this study is the first to examine methylation of all seven of the major flavonolignans in silymarin. All the derivatives showed cytotoxic effects against human hepatoma cells. The most potent analogue was 7-O-methylsilybin A, which exhibited an IC50 value of 9.5 μM compared with parent silybin A with an IC50 value of 80 μM. In terms of anti-HCV activity, parent compounds with minimal to no antiviral activity (i.e., 9, 11, 13) showed increased antiviral activity when converted to the 7-O-methyl derivatives. For these compounds, acquisition of antiviral activity by 7-O-methylation was associated with enhanced cytotoxicity. In contrast, parent compounds with anti-HCV activity either retained (1), decreased (3, 5), or slightly increased (7) activity upon methylation. 7-O-Methylation differentially modified the inhibitory potency of flavonolignans towards major drug metabolizing enzymes, with compounds demonstrating increased, decreased, or no change in potency depending upon the enzyme system investigated. Despite inconsistent patterns, the differences observed between parent and analogue suggested the importance of the 7-position. Cumulatively, these data suggested that silymarin-derived compounds exert pleiotropic effects on cells. Ongoing efforts are focusing on how these compounds affect cellular metabolic processes that culminate in cytotoxicity, antiviral activity, and drug metabolism inhibition.
4. Experimental
4.1. General experimental procedures
Optical rotation and UV data were acquired on a Rudolph Research Autopol® III polarimeter and a Varian Cary 3 UV–vis spectrophotometer, respectively. All NMR experiments were conducted in DMSO-d6 at 30 °C using a JEOL ECA-500 (operating at 500 MHz for 1H and 125 MHz for 13C). HRESIMS data were measured using an electrospray ionization (ESI) source coupled to a QTOF Premier mass spectrometer (Waters Corp., Milford, MA, USA) in negative ionization mode via a liquid chromatographic/autosampler system that consisted of an Acquity UPLC system (Waters Corp.). HPLC was carried out on Varian Prostar HPLC systems equipped with Prostar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2). For preparative HPLC, a YMC ODS-A (5 μm, 250 × 20 mm; Waters Corp.) column was used at a 7 mL/min flow rate and a Phenomenex PFP (pentafluorophenyl propyl; 5 μm; 250 × 21 mm) column was used at a 21.2 mL/min flow rate. For analytical HPLC, a YMC ODS-A (5 μm; 150 × 4.6 mm) column and a PFP (5 μm; 150 × 4.6 mm) column were used, both at a 1 mL/min flow rate. All reactions were carried out in a N2 atmosphere under anhydrous conditions.
4.2. Chemistry, synthesis and purification
The individual flavonolignans were isolated from milk thistle extract (silymarin) in >98% purity as described in detail previously. 11 To purify the reaction mixtures, two different reverse-phase columns [ODS-A C18 (5 μm; 250 × 20 mm) and PFP (5 μm; 250 × 21 mm)] were examined; the latter gave the best results according to peak symmetry and resolution when using acetonitrile as the organic modifier (data not shown). Accordingly, each 7-O-methylated flavonolignan analogue was purified until >98% pure, as measured by analytical HPLC (Fig. S1; Supplementary data). 7-O-methylsilybin A (2) and 7-O-methylisosilybin A (6) were purified using a gradient of 30:70 to 50:50 CH3CN/H2O (0.1% formic acid) over 30 min. 7-O-methylsilybin B (4) was purified using a gradient of 20:80 to 40:60 CH3CN/H2O (0.1% formic acid) over 30 min. 7-O-methylisosilybin B (8) was purified using 20:80 to 60:40 CH3CN/H2O (0.1% formic acid) over 40 min. 7-O-Methylsilychristin (10) and 7-O-methylsilydianin (12) were purified using a gradient of 20:80 to 42:58 CH3CN/H2O (0.1% formic acid) over 45 min. 7-O-Methylisosilychristin (14) was purified using a gradient of 15:85 to 36:64 CH3CN/H2O (0.1% formic acid) over 50 min.
