

Abstract
The order and timing of cell cycle events is controlled by changing substrate specificity and different activity thresholds of cyclin-dependent kinases (CDK). However, it is not understood how a single protein kinase can trigger hundreds of switches in a sufficiently time-resolved fashion. We show that the cyclin-Cdk1-Cks1-dependent phosphorylation of multisite targets in Saccharomyces cerevisiae is controlled by key substrate parameters including distances between phosphorylation sites, the distribution of serines and threonines as phospho-acceptors, and the positioning of cyclin-docking motifs. The component mediating the key interactions in this process is Cks1, the phospho-adaptor subunit of the cyclin-Cdk1-Cks1 complex. We propose that variation of these parameters within the networks of phosphorylation sites in different targets provides a wide range of possibilities for the differential amplification of Cdk1 signals, providing a mechanism to generate a wide range of thresholds in the cell cycle.
Introduction
Cyclin-dependent kinases (CDKs) are the master regulators of the eukaryotic cell cycle 1. CDKs catalyze an ordered phosphorylation of hundreds of targets that trigger a sequence of coordinated molecular events, which drive the cell through the stages of cell division 2-5. Several individual CDK-driven switches have been identified 6, but our understanding of CDK as a general coordinator of the entire process of cell division is not clear.
The existing model states that as levels of CDK activity rise during the cell cycle, specific molecular events are executed at each of a series of activity thresholds 7-11. This model requires that substrates are phosphorylated with a wide range of efficiencies, such that very good substrates are phosphorylated early, at a low CDK activity threshold, while poor substrates are phosphorylated only when higher CDK activity is achieved later in the cell cycle. CDK targets contain both optimal consensus phosphorylation motifs (S/T-P-x-K/R) 12, 13 and suboptimal consensus sites (S/T-P) that are phosphorylated much less efficiently 12, 14. Therefore, a mechanism in which optimal motifs are used for the ‘early’ switches and suboptimal motifs for the ‘later’ switches is conceivable. However, if the system relied entirely on the distinction between optimal and suboptimal motifs, there could be, in principle, only two robustly resolved thresholds. In fact, several studies have shown that even within the narrow window of time at the onset of mitosis there is a finely resolved order of discrete events that are triggered in response to different levels of accumulating mitotic cyclins 15-17. How do relatively small changes in total CDK levels provide temporally resolved triggering of these events?
Cyclin specificity in substrate recognition has provided some insight into the mechanism by which CDKs coordinate cell cycle events 18-22. In budding yeast, the intrinsic activities of different cyclin-Cdk1 complexes increase during the cycle, and the low activity of early complexes is compensated by cyclin-dependent docking sites 14, 23. Docking interactions enhance the abrupt phosphorylation of selected early targets in late G1 and early S phases without interference from later targets. Although this specificity allows early cyclins to efficiently phosphorylate early targets, they cannot drive the later stages of the cell cycle in budding yeast 24. This difference in cyclin specificities and the overall modulation of CDK activity due to its own regulatory phosphorylations provides a coarse mechanism to separate early and late events, but it still fails to explain how relatively small changes in CDK activity can trigger the ordered sequence of discrete events observed on a finer time scale.
The complexity of CDK function is further increased by the multisite nature of most of its targets. The input CDK signal is often processed through several phosphorylation steps to yield a multi-phosphorylated output state of the target 25-29. Recently, we demonstrated that a network of Cdk1 phosphorylation sites in budding yeast Sic1 is phosphorylated in semi-processive cascades22. The output of the cascade is the phosphorylation of two diphosphodegrons that direct Sic1 to degradation via the SCF-proteasome pathway 30, 31. The degron phosphorylation sites are not specific enough to be efficiently phosphorylated by Cdk1 directly, because they contain suboptimal sets of recognition elements. Instead, the suboptimal degron sites become phosphorylated due to docking-dependent amplification of Cdk1 specificity via sequential steps of priming phosphorylations and interactions with the phospho-adaptor subunit Cks1.
These semi-processive cascades, whose net phosphorylation depends on parameters other than consensus motifs, lead us to a hypothesis that the relative positions of phosphorylation sites and cyclin-dependent docking sites within multisite clusters govern the net phosphorylation rate through the cascades and thereby can generate thresholds for phosphorylation of functional sites at different CDK activity levels. Variation of these parameters provides a wide range of possibilities for the differential outputs in response to changes of CDK input strengths.
The central element defining the output signal strength of these multisite processor systems is the phospho-adaptor Cks1. Cks1 was first discovered in S. pombe as a high-copy suppressor of a defective Cdk1 mutant 32. In budding yeast, Cks1 is essential for both G1/S and G2/M transitions 33, 34 and associates with cyclin-Cdk1 complexes at close to stoichiometric ratios in vivo 35. An in vitro study with the Xenopus version of Cks1 demonstrated its possible role in promoting the multisite phosphorylation of the CDK regulators Wee1, Myt1, and Cdc25, as well as the APC 36, 37, while in budding yeast the G1-specific complex Cln1,2-Cdk1 requires the Cks1 subunit for activity 38. However, the function of Cks1 as a phospho-adaptor protein in the mechanism of CDK-driven switches has been largely overlooked. Also, recent large-scale proteomic screens have identified hundreds of CDK targets 2, 4, but these studies have not touched the level of complexity that the multisite nature of these targets may present for the mechanism of CDK signal processing.
In the present study we set out to analyze the biochemical parameters that control Cdk1 signal flux through multisite phosphorylation networks. We explored how different combinations of these parameters influence the net output of the signal, and we analyzed the mechanism of Cks1-dependent multiphosphorylation cascades and how processivity can be used for differential amplification of output signals. Based on these data, we propose a new mechanism that provides a solution to the question of how accumulating CDK activity triggers the correct timing and sequence of cell cycle events.
Results
Cks1-dependent multisite phosphorylation of Cdk1 targets
Most known Cdk1 targets contain multiple phosphorylation sites, which tend to be clustered in regions of predicted disorder 3. To obtain an overview of the importance of Cks1 in promoting multisite phosphorylation cascades in different targets, we analyzed a set of targets in phosphorylation assays involving either wild type Cks1 (Cks1wt) or Cks1 with a mutated phosphate-binding pocket (Cks1mut) 22, 39. The autoradiographs of the phosphorylation patterns reveal that there are wide variations in Cks1 dependence among the targets (Figure 1a). In some cases, the appearance of multi-phosphorylated patterns is highly dependent on Cks1. In other cases, phosphorylation is not affected by Cks1. Also, there are targets that display intermediate effects, suggesting that only a subset of sites is enhanced by Cks1. We also found differences among cyclin-Cdk1 complexes: for example, compare phosphorylation of the G1 transcriptional regulator Whi5 by G1-specific Cln2-Cdk1 and M-phase specific Clb2-Cdk1 complexes. Note that Cks1 had little effect on the phosphorylation of a substrate containing a single Cdk1 site (T5-Sic1ΔC; Fig. 1b).
Figure 1. Cks1-dependent multisite phosphorylation of Cdk1 targets.

(a) Demonstration of Cks1-dependent accumulation of multi-phosphorylated forms in selected targets of Cdk1. The kinase assays were performed using purified Cln2-, Clb5-, and Clb2-Cdk1 complexes, which were preincubated with either wild-type Cks1 or Cks1mut (phospho-pocket mutant). The radioactively labeled multi-phosphorylated forms were separated using Phos-Tag SDS PAGE. The number of optimal and suboptimal consensus motifs, together with an indication of whether the sites have serine or threonine residue as the phospho-acceptor, are provided below the panels; (b) Phosphorylation of a substrate construct containing a single CDK consensus phosphorylation site (T5-Sic1ΔC) is not influenced by Cks1-dependent phospho-docking. Additionally, the phosphorylation assays using a standard substrate histone H1 are shown; (c) A schematic diagram indicating three pockets in cyclin-Cdk1-Cks1 complex whose local site-specificity and positioning geometry could potentially control the phosphorylation dynamics of multisite targets. The uncropped autoradiography scans are provided in Supplementary Figure 8.
