

Abstract
Aims/hypothesis
Diabetic retinopathy is a major complication of type 2 diabetes and the leading cause of blindness in adults of working age. Neuronal defects are known to occur early in disease, but the source of this dysfunction is unknown. The aim of this study was to examine differences in the retinal membrane proteome among non-diabetic mice and mouse models of diabetes either with or without metformin treatment.
Methods
Alterations in the retinal membrane proteome of 10-week-old diabetic db/db mice, diabetic db/db mice orally treated with the anti-hyperglycaemic metformin, and congenic wild-type littermates were examined using label-free mass spectrometry. Pathway enrichment analysis was completed with Genomatix and Ingenuity. Alterations in Slc17a7 mRNA and vesicular glutamate transporter 1 (VGLUT1) protein expression were evaluated using real-time quantitative PCR and immunofluorescence.
Results
A total of 98 proteins were significantly differentially abundant between db/db and wild-type animals. Pathway enrichment analysis indicated decreases in levels of proteins related to synaptic transmission and cell signalling. Metformin treatment produced 63 differentially abundant proteins compared with untreated db/db mice, of which only 43 proteins were found to occur in both datasets, suggesting that treatment only partially normalises the alterations induced by diabetes. VGLUT1, which is responsible for loading glutamate into synaptic vesicles, was found to be differentially abundant in db/db mice and was not normalised by metformin. The decrease in Slc17a7/VGLUT1 was confirmed by transcriptomic and immunocytochemical analysis.
Conclusions/interpretation
These findings expand the knowledge of the protein changes in diabetic retinopathy and suggest that membrane-associated signalling proteins are susceptible to changes that are partially ameliorated by treatment.
Electronic supplementary material
The online version of this article (doi:10.1007/s00125-013-3070-2) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Keywords: Complex I, Diabetes, Label-free mass spectrometry, Membrane, Metformin, Proteome, Retinopathy, VGLUT1
Introduction
Type 2 diabetes is emerging as a major health issue, with the global prevalence estimated to reach 300 million people by the year 2025 [1]. This increase has been linked to an ageing population, lifestyle changes and an increase in global obesity. At present, the oral anti-hyperglycaemic drug metformin is a first-line treatment for type 2 diabetes, and predominantly acts through decreasing glucose production by the liver [2]. In addition to reducing hyperglycaemia, metformin treatment in diabetic patients has been linked to decreased rates of macrovascular disease and mortality [3]. Despite receiving treatment, however, many diabetic patients are still at increased risk of developing complications associated with the disease [4, 5].
Diabetic retinopathy is a major complication of types 1 and 2 diabetes [6]. Characterised and diagnosed by alterations in the retinal vasculature [7], studies have shown that changes to neurons and glia occur during diabetic retinopathy [8–10], with functional tests in humans and animal models indicating that neuronal dysfunction is present prior to overt vascular damage [11–13]. The underlying mechanism of neuronal dysfunction is unknown, but connections have been made to abnormal Na/K-ATPase activity [14], shortened photoreceptor segments [15] and differences in the light and dark vs colour visual pathways [16]. More than 90% of type 1 and 60% of type 2 diabetes patients develop some form of retinopathy following two decades of diabetes [6], despite receiving treatment for hyperglycaemia. This implies that hyperglycaemia-associated pathological retinal changes are not fully ameliorated by treatment. As such, it is necessary to understand the changes in the retina induced by diabetes, which pathways are affected by disease and the degree to which treatment ameliorates these alterations.
This study examined the retinal membrane proteome of the db/db mouse model of type 2 diabetes, and compared it with the proteome of non-diabetic mice and diabetic mice treated with metformin (dbM). Diabetic db/db mice have a mutation in the Lepr gene (encoding the leptin receptor), resulting in excessive food consumption and the development of obesity, hyperglycaemia and insulin resistance [17]. Pathological changes in the retinas of db/db mice include breakdown of the blood–retina barrier, acellular capillaries and neuronal dysfunction [18–20]. The membrane proteome is of particular interest in understanding changes in the diabetic retina, as it consists of transporters, receptors and signalling proteins that might be altered in disease or function as targets of drug treatment.
Methods
Experimental animals
Mice (db/db [BKS.Cg-m+/+Lepr db/db] and wild-type [BKS.Cg-m+/+Lepr m/m]; Jackson Laboratories, Bar Harbor, ME, USA) were bred and housed in single ventilated cages (SealsafePlus AERO IVC Greenline, Tecniplast, Hohenpeissenberg, Germany) at 22 ± 1°C, with a 12:12 h light:dark cycle and controlled humidity, in the German Mouse Clinic