4.2.1. 7-O-Methylsilybin A (2)
To a 150 mL three-neck round-bottom flask, silybin A (122 mg, 0.253 mmol) and K2CO3 (293 mg, 2.03 mmol) were dissolved in acetone (80 mL, 0.003 M). After stirring for 10 min, CH3I (244 μL, 3.95 mmol) was added dropwise. The reaction mixture was heated at reflux under N2 for 3 h and monitored by HPLC. After cooling to rt, the reaction was quenched by the addition of 2% aqueous HCl (pH 1.5) and diluted with H2O (125 mL). The mixture was extracted with EtOAc (2 × 80 mL). The organic phases were combined and dried over Na2SO4, filtered, and then the solvent was removed under reduced pressure. The compounds were purified as described in Section 2.2. Yield: 51 mg, 41%; white solid; +20 (c 0.1, MeOH); UV (MeOH) λmax (logε) 218 (4.2), 287 (4.1) nm; CD (MeOH) λext (Δε) 237 (−3.3), 296 (−9.2), 330 (1.7) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S2 and S3; HMBC data, see Figs. 1 and S4; HRESIMS m/z 495.1281 [M–H]− (calcd for C26H23O10 495.1297).
4.2.2. 7-O-Methylsilybin B (4)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 3 was reacted with 15 equiv of CH3I at reflux for 3.5 h to yield compound 4. Yield: 10 mg, 33%; white solid; +7.7 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 230 (4.0), 287 (4.0) nm; CD (MeOH) λext (Δε) 230 (8.6), 296 (−9.2), 334 (1.3) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S5 and S6; HMBC data, see Figs. S7 and S23; HRESIMS m/z 495.1290 [M–H]− (calcd for C26H23O10 495.1297).
4.2.3. 7-O-Methylisosilybin A (6)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 5 was reacted with 2 equiv of CH3I at 30 ®C for 2.5 h to yield compound 6. Yield: 22 mg, 21%; white solid; + 46 (c 0.1, MeOH); UV (MeOH) λmax (logε) 214 (4.1), 287 (4.0) nm; CD (MeOH) λext (Δε) 236 (−0.9), 296 (−5.7), 331 (0.7) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S8 and S9; HMBC data, see Figs. S10 and S23; HRESIMS m/z 495.1292 [M–H]− (calcd for C26H23O10 495.1297).
4.2.4. 7-O-Methylisosilybin B (8)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 7 was reacted with 4 equiv of CH3I at 40 °C for 2.5 h to yield compound 8. Yield: 12 mg, 24%; white solid; −17.4 (c 0.09, MeOH); UV (MeOH) λmax (logε) 211 (4.1), 287 (3.8) nm; CD (MeOH) λext (Δε) 229 (1.8), 294 (−4.2), 331 (−0.1) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S11 and S12; HMBC data, see Figs. S13 and S23; HRESIMS m/z 495.1294 [M–H]− (calcd for C26H23O10 495.1297).
4.2.5. 7-O-Methylsilychristin (10)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 9 was reacted with 12 equiv of CH3I at reflux for 2 h to yield compound 10. Yield: 41 mg, 40%; white solid; +56.6 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 215 (4.0), 288 (3.8) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S14 and S15; HMBC data, see Figs. S16 and S23; HRESIMS m/z 495.1284 [M–H]− (calcd for C26H23O10 495.1297).
4.2.6. 7-O-Methylisosilychristin (12)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 11 was reacted with 24 equiv of CH3I at 40°C for 2 h to yield compound 12. Yield: 15 mg, 29%; white solid; +144.4 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 212 (4.1), 288 (3.7) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S17 and S18; HMBC data, see Figs. S19 and S23; HRESIMS m/z 495.1284 [M–H]− (calcd for C26H23O10 495.1297).
4.2.7. 7-O-Methylsilydianin (14)
In a manner that was otherwise consistent with the preparation and purification of 2, compound 13 was reacted with 12 equiv of CH3I at reflux for 2 h to yield compound 14. Yield: 20 mg, 33%; white solid; +157.9 (c 0.06, MeOH); UV (MeOH) λmax (logε) 207 (3.9), 288 (3.6) nm; 1H and 13C NMR data, see Tables 1 and 2 and Figs. S20 and S21; HMBC data, see Figs. S22 and S23; HRESIMS m/z 495.1284 [M–H]− (calcd for C26H23O10 495.1297).