The observed differences indicate that the multisite networks may have functionally different patterns. What are the parameters that determine Cdk1 activity through the networks? The specificity of cyclin-Cdk1 complexes is controlled at three different levels: first, by the active site specificity of Cdk1, second, by cyclin-specific docking interactions, and third, by the specificity of Cks1 (Fig. 1c). Using a non-inhibitory form of Sic1 (Sic1ΔC) and other Cdk1 targets, we investigated how these three factors control the Cdk1-dependent phosphorylation of multisite networks.
Cks1 binds phospho-threonines but not phospho-serines
One source of variation among the targets in Figure 1 is the ratio of threonines to serines as phospho-acceptor residues. Interestingly, we noticed that the targets that do not display Cks1 dependence (Bop3, Sld2ΔC and Ypr174) had only serine residues within optimal consensus motifs (S-P-x-K/R). Therefore, we questioned if Cks1 may prefer phospho-threonines over phospho-serines. Of the nine CDK consensus motifs in Sic1, five are threonines and four are serines (Fig. 2a). The N-terminal residues T2, T5, and T33 were shown previously to serve as phospho-docking sites for Cks1, thereby promoting fast phosphorylation of the C-terminal phospho-degrons22. To test if serines are equally able to mediate Cks1-dependent phosphorylation, we replaced the threonines in the CDK consensus sites of Sic1 with serines. Strikingly, the abrupt accumulation of multiply phosphorylated species was severely suppressed (Fig. 2b, Supplementary Fig. 1a,b). The effect was as strong as that of the Cks1mut (Figure 1a), indicating that the entire phospho-binding capacity of Cks1 was lost. The effect was entirely due to Cks1, because the Cks1-independent phosphorylation of the all-serine mutant was not affected (Supplementary Fig. 1c,d).
Figure 2. Exclusive preference of threonine over serine residues as the priming sites for Cks1-dependent docking and phosphorylation steps.

(a) A diagram indicating the Thr-to-Ser substitutions made in Sic1 for the experiments in panels ‘b’ and ‘d’; (b) The kinase assay demonstrating the loss of abrupt multisite phosphorylation caused by Thr-to-Ser substitution of the Cdk1 phosphorylation sites in Sic1ΔC; (c) The kinase assays demonstrating the Thr-to-Ser substitution effect on the accumulation rate of doubly phosphorylated forms in T33-T48-Sic1ΔC construct; (d) Full-length Sic1 versions were overexpressed under the galactose promoter to assay the ability of cells to degrade Sic1; (e) Representative ITC experiment to analyze the binding of pT33-Sic1ΔC and pS33-Sic1ΔC to Cks1. No binding heat was detected in the case of pS33-Sic1ΔC; (f) The in vivo demonstration of the requirement of Thr as a priming site of multiphosphorylation using western blotting of Phos-Tag SDS-PAA gels. The dynamics of phosphorylation shifts of the non-destructible Sic1ΔC-derived substrate constructs was followed after the release of cells from α-factor arrest. *Partial phosphorylation of sites by an unknown kinase activity in G1; (g) Mutation of threonines in the CDK consensus sites to serines suppressed the Cks1-dependent multisite phosphorylation of Srl3; (h) Mutation of serines of the CDK consensus sites to threonines induced Cks1-dependent potentiation of multisite phosphorylation of Swi5. The kinase assays in ‘g’ and ‘h’ were performed according to the same protocol as in case of the ones presented in Fig. 1a. The uncropped scans are provided in Supplementary Figure 8.
To directly confirm the inability of a serine to prime a single Cks1-dependent phosphorylation step we constructed a version of Sic1ΔC containing only two sites: the optimal Cdk1 target site T33 (or S33) served as a priming site and a suboptimal site T48, served as a secondary site. Indeed, the threonine was required for the accumulation of the doubly phosphorylated species (Fig. 2c, Supplementary Fig. 1e).
Cells overexpressing the all-Ser form of Sic1 are inviable, unlike cells overexpressing wild-type Sic1 (Fig. 2d). This result together with the analysis of the protein levels and phosphorylation shifts (Supplementary Fig. 2a,b) indicate that Cdk1 is not able to phosphorylate Sic1-Ser to a sufficient level to cause its proper degradation. It is most likely that inviability is caused by weak binding of pSer sites to Cks1 because no phospho-serine versus phosphothreonine specificity has been observed for SCF-Cdc4 phospho-degrons31. Additionally, a single serine substitution in the crucial priming site at position 33 caused a partial loss of viability (Fig. 2d). Thus, it is possible to disrupt the docking connections of the Cks1-dependent cascade by replacing threonines in CDK sites with serines. The resulting construct follows a phosphorylation mode in which phosphorylation of one site is not dependent on previous phosphorylation of the other. In fact, such a random distributive mode was the basis of an earlier model of Sic1 phosphorylation40, and these results provide an additional argument supporting the semi-processive Cks1-dependent cascade model22.
To measure the direct binding affinity between the phosphorylated sites of Sic1 and Cks1 we used an isothermal calorimetry (ITC) assay with purified proteins. We produced stoichiometrically phosphorylated versions of Sic1ΔC containing single phosphorylation sites. The data obtained further confirmed the exclusive preference of phospho-threonine over phospho-serine in Cks1 binding (Fig. 2e, Supplementary Table 1).
Finally, we demonstrated the requirement for threonine as the crucial priming residue in vivo. The replacement of the threonine at the crucial N-terminal priming site T5 in Sic1 resulted in a severe reduction in the multi-phosphorylated forms of Sic1 substrate construct containing five evenly-paced serine consensus motifs (Fig. 2f, Supplementary Fig. 2c).
To confirm that the effects observed in the threonine-to-serine mutations in the Sic1 constructs were reflecting a general phenomenon, and were not specific for the Sic1 protein, we investigated another multisite target, Srl3. Similarily, the mutation of threonines to serines in CDK consensus sites suppressed the Cks1-dependence of the multisite phosphorylation (Fig. 2g). Furthermore, we also tested a reverse approach: mutation of serines to threonines in CDK sites of Swi5, a substrate which has only serines in the optimal CDK sites and showed little Cks1-dependence, created a strong Cks1-dependent stimulation of the phosphorylation pattern (Fig. 2h).
We also performed a set of positional variations by introducing basic and hydrophobic residues around the T33 site. We found that a proline residue at position −2 relative to phosphothreonine enhanced the interaction of the phospho-epitope with Cks1 (Supplementary Table 1, Supplementary Fig. 3, and 41).
Positions of sites affect the rate of Cks1-dependent step
The second parameter that is likely to control signal flux through multisite cascades is the distance and relative positioning between the priming site and the secondary phosphorylation site. To analyze the impact of this parameter on the rate of Cks1-dependent phosphorylation steps, we created a series of Sic1ΔC-based substrate constructs containing two phosphorylation sites at different distances from each other. Due to its intrinsically disordered nature,42, 43 the Sic1 polypeptide is an excellent system to study such distance requirements. By varying the distance, we aimed to measure the optimal distances between the sites in terms of the number of amino acids in the polypeptide chain.
We constructed a series of Sic1 proteins carrying the site T33, which contains the optimal CDK consensus motif (TPQK), as the primary phosphorylation site. The secondary site was a short sequence bearing a suboptimal CDK motif (TPQA), which was placed at various distances from T33 (see Online Methods). The priming site T33 was efficiently phosphorylated by cyclin-Cdk1s, while the suboptimal motif used in the secondary site was not phosphorylated (Supplementary Fig. 1f). Therefore, when doubly-phosphorylated species were detected, the sequence of the two-step cascade was always primary phosphorylation of T33 first, followed by the phosphorylation of the secondary site (Fig. 3a, see also Supplementary Fig. 1f).
Figure 3. Analysis of the influence of distance between the priming phosphorylation site and the secondary phosphorylation site.

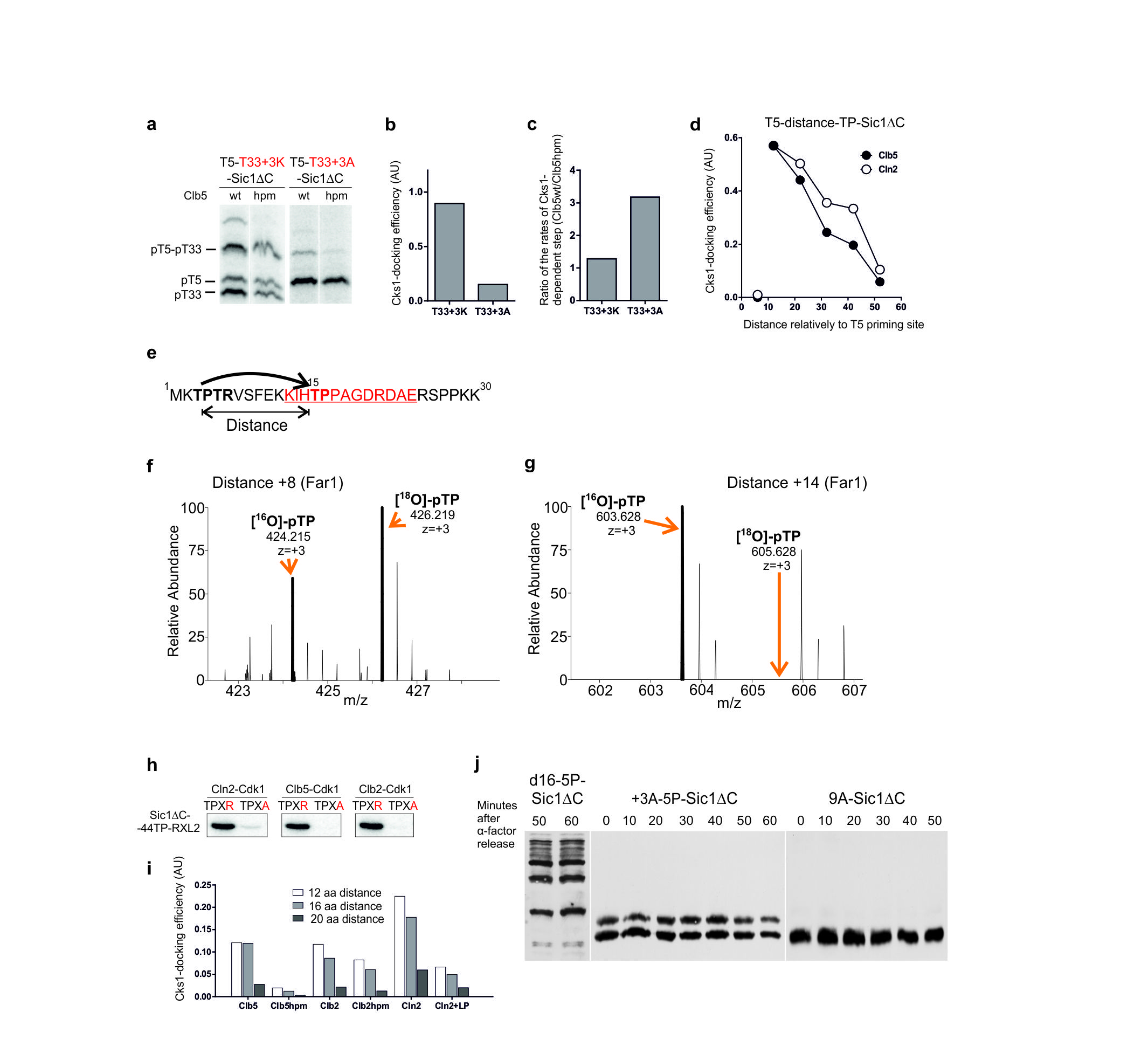
(a) A scheme explaining the positional variation of the secondary site along the Sic1 polypeptide; (b) The autoradiographs of Phos-Tag gels showing the phosphorylation of constructs with varied distances between the priming site (T33) and the secondary site using Clb5-Cdk1. The last lane of the left panel shows the construct containing only the priming site T33; (c-e) The quantified profiles of the relative accumulation rates of doubly phosphorylated forms at different distances between the priming sites and the secondary sites using Clb5-Cdk1 (c), Clb2- Cdk1 (d), and Cln2-Cdk1 (e); (f) The demonstration of the requirement of optimal distances between the phosphorylation sites in vivo by following the phosphorylation shifts in Sic1ΔC-derived constructs by western blotting after the release of cells from α-factor arrest; (g) Distance-dependence in phosphorylation of Cnn1. A secondary phosphorylation site based on site T42 was introduced at different distances from the potential priming site T3; (h) Distance-dependence in phosphorylation of Far1 analyzed by mass spectrometry. A segment containing a secondary phosphorylation site was introduced at different positions relatively to a potential priming site T3 (see Supplementary Fig. 4e-g, Supplementary Table 2). The asterisk indicates that the +14 phosphopeptide of the Cks1mut incubation was not detected, and therefore, the Cks1wt/Cks1mut ratio is large but not precisely measurable; (i) A scheme explaining the optimal distance requirements and the N-to-C-directionality in the Cks1-dependent phosphorylation step. The uncropped scans are provided in Supplementary Figure 8.
A distance dependence in the rates of Cks1-dependent secondary steps was observed (Fig. 3b). A surprisingly sharp change in rate was observed in the step from 10 to 12 amino acids from the T33 priming site: a distance of 10 amino acids yielded no secondary phosphorylation, while a distance of 12 amino acids showed very rapid accumulation of the doubly phosphorylated form. This drastic distance cut-off was similar for all three cyclin-Cdk1 complexes tested (Fig. 3c-e), indicating that the Cks1-Cdk1 module has a similar architecture and functional capability that does not depend on cyclin specificity. In all three complexes tested, a sharp peak value of 12-16 amino acids in the secondary phosphorylation rates was followed by rapid decline around the distance of 20-30 amino acids downstream from the priming site.
A surprising feature of the Cks1-dependent step was that the cascade operates exclusively in the N-to-C direction (Fig. 3c-e). We did not observe any docking-enhanced secondary phosphorylation even when the priming site T33 was moved further downstream, yielding distances of −40, −60 and −80 amino acids (Fig. 3c-e).
Interestingly, the relatively sharp distance optimum was broadened when the Cks1 specificity was improved by introducing a proline at position −2 from the T33 priming site (Fig. 3c-e). This suggests that the negative effect of above-optimal distances can be compensated by improved binding of the phosphorylated site to Cks1. The improved Cks1 specificity, however, did not improve activity with a distance shorter than the 10 amino acid minimal cut-off. Additionally, it is possible to increase the effective distance window for secondary phosphorylation by introducing the optimal CDK consensus site in the secondary site (Supplementary Fig. 4a-c). We confirmed that the distance relationship was not specific for the particular sequence context downstream from the site T33. For this we created a control set of similar constructs with the priming site T5 bearing an optimal motif PSTPPR (Supplementary Fig. 4d).
To test if the distances between sites are also important for multisite phosphorylation in vivo, we varied the positions of phosphorylation sites in a non-destructible version of Sic1ΔC and analyzed phosphorylation by western blotting, as in the experiment presented in Figure 2f. We made one artificial substrate with 5 phosphorylation sites in their original positions, but lacking the intermediate sites T33 and T45 to remove the possibility for Cks1-dependent docking. This substrate was compared to a construct that was identical, except that three serine Cdk1 sites were repositioned to be 16 amino acids apart (Figure 3f) (d16-Sic1ΔC). These constructs contain a single optimal priming site T5, while the other sites were serine-based suboptimal sites. The first construct, which has sites either amino-terminal of T5 or greater than 64 amino acids downstream, showed almost no change in phosphorylation profiles after entry into S phase. In contrast, the construct with the sites repositioned at accessible distances from the T5 priming site displayed mobility shifts of singly- and doubly-phosphorylated species. The phosphorylation pattern of this construct fits well within the distance relationship observed in the case of the optimal priming site containing a proline in position −2 (Fig. 3c-e, Supplementary Fig. 4d). The sites at distances of +16 and +32 are within the range of relatively fast rates while the longer distances apparently fail to gain any enhancement from Cks1-dependent docking.
Next we analyzed the distance requirements for the Cks1-dependent phosphorylation of two additional representative Cdk1 targets. First, the distance between a potential priming site and a secondary site was varied in the kinetochore protein Cnn1 (Fig. 3g). The obtained activity profile closely matched the one obtained using the detailed approach with the Sic1-based constructs.
Second, to analyze the distance requirement between a single pair of sites in the context of many other sites in a multisite target we designed a quantitative mass-spectrometry approach (see Supplementary Fig. 4e-g), and used it to analyze the distance requirements in Far1, a Cdk1 target in the pheromone pathway. Again, the obtained distance profile closely resembled that obtained using the Sic1-based constructs (Fig. 3h, Supplementary Table. 2).
These experiments reveal a strikingly sharp minimal distance cut-off and strict N-to-C-directionality both in vitro and in vivo. From this we conclude that the underlying mechanism involves simultaneous binding of the phosphorylated priming site to the Cks1 pocket and the secondary site to the active site pocket of Cdk1 (Fig. 3i).
Distances between phosphorylation sites are critical in vivo
To confirm that the distances between sites encode biological information in multisite networks, we performed a series of viability assays as described in Figure 2d. To increase the sensitivity of the critical distance variations we used a version of Sic1 with a minimally viable set of 5 phosphorylation sites (Fig. 4a). Surprisingly, moving the priming site T33 in these constructs by only 2 amino acids upstream or downstream caused inviability. Apparently, in these constructs a docking distance that perfectly fits the optimum of 12-16 amino acids (Fig. 3c-e) is required for efficient phosphorylation of both sites of the diphosphodegron (T45 and T48). One of the acceptor sites of the diphosphodegron is a ‘non-CDK’ consensus site (T48) whose phosphorylation could be even more sensitive to the docking distance compared with the consensus sites. We also moved the position of the diphosphodegron T45-T48 by 10 amino acids in a version of Sic1 containing all physiological sites. Similarly, this moderate shift of the position caused a severe reduction of viability (Fig. 4b). These data indicate that site positioning in multisite networks in Cdk1 targets is not random, but involves critical distances between primer and acceptor sites that must fit the distance between Cks1 phosphate binding pocket and the active site of Cdk1.
Figure 4. The optimal distances between the sites of Sic1 are critical for its degradation in vivo.

(a) The viability assay overexpressing Sic1 versions containing a set of phosphorylation sites T33-T45-T48-S76-S80. The position of the priming site T33 was shifted by 2 amino acid steps upstream and downstream of its original position. Because the strain expressing T33-T45-T48-S76-S80-Sic1 grows slightly slower compared with the strain expressing wild type Sic1, the colonies were grown for a longer time compared with the assay presented below in panel ‘b’ involving the wild type version of Sic1; (b) Similar viability assay as in ‘a’ showing the effect of shifting the position of a critical di-phosphodegron T45-T48 in context of wild type Sic1.
Clb5 directs phosphorylation to a specific C-to-N distance
Next we studied the effect of altering the distance of phosphorylation sites from cyclin-specific docking sites. In Sic1 there are two Clb5-specific docking sites (RXL)18-20, 44-49, and a single Cln2-specific docking motif (LLPP)14, 50. We analyzed constructs containing only one of the RXL motifs and the LLPP motif (Fig. 5a). The position of an optimal CDK consensus motif based on site T5 was varied over a wide range in both C- and N-terminal directions. We tested phosphorylation of these constructs with wild-type Clb5-Cdk1 or Clb2-Cdk1, as well as with kinase complexes with mutated hydrophobic patch docking site on cyclin (hpm) 18, 19. In Figure 5b and c the distance profiles are plotted as ratios of the phosphorylation rates obtained with wild-type and hpm kinase complexes. For Clb5, a striking increase of docking-enhanced phosphorylation rate was observed when the phosphor-acceptor site was 16-20 amino acids N-terminal from the RXL motif. No strong potentiation was observed when the phospho-acceptor site was C-terminal to the RXL (Fig. 5b, e). For Clb2, only a small increase in activity was observed at the same distance. These data are consistent with our previous results that Clb2 has much weaker hydrophobic patch specificity towards its substrates compared with Clb5 20.
Figure 5. Analysis of the influence of distance between the phosphorylation site and the cyclin-specific docking sites.

(a) The scheme of varied phosphorylation site positions along the polypeptide chain of constructs based on Sic1ΔC with all CDK sites mutated to alanines and containing a single RXL motif and a Cln2-specific LLPP motif. In these constructs a single CDK consensus motif based on T5 was placed at various distances from the Clb5 docking site RTL and Cln2 docking site LLPP; (b) The specificity profiles obtained by using substrate constructs with different distances from the RXL motif are plotted as a ratio of phosphorylation rates of wild type and hpm version of Clb5-Cdk1 complex; (c) Analogous specificity profile as in ‘b’ plotted for Clb2; (d) Analogous specificity profile as in ‘b’ plotted for Cln2-Cdk1 when assayed in the absence and in the presence of the competitor peptide containing the cyclin docking motif ‘LLPP’; (e) Schematic representation of the cyclin-dependent docking modes; (f) The three interaction sites in the cyclin-Cdk1-Cks1 complex are the key for processing of Cdk1 signal through different multisite phosphorylation networks. A structural model showing the arrangement of the three key pockets in the complex. The model was created by superimposing domains from crystal structures (PDB codes: 1BUH, 2CCI, 4LPA) solved in the presence of the relevant substrate peptide bound to the pocket 41, 45, 55.
Cln2 directs phosphorylation to both N- and C-terminal sites
The Figure 5d presents a docking specificity profile obtained for Cln2-Cdk1-Cks1 using a competitor peptide containing the Cln2-specific LLPP docking site. A gradual increase in docking-dependent phosphorylation of N-terminally located sites was observed, as well as, a large potentiation towards a distant C-terminal site. These data suggest that the LLPP binding site, whose location in Cln2 is not known, is less directionally constrained than the Cks1 phospho-docking or hydrophobic patch-RXL docking interactions (Fig. 3e, Fig. 5d,e).
The data for both Cks1- and cyclin-dependent docking suggest that cyclin-Cdk1-Cks1 complexes can use a triple-docking mechanism for multisite phosphorylation. The combination of optimal and suboptimal distances and other specificity parameters provides a way to adjust the overall phosphorylation rate (see also Supplementary Fig. 4a-d, h-j). The scaffold that docks the disordered peptide chain, and processes the phosphorylation of its multiple sites according to specific distance constraints, is the binding surface created by the fixed architecture of the cyclin-Cdk1-Cks1 complex. The model in Figure 5f, constructed from crystal structures of domains in complex with substrate peptides 41, 45, 55, shows the positioning of the three key substrate pockets in the complex. In addition, the directionality requirements observed for the Cks1 and hydrophobic path-docking are consistent with the directionality of the peptides in the crystal structures. As shown, one can model a continuous polypeptide chain that has an N-terminal Cks1-docking phosphate, a middle phospho-acceptor site, and a C-terminal RxLxF cyclin-docking sequence. Assuming a substrate in an extended conformation, the minimum sequence lengths between these elements roughly correspond to the distance requirements found in our experiments.
Processivity of Cks1-dependent multisite phosphorylation
Our observations suggest that multisite phosphorylation of many Cdk1 substrates depends on multiple interactions between the substrate and kinase. It is likely that these interactions enable processive or semi-processive attachment of multiple phosphates during a single substrate-binding event (Supplementary Fig. 5).
In the case of Cks1-dependent cascades, there are two mechanisms by which the processive step could take place. The first mechanism allows that the phosphorylated priming site withdraws from the active site of Cdk1 and subsequently reassociates with the phosphate-binding pocket of Cks1 without dissociation from the enzyme (Fig. 6a). Support from cyclin docking sites would be required to maintain the complex during such a displacement. To explore the existence of such a mechanism, we used a Sic1ΔC-based construct containing a pair of phosphorylation sites (Fig. 6b). We measured the accumulation of singly and doubly phosphorylated forms, which were resolved in Phos-Tag gels (Fig. 6c). Interestingly, we observed a considerable degree of processivity when either Clb5- or Cln2-Cdk1 was used, as the relative abundance of phosphorylated forms was high and constant at the early stages of the reaction (Fig. 6c-e). Additionally, in the same time window, we did not see any change in the ratios of doubly and singly phosphorylated species at different enzyme concentrations, which further confirms the semi-processivity. (Fig. 6d,e, Supplementary Fig. 6,7a-d). Explanation of the analysis is presented by the simulations of a mathematical model in Supplementary Fig. 6. The processivity factors obtained for the Clb5 complex suggest that after the phosphorylation of the first site there is about a 40% chance to add the secondary phosphate without dissociation. Intriguingly, in the case of the hpm version of Clb5, the appearance of the doubly phosphorylated form was not immediate, and apparent processivity factor was very low (Fig. 6d). These data suggest that support from the cyclin docking site is necessary for processivity, as depicted in Figure 6a. In addition, the Clb5-dependent reaction lost its processivity when Cks1mut was used in the assay (Fig 6c,d, Supplementary Fig. 7c,d). Furthermore, while Cln2-Cdk1 showed only a slightly lower level of processivity compared with Clb5-Cdk1, the mitotic Clb2-Cdk1 complex showed almost no processivity. This finding correlates with the fact that the hydrophobic patch docking of Clb2 is weak (Fig. 5c). Thus, the earlier cyclin complexes show higher processivity because they can use cyclin docking sites.
Figure 6. Analysis of the processivity of multi-phosphorylation.

(a) A mechanism in which the phosphorylation of a priming site is followed by the phosphorylation of the secondary site without dissociation of the substrate from the enzyme; (b) Scheme of the substrate construct T5-T33-Sic1ΔC; (c) The time courses showing early stages of the accumulation of doubly phosphorylated forms, separated by Phos-Tag. * unidentified phospho-forms; (d, e) Graphs showing the relative accumulation patterns of doubly phosphorylated forms in the experiments exemplified in panel ‘c’. The y-axis (multi-phosphorylation factor, XMP) shows the ratio of 2P/(1P+2P), where 2P and 1P are the quantified amounts of the doubly and singly (pT5-T33-Sic1ΔC) phosphorylated species, respectively. The y-intercepts are the estimates of the processivity factors, representing the immediate (without a lag period) appearance of doubly phosphorylated forms (see Supplementary Fig. 6). For Clb5 and Cln2 the XMP value does not depend on enzyme concentration, which is another hallmark of processivity (Supplementary Fig. 6). A similar experiment was performed for a two-site substrate construct, T33+12TP-Sic1ΔC (Supplementary Fig.7b). (f) A scheme for the second major processive mechanism. The phosphorylated priming site when docked to the Cks1 pocket can processively phosphorylate several secondary sites without dissociation; (g-j) Analysis of Mechanism II using a construct T33-S69-S76-S80-Sic1ΔC, in which T33 is a priming site. The full time course was used in this case to facilitate broader analysis of the phosphorylated forms. The intercepts provide the estimates for the processivity factors at each step. The uncropped scans are provided in Supplementary Figure 8.
The second possible mechanism for Cks1-dependent processivity is the sequential addition of phosphates without dissociation of the primed phosphate from Cks1 (Fig. 6f). To analyze this mechanism, we used a construct containing T33 as a priming site and a triple-serine cluster S69-S76-S80 as secondary sites (Fig. 6g). These experiments showed that this type of processivity exists as well. The processivity factors at subsequent steps showed a 20-40% probability to continue with the next phosphorylation step without dissociation (Fig. 6h-j).
Thus, the degree of processivity may be an additional factor that can differentiate the output signals in different Cdk1 substrates. We tested this possibility on two additional physiological targets: Whi5 and Fin1. Our results (Supplementary Fig. 7e-j) suggest that various multisite networks can provide different degrees of processivity for each cyclin-Cdk complex, illustrating the potential for broad dynamic range in the processing of Cdk1 signals by different substrates.
Multisite clusters form Cks1-dependent docking networks
To detail the role of Cks1 in other multisite targets, we analyzed the phosphorylation of Stb1, Ndd1 and Swi5 using a quantitative mass-spectrometry based approach (Fig. 7). The Cln2-specific target Stb1 showed a number of Cks1 docking connections that are in agreement with the N-to-C directionality rule, threonine versus serine specificity, and the distance rules. Similarly, the Clb2-Cdk1 target Ndd1 showed Cks1-dependent phosphorylation enhancements that can be explained by the network rules. Finally, the threonine vs serine rule was well exemplified in Swi5, which contains 8 serine-based optimal CDK sites, with no threonine-based optimal priming sites at required distances. None of these SP sites showed defects in phosphorylation when using the Cks1mut, whereas Cks1 did enhance phosphorylation at sites downstream of suboptimal threonine-based sites. In conclusion, the phosphorylation of these three representative substrates followed the rules of Cks1-dependent phosphorylation derived from the Sic1 model experiments (See also Suppl. Tables 3,4).
Figure 7. Cks1 differentially stimulates the phosphorylation of various sites in Stb1, Ndd1 and Swi5.

Purified cyclin-Cdk1 complexes with either Cks1wt or Cks1mut were used for phosphorylation of recombinant Stb1, Ndd1 and Swi5. The relative abundance of proteolytic phosphopeptides was analyzed using quantitative mass-spectrometry. The kinase reactions with Cks1wt or Cks1mut were supplemented with [16O]-ATP or [18O]-ATP, respectively, which allowed the detection of the phospho-peptides by the isotope shift (for further experimental details see the Online Methods section). (a, b, e, f, i, j) Examples of spectra showing the relative phosphorylation intensity of various phosphopeptides in reactions with Cks1wt or Cks1mut ([16O] and [18O], respectively); (c, g, k) Diagrams outlining the relative patterns of phosphopeptides obtained in the reactions with Cks1wt and Cks1mut; (d, h, l) Diagrams outlining Cks1-dependent docking networks in Stb1, Ndd1 and Stb1.
Discussion
In this study, we analyzed parameters that control the phosphorylation of multisite targets of Cdk1. We propose that the ability of Cdk1 to produce the multiphosphorylated output form of its targets depends on the overall spatial pattern of the multisite cluster, the distances between the sites, the direction of docking connections, the composition of the network with respect to serine versus threonine residues, Cks1-phosphoepitope specificity, and the processivity at each step.
These mechanisms may have broad importance in cell cycle regulation, as the majority of Cdk targets contain clusters of multiple phosphorylation sites within regions that are predicted to be intrinsically disordered 3. Within these clusters many potential Cks1-dependent docking connections can be predicted. Among the 74 in vivo proven Cdk1 targets listed in a recent review 6, 53 contain potential docking connections between pairs of sites (data not shown). Altogether, the number of possible docking connections in these targets was 151, and 75% of these pairs of sites were located in the same disordered region of the substrate.
It is important to note that besides the parameters controlling the cyclin-Cdk1-Cks1-dependent phosphorylation of multisite clusters, there is also a possibility that dephosphorylation specificity contributes to the timing of CDK-dependent phosphorylation, with earlier targets being more resistant to phosphatases. Gradually changing phosphatase specificity has been shown to play a role in ordered dephosphorylation of Cdk1 targets in mitotic exit by Cdc14 51. However, it is not clear if the same is true for phosphatases counteracting the rising Cdk1 activity before mitotic exit. Instead, a phosphatase was shown to have highly specific interaction with a mammalian early CDK target, the pRb protein 52. Indeed, different phosphatase specificity at each step would further increase the possible complexity of the networks.
Similar principles of stepwise modulation of the output signal of Cdk1 targets can be extended to other kinases that use phosphorylated CDK sites as priming sites (e.g. GSK, Cdc7 and Cdc5), as demonstrated recently 53, 54. Future studies of the mechanisms of multisite processor systems will hopefully uncover the intricate complexity of Cdk1 switches and will finally provide a full understanding of the general mechanism of the CDK-controlled cell cycle clock.
Online Methods
Protein purification
TAP-purification of cyclin-Cdk1 complexes (Clb5- and Clb2-Cdk1) was performed as described previously 2, 56 using C-terminally TAP-tagged cyclin constructs cloned into 2 micron vectors and overexpressed from the GAL1 promoter. For purification of 3HA-Cln2-Cdk1, a yeast strain was used (a kind gift from Dr. Doug Kellogg, UCSC) with the GAL1 promoter introduced along with the N-terminal 3HA tag in the chromosomal locus of the CLN2 gene. The overexpressed 3HA-Cln2-Cdk1 complex was purified as previously described 57, using immunoaffinity chromatography with a rabbit polyclonal antibody against the HA epitope (purchased from Labas, Estonia). N-terminally 6His-tagged recombinant Sic1ΔC constructs and 6His-tagged substrates in Figure 1 were purified by standard cobalt affinity chromatography with 200 mM imidazole used for elution. GST-tagged substrates used in the kinase assay presented in Figure 1 were purified on glutathione agarose columns. Far1 constructs, used in MS analysis, contained GST-6His tags and were purified by affinity chromatography via GST-tag.
Kinase assays
For the phosphorylation assays of Sic1ΔC constructs, substrate concentrations were kept in the range of 0.5–2 μM (in the linear [S] versus v0 range, several times below the estimated KM value). About 0.1-10 nM of purified kinase complex was used, reaction aliquots were taken at two or more time points, and the reaction was stopped using an SDS-PAGE sample buffer. The basal composition of the assay mixture contained 50 mM Hepes, pH 7.4, 180 mM NaCl, 5 mM MgCl2, 20 mM imidazole, 0.1 mg/ml 2HA peptide, 2% glycerol, 2 mM EGTA, 0.2 mg/ml BSA, 500 nM Cks1, and 500 μM ATP (with added γ-32P-ATP (Perkin Elmer)). For the phosphorylation assay with mutant Cks1 (Cks1mut), purified kinase complexes were preincubated for 45 minutes with Cks1wt or Cks1mut to compensate for differences in the amounts of Cks1 already present in the preparations. The optimal working concentration for Cks1 (500 nM) was based on optimizations decribed previously 22. The general composition of the preincubation mixture was: 50mM Hepes pH 7.4, 5 mM MgCl2, 150 mM NaCl, 0.4 mg/ml BSA, 500 μM ATP, 1 mM Cks1. In the assays with LP peptide (VLLPPSRPTS), 4 mM peptide was used. To separate the phosphorylated versions of Sic1, 10% SDS-PAGE was used, supplemented with the Phos-TagTM reagent according to the instructions from the manufacturer.
The kinase assays for determination of docking efficiencies (Fig. 3b-e, Supplementary Fig. 4d,i) were performed under conditions below 10-30% of initial substrate turnover. The phosphorylation of the substrate was followed in a conventional kinase assay and singly and doubly phosphorylated species were resolved using Phos-Tag SDS-PAGE and quantified by ImageQuant. The quantified values expressing the docking efficiencies (Fig. 3c-e) were calculated as the ratio of observed fraction of doubly phosphorylated form and the sum of observed fraction of doubly phosphorylated form and of the form with a single phosphate. The obtained value was divided by enzyme concentration. In this way, quantitative and comparable indicators showing the ability of Cks1-dependent docking steps were obtained. The determination of real kinetic constants in this system would become complicated due to the mixed mechanism of the two-step process. The reaction aliquot was taken at two consecutive time points (i.e. 8 and 16 minutes) and the average value for the docking efficiency of the two time points was calculated. For processivity analysis lower enzyme concentrations were used to minimize the impact of the distributive/cooperative mechanism in the formation of the multi-phosphorylated form(s). The range of enzyme concentration for observation of purely processive process was chosen so that the difference in relative ratios of multiply phosphorylated forms at two different enzyme concentrations was lower than 20%. The processivity factors were calculated similarly to docking efficiencies, except that the obtained value was not divided by enzyme concentration. For all the phosphorylation assays presented in the paper the concentration of the Phos-Tag in 10% SDS-PAGE was 100 μM for Sic1, Fin1, Ypr174c, or 20 μM for Stb1, Whi5, Nrm1, Bop3, Xbp1, Hcm1, Orc6, Cdc6, Cnn1, Sld2(1-185aa N-terminal fragment), Ndd1, Plm2, Yox1 and Swi5. Ash1, and Fir1 phosphorylation assays were separated in 8% SDS-PAGE, supplemented with 20 μM Phos-Tag reagent. 7% SDS-PAGE supplemented with 100 μM Phos-Tag reagent was used for Far1 protein.
Isothermal calorimetry
Dissociation constants were obtained using ITC binding experiments. Recombinant 6His-Sic1ΔC-based constructs were phosphorylated with purified Clb2-Cdk1 (no Cks1 was added). Stoichiometric phosphorylation was confirmed by following the phosphorylation shift by Phos-Tag SDS-PAGE. Phosphorylated constructs were repurified on chelating chromatography like described above with the exception that the elution was performed using 10mM EDTA. Purified proteins were dialyzed overnight at 4 °C in 25 mM Tris (pH 8.0) and 150 mM NaCl. Cks1 at a concentration of ~ 0.2–1 mM was titrated into a 20–90 μM solution of phospho-Sic1. Experiments were performed at 25 °C using a VP-ITC instrument (Microcal). When binding was detected, experiments were performed in duplicate. Binding constants were calculated by averaging the Kds, and the error is the standard deviation of the Kds.
Western blotting and viability assays
For western blotting experiments, versions of Sic1ΔC-3HA were cloned into vector pRS315 and constitutively expressed under the ADH1 promoter. The cells were treated for 2.5 hr with 1 μg/ml α factor and released from the arrest by removing the α factor by washing. The cells were lysed by bead beating in lysis buffer containing urea. Blotting of Phos-Tag SDS-PAGE gels was performed using a dry system iBlot (Invitrogen). Covance HA.11 Clone 16B12 monoclonal antibody (1:500) (Catalog Number: MMS-101P) and HRP-conjugated anti-mouse antibody (1:7500) from Labas, Estonia were used for detection of HA-tagged proteins by western blot. For the viability assays the SIC1 versions were cloned into the pRS413 vector under the control of GAL1 promoter.
Sic1- and Cnn1-based substrate constructs
For studies of docking distances we used substrate constructs based on a non-inhibitory version of Sic1 with truncation of the inhibitory domain at position 216-284 (Sic1ΔC). For the constructs used for experiments presented in Figure 3b-e the sequence motif QATPQAAAQ (based on the surroundings of T33 with the lysines, prolines and serines mutated to alanines) was introduced in positions 8, 13, 23, 39, 41, 43, 45, 47, 49, 51, 53, 65, 75, 85, 95 and 105 (the number designates the position of phospho-acceptor threonine). The priming site T33 was left unchanged in this set of the constructs. At the positions of other physiological phosphorylation sites the double alanine replacements were introduced at the S/TP motifs in case of all Sic1ΔC constructs. In order to analyze the potential N-terminally directed docking-enhanced phosphorylation steps over longer distances the T33 motif (QKTPQKPSQ) was replaced into the positions 47, 67, and 87, while the secondary site motif was introduced at position 8. For studies of distance requirements of cyclin-dependent docking sites the substrate constructs contained only a single RXL motif (positions 89-91), while the other motifs were mutated to alanines as described previously 14. The Cln2-specific LLPP motif (in positions 137-140) was left intact. In these constructs the sequence motif PSTPPRSRG based on the site T5 was introduced at positions 25, 45, 67, 69, 73, 77, 81, 103, 115, 135 and 155. In case of the construct used in Supplementary Fig. 4h QATPQAAAQ phosphorylation site was introduced at position 45.
The substrate constructs used in the western blotting experiments presented in Figure 2f were based on non-inhibitory version of Sic1 containing a 3HA tag in its C-terminus (Sic1 ΔC-3HA). The sequence fragments AMSPSA, LTSPQA, and QRSPFP, based on the primary structure surrounding the physiological sites T2, S76, and S80 (with exceptions that the lysine in position +3 from the site S76 was mutated to alanine and the threonine in position 2 was mutated to serine), were replaced into positions 21, 37, and 53 (indicating the new positions of the phospho-acceptor residue). The sites T5 and S69 were left in their original positions. These replacements yielded constructs with 5 CDK consensus sites with 16 amino acids intervals between the sites. For testing serine as the priming site, the T5S mutation was introduced. In case of these constructs the RXL motifs and the LLPP motif was mutated to alanines as described in 22, due to the observed slight RXL-LLPP-dependent preference of serine residues over the threonine in the phosphorylation efficiencies in vivo. In the western blotting experiment presented in Figure 3f the construct with the five physiological sites in their original positions was made by mutating the sites T33, T45, T173, and S191 to alanines; the site T2 to serine; the lysine in position +3 from the site S76 to alanine. The construct with the changed distances between the sites was made by similar rearrangements of sequence motifs as described above for the constructs used in Figure 2f. For the western blotting experiments presented in the Supplementary Figure 4j the constructs with suboptimal sites had similar positioning of the sequence fragments as the constructs with equal distance interval shown in Figures 2f and 3f with the exception that the arginine in position +3 from the phosphorylation site T5 or S5 was mutated to alanine.
For the Cnn1 constructs used in studies of docking distances in Figure 3h the sequence motif of RNTPGY (based on the surroundings of T42) was introduced in positions 8, 12, 16, 16 and 39. In these constructs the CDK phosphorylation site T21 was mutated to alanine.
Quantitative mass spectrometry
To quantitatively determine how Cks1 stimulates phosphorylation of different Stb1, Ndd1, Swi5, and Far1 phosphorylation sites, equal amounts of Stb1, Ndd1, Swi5 or Far1 proteins were phosphorylated by Cln2- (Stb1), Clb5- (Far1) or Clb2-Cdk1 (Ndd1, Swi5) supplemented with normal isotopic ATP [(16O)-ATP] or heavy ATP [(18O)-ATP] (Cambridge Isotope Laboratories, Inc.). Kinase assays were performed to achieve 20-30% of total substrate turnover. Aliquots from reactions with Cks1wt and Cks1mut were pooled together in a 1:1 ratio in SDS-PAGE sample buffer. The proteins were separated by 10% SDS-PAGE and the gels were stained with Coomassie brilliant blue G-250 (Sigma) and protein bands were excised from the stained gels. Trypsin/P (20 ng μl–1) was used for in-gel digestion of proteins and peptides were purified using C18 StageTips. Peptides were separated by Agilent 1200 series nanoflow system (Agilent Technologies) connected to a LTQ Orbitrap classic mass-spectrometer (Thermo Electron) equipped with nanoelectrospray ion source (Proxeon). Purified peptides were loaded on a fused silica emitter (75 μm x 150 mm) (Proxeon) packed in-house with Reprosil-Pur C18-AQ 3 μm particles. Peptides were separated with 30 minute 3–40% B gradient (A: 0.5% acetic acid, B: 0.5% acetic acid/80% acetonitrile) at a flow-rate of 200 nl/min, eluted peptides were sprayed directly into an LTQ Orbitrap mass-spectrometer with a spray voltage of 2.2 kV. The MS scan range was m/z 300–1800 and the top 5 precursor ions were selected for subsequent MS/MS scans. A lock-mass was used for the LTQ-Orbitrap to obtain constant mass-accuracy during the gradient analysis.
Peptides were identified with Mascot 2.3 (www.matrixscience.com) search engine (see Supplementary Table 2 and 3). Peptide mass tolerance of 7 ppm and fragment ion mass tolerance of 0.6 Da were used. Two missed cleavage sites for Trypsin/P were allowed. The carbamidomethylation of cysteine was set as fixed modification. The oxidation of the methionine and the phosphorylation of serine and threonine were set as variable modifications. The relative intensities of phosphorylated peptides (Cks1wt/Cks1mut) were standardized by the intensity of the phosphorylated peptide containing the most N-terminal optimal Cdk1 phosphorylation site (S72 of Stb1, T179/T183 of Ndd1 and S225 of Swi5). Two independent phosphorylation experiments were performed and the peptide intensities from at least three retention times in case of each experiment were quantified and used for calculation of the Cks1wt/Cks1mut effect.
Search criteria for potential docking connections in physiological targets
12-32 amino acid distance for any TP motif, or 12-52 amino acid distance for optimal Cks1-binding motif (F/I/L/P/V/W/YxTP 41(cosubmitted manuscript). The Yeast Genome Pattern Matching (http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch) was used for the search of docking connections and PSIPRED v3.3 (http://bioinf.cs.ucl.ac.uk/psipred/) was used for the prediction of disordered regions in the targets.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figure 4
{kind=link}
Supplementary Figure 5
{kind=link}
Supplementary Figure 6
{kind=link}
Supplementary Figure 7
{kind=link}
Supplementary Figure 8
{kind=link}
Includes Supplementary Figures 1-8 (with captions), Supplementary Tables 1-6 and Supplementary Note.
Acknowledgements
We would like to thank Liam Holt and David Morgan for valuable comments on the manuscript. We thank Doug Kellogg (University of California, Santa Cruz) for kindly providing the strains and Jevgeni Mihhejev for excellent technical assistance. This work was supported by International Senior Research Fellowship No. 079014/Z/06/Z from the Wellcome Trust (M.L.), an installation grant from EMBO and HHMI, No. 1253 (M.L.), a targeted financing scheme and an institutional grant IUT2-21 from the Estonian government (M.L.), and funding to S.M.R. from the American Cancer Society (RSG-12-131-01-CCG).
References
- 1.Morgan DO. The Cell Cycle: Principles of Control. New Science Press Ltd; 2007. [Google Scholar]
- 2.Ubersax JA, et al. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- 3.Holt LJ, et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Errico A, Deshmukh K, Tanaka Y, Pozniakovsky A, Hunt T. Identification of Substrates for Cyclin Dependent Kinases. Adv. Enzyme Regul. 2009 doi: 10.1016/j.advenzreg.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Pagliuca FW, et al. Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Mol. Cell. 2011;43:406–417. doi: 10.1016/j.molcel.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell. Div. 2010;5 doi: 10.1186/1747-1028-5-11. 11-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stern B, Nurse P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996;12:345–350. [PubMed] [Google Scholar]
- 8.Coudreuse D, Nurse P. Driving the cell cycle with a minimal CDK control network. Nature. 2010;468:1074–1079. doi: 10.1038/nature09543. [DOI] [PubMed] [Google Scholar]
- 9.Uhlmann F, Bouchoux C, Lopez-Aviles S. A quantitative model for cyclin-dependent kinase control of the cell cycle: revisited. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011;366:3572–3583. doi: 10.1098/rstb.2011.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher D, Krasinska L, Coudreuse D, Novak B. Phosphorylation network dynamics in the control of cell cycle transitions. J. Cell. Sci. 2012;125:4703–4711. doi: 10.1242/jcs.106351. [DOI] [PubMed] [Google Scholar]
- 11.Fisher DL, Nurse P. A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1 cyclins. EMBO J. 1996;15:850–860. [PMC free article] [PubMed] [Google Scholar]
- 12.Songyang Z, et al. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 13.Mok J, et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koivomagi M, et al. Dynamics of Cdk1 Substrate Specificity during the Cell Cycle. Mol. Cell. 2011;42:610–623. doi: 10.1016/j.molcel.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deibler RW, Kirschner MW. Quantitative reconstitution of mitotic CDK1 activation in somatic cell extracts. Mol. Cell. 2010;37:753–767. doi: 10.1016/j.molcel.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oikonomou C, Cross FR. Rising cyclin-CDK levels order cell cycle events. PLoS One. 2011;6:e20788. doi: 10.1371/journal.pone.0020788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cross FR, Yuste-Rojas M, Gray S, Jacobson MD. Specialization and targeting of B-type cyclins. Mol. Cell. 1999;4:11–19. doi: 10.1016/s1097-2765(00)80183-5. [DOI] [PubMed] [Google Scholar]
- 19.Cross FR, Jacobson MD. Conservation and function of a potential substrate-binding domain in the yeast Clb5 B-type cyclin. Mol. Cell. Biol. 2000;20:4782–4790. doi: 10.1128/mcb.20.13.4782-4790.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- 21.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat. Rev. Mol. Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 22.Koivomagi M, et al. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature. 2011;480:128–131. doi: 10.1038/nature10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koivomagi M, Loog M. Cdk1: a kinase with changing substrate specificity. Cell. Cycle. 2011;10:3625–3626. doi: 10.4161/cc.10.21.17854. [DOI] [PubMed] [Google Scholar]
- 24.Mendenhall MD, Hodge AE. Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998;62:1191–1243. doi: 10.1128/mmbr.62.4.1191-1243.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim SY, Ferrell JE., Jr. Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell. 2007;128:1133–1145. doi: 10.1016/j.cell.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 26.Trunnell NB, Poon AC, Kim SY, Ferrell JE., Jr. Ultrasensitivity in the Regulation of Cdc25C by Cdk1. Mol. Cell. 2011;41:263–274. doi: 10.1016/j.molcel.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi S, Pryciak PM. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr. Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner MV, et al. Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS One. 2009;4:e4300. doi: 10.1371/journal.pone.0004300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012;26:1156–1166. doi: 10.1101/gad.189837.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma R, Feldman RM, Deshaies RJ. SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol. Biol. Cell. 1997;8:1427–1437. doi: 10.1091/mbc.8.8.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol. Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 32.Hayles J, Beach D, Durkacz B, Nurse P. The fission yeast cell cycle control gene cdc2: isolation of a sequence suc1 that suppresses cdc2 mutant function. Mol. Gen. Genet. 1986;202:291–293. doi: 10.1007/BF00331653. [DOI] [PubMed] [Google Scholar]
- 33.Hadwiger JA, Wittenberg C, Richardson HE, de Barros Lopes M, Reed SI. A family of cyclin homologs that control the G1 phase in yeast. Proc. Natl. Acad. Sci. U. S. A. 1989;86:6255–6259. doi: 10.1073/pnas.86.16.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Y, Reed SI. The Cdk-associated protein Cks1 functions both in G1 and G2 in Saccharomyces cerevisiae. Genes Dev. 1993;7:822–832. doi: 10.1101/gad.7.5.822. [DOI] [PubMed] [Google Scholar]
- 35.Kito K, Kawaguchi N, Okada S, Ito T. Discrimination between stable and dynamic components of protein complexes by means of quantitative proteomics. Proteomics. 2008;8:2366–2370. doi: 10.1002/pmic.200800182. [DOI] [PubMed] [Google Scholar]
- 36.Patra D, Wang SX, Kumagai A, Dunphy WG. The xenopus Suc1/Cks protein promotes the phosphorylation of G(2)/M regulators. J. Biol. Chem. 1999;274:36839–36842. doi: 10.1074/jbc.274.52.36839. [DOI] [PubMed] [Google Scholar]
- 37.Patra D, Dunphy WG. Xe-p9, a Xenopus Suc1/Cks protein, is essential for the Cdc2-dependent phosphorylation of the anaphase- promoting complex at mitosis. Genes Dev. 1998;12:2549–2559. doi: 10.1101/gad.12.16.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynard GJ, Reynolds W, Verma R, Deshaies RJ. Cks1 is required for G(1) cyclin cyclin-dependent kinase activity in budding yeast. Mol. Cell. Biol. 2000;20:5858–5864. doi: 10.1128/mcb.20.16.5858-5864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourne Y, et al. Crystal structure and mutational analysis of the Saccharomyces cerevisiae cell cycle regulatory protein Cks1: implications for domain swapping, anion binding and protein interactions. Structure. 2000;8:841–850. doi: 10.1016/s0969-2126(00)00175-1. [DOI] [PubMed] [Google Scholar]
- 40.Nash P, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 41.McGrath D, et al. Cks Confers Specificity to Phosphorylation-Dependent Cdk Signaling Pathways. Cosubmitted. [DOI] [PMC free article] [PubMed]
- 42.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 43.Brocca S, et al. Order propensity of an intrinsically disordered protein, the cyclin-dependent-kinase inhibitor Sic1. Proteins. 2009;76:731–746. doi: 10.1002/prot.22385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulman BA, Lindstrom DL, Harlow E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U. S. A. 1998;95:10453–10458. doi: 10.1073/pnas.95.18.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 46.Takeda DY, Wohlschlegel JA, Dutta A. A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 2001;276:1993–1997. doi: 10.1074/jbc.M005719200. [DOI] [PubMed] [Google Scholar]
- 47.Wohlschlegel JA, Dwyer BT, Takeda DY, Dutta A. Mutational analysis of the Cy motif from p21 reveals sequence degeneracy and specificity for different cyclin-dependent kinases. Mol. Cell. Biol. 2001;21:4868–4874. doi: 10.1128/MCB.21.15.4868-4874.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilmes GM, et al. Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev. 2004;18:981–991. doi: 10.1101/gad.1202304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowe ED, et al. Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry. 2002;41:15625–15634. doi: 10.1021/bi0268910. [DOI] [PubMed] [Google Scholar]
- 50.Bhaduri S, Pryciak PM. Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr. Biol. 2011;21:1615–1623. doi: 10.1016/j.cub.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouchoux C, Uhlmann F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell. 2011;147:803–814. doi: 10.1016/j.cell.2011.09.047. [DOI] [PubMed] [Google Scholar]
- 52.Hirschi A, et al. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 2010;17:1051–1057. doi: 10.1038/nsmb.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikui AE, Rossio V, Schroeder L, Yoshida S. A Yeast GSK-3 Kinase Mck1 Promotes Cdc6 Degradation to Inhibit DNA Re-Replication. PLoS Genet. 2012;8:e1003099. doi: 10.1371/journal.pgen.1003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyons NA, Fonslow BR, Diedrich JK, Yates JR, 3rd, Morgan DO. Sequential primed kinases create a damage-responsive phosphodegron on Eco1. Nat. Struct. Mol. Biol. 2013 doi: 10.1038/nsmb.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bourne Y, et al. Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle-regulatory protein CksHs1. Cell. 1996;84:863–874. doi: 10.1016/s0092-8674(00)81065-x. [DOI] [PubMed] [Google Scholar]
- 56.Puig O, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 57.McCusker D, et al. Cdk1 coordinates cell-surface growth with the cell cycle. Nat. Cell Biol. 2007;9:506–515. doi: 10.1038/ncb1568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Includes Supplementary Figures 1-8 (with captions), Supplementary Tables 1-6 and Supplementary Note.