4.3. Biological assays
4.3.1. Antiproliferative assay
The antiproliferative/cytotoxic activities of all compounds were examined against a human hepatoma cell line, Huh7.5.1,19 as described previously.10 Briefly, 10,000 cells per well were plated in 96-well plates, and following overnight incubation, were incubated with increasing concentrations of compounds. Cell viability was measured 72 h later using the ATPlite kit, as described previously.12 Each IC50 value was evaluated via the measurement of three replicates over nine different concentrations, ranging from 0 to 62.5 μM, with the IC50 values calculated by linear regression using GraphPad Prism.
4.3.2. Antiviral assay
Cells (150,000) were plated in 12-well plates, and the next day cells were infected with JFH-1, a HCV that grows well in Huh7.5.1 cells, at a multiplicity of infection of 0.05 focus forming units per cell. Virus inocula were removed 5 h post-infection and replaced with fresh medium containing test compounds. Protein lysates were harvested 72 h later, and HCV proteins were detected by Western blot analyses as described previously.12 Importantly, total protein content of each sample was determined, and equal amounts of total protein lysates were run on protein gels prior to Western blotting.
4.3.3. Inhibition of drug metabolizing enzymes
Milk thistle flavonolignans and 7-O-methylated analogues were evaluated as inhibitors of CYP2C9, CYP3A4/5, and UDP-glucuronosyltransferase (UGT) activity using the probe substrates (S)-warfarin, midazolam, and 4-methylumbelliferone (4-MU), respectively.
Inhibition of CYP2C9 activity
Incubation mixtures consisted of pooled HLM (0.1 mg/mL microsomal protein), (S)-warfarin (4 μM), flavonolignan or 7-O-methylated analogue (10 or 100 μM), and potassium phosphate buffer (100 mM, pH 7.4). The CYP2C9 inhibitor sulfaphenazole (1 μM) was used as a positive control. Control incubation mixtures contained 0.75% MeOH (v/v) in place of flavonolignan/7-O-methylated flavonolignan or sulfaphenazole. Incubation mixtures were analyzed for 7-hydroxywarfarin by LC/MS–MS as described previously.20,21
Inhibition of CYP3A4/5 activity
Incubation mixtures consisted of pooled HIM (0.05 mg/mL microsomal protein), midazolam (4 μM), flavonolignans or 7-O-methylated analogue (10 or 100 μM), and potassium phosphate buffer (100 mM, pH 7.4) supplemented with magnesium chloride (3.3 mM). The CYP3A4/5 inhibitor ketoconazole (1 μM) was used as a positive control. Control incubation mixtures contained 0.1% DMSO (v/v) in place of flavonolignan/7-O-methylated flavonolignan or ketoconazole. Incubation mixtures were analyzed for 1′-hydroxymidazolam by LC/MS–MS as described previously.22,23
Inhibition of UGT activity
Incubation mixtures consisted of pooled HLM (0.4 mg/mL microsomal protein), 4-MU (100 μM), flavonolignan or 7-O-methylated analogue (10 or 100 μM), bovine serum albumin (BSA, 0.05%), and Tris–HCl buffer (0.1 M, pH 7.5) supplemented with magnesium chloride (5 mM). The UGT inhibitor diclofenac (400 μM) was used as a positive control. Control incubation mixtures contained 0.2% MeOH (v/v) in place of flavonolignan/7-O-methylated flavonolignan or diclofenac. Incubation mixtures were analyzed for 4-MU depletion by fluorescence as described previously.24
Data were analyzed statistically using SigmaStat (version 3.5; Systat Software, Inc., San Jose, CA) and are presented as means ± SDs of triplicate incubations. Concentration-dependent inhibition of each flavonolignan/7-O-methylated analogue and comparisons between flavonolignan and 7-O-methylated analogue at 100 μM were evaluated by two-way analysis of variance (ANOVA) with Bonferroni adjustment for multiple comparisons; p <0.05 was considered significant.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health by both the National Institute of General Medical Sciences via Grant R01 GM077482 and the National Center for Complementary and Alternative Medicine via Grant R01 AT006842. H.S.A. was supported by the Ministry of Higher Education of Saudi Arabia, King Abdullah Scholarships Program.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.04.017.
References and notes
- 1.Polyak SJ, Ferenci P, Pawlotsky JM. Hepatology. 2013;57:1262. doi: 10.1002/hep.26179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polyak SJ, Oberlies NH, Pecheur EI, Dahari H, Ferenci P, Pawlotsky JM. Antivir Ther. 2013;18:141. doi: 10.3851/IMP2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abenavoli L, Capasso R, Milic N, Capasso F. Phytother Res. 2010;24:1423. doi: 10.1002/ptr.3207. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer Res. 2006;26:4457. [PubMed] [Google Scholar]
- 5.Deep G, Agarwal R. Cancer Metastasis Rev. 2010;29:447. doi: 10.1007/s10555-010-9237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramasamy K, Agarwal R. Cancer Lett. 2008;269:352. doi: 10.1016/j.canlet.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comelli MC, Mengs U, Schneider C, Prosdocimi M. Integr Cancer Ther. 2007;6:120. doi: 10.1177/1534735407302349. [DOI] [PubMed] [Google Scholar]
- 8.Gazak R, Walterova D, Kren V. Curr Med Chem. 2007;14:315. doi: 10.2174/092986707779941159. [DOI] [PubMed] [Google Scholar]
- 9.Kroll DJ, Shaw HS, Oberlies NH. Integr Cancer Ther. 2007;6:110. doi: 10.1177/1534735407301825. [DOI] [PubMed] [Google Scholar]
- 10.Sy-Cordero AA, Graf TN, Runyon SP, Wani MC, Kroll DJ, Agarwal R, Brantley SJ, Paine MF, Polyak SJ, Oberlies NH. Bioorg Med Chem. 2013;21:742. doi: 10.1016/j.bmc.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graf TN, Wani MC, Agarwal R, Kroll DJ, Oberlies NH. Planta Med. 2007;73:1495. doi: 10.1055/s-2007-990239. [DOI] [PubMed] [Google Scholar]
- 12.Polyak SJ, Morishima C, Lohmann V, Pal S, Lee DY, Liu Y, Graf TN, Oberlies NH. Proc Natl Acad Sci USA. 2010;107:5995. doi: 10.1073/pnas.0914009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deep G, Raina K, Singh RP, Oberlies NH, Kroll DJ, Agarwal R. Int J Cancer. 2008;123:2750. doi: 10.1002/ijc.23879. [DOI] [PubMed] [Google Scholar]
- 14.Davis-Searles PR, Nakanishi Y, Kim NC, Graf TN, Oberlies NH, Wani MC, Wall ME, Agarwal R, Kroll DJ. Cancer Res. 2005;65:4448. doi: 10.1158/0008-5472.CAN-04-4662. [DOI] [PubMed] [Google Scholar]
- 15.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Carcinogenesis. 2007;28:1533. doi: 10.1093/carcin/bgm069. [DOI] [PubMed] [Google Scholar]
- 16.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Oncogene. 2008;27:3986. doi: 10.1038/onc.2008.45. [DOI] [PubMed] [Google Scholar]
- 17.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Int J Cancer. 2008;123:41. doi: 10.1002/ijc.23485. [DOI] [PubMed] [Google Scholar]
- 18.Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. Drug Metab Dispos. 2006;34:880. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Proc Natl Acad Sci USA. 2005;102:9294. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brantley SJ, Oberlies NH, Kroll DJ, Paine MF. J Pharmacol Exp Ther. 2010;1081:332. doi: 10.1124/jpet.109.161927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ngo N, Brantley SJ, Carrizosa DR, Kashuba AD, Dees EC, Kroll DJ, Oberlies NH, Paine MF. J Exp Pharmacol. 2010;2:83. doi: 10.2147/JEP.S11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang MZ, Wu JQ, Bridges AS, Zeldin DC, Kornbluth S, Tidwell RR, Hall JE, Paine MF. Drug Metab Dispos. 2007;35:2067. doi: 10.1124/dmd.107.016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngo N, Yan Z, Graf TN, Carrizosa DR, Kashuba AD, Dees EC, Oberlies NH, Paine MF. Drug Metab Dispos. 2009;37:514. doi: 10.1124/dmd.108.024968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collier AC, Tingle MD, Keelan JA, Paxton JW, Mitchell MD. Drug Metab Dispos. 2000;28:1184. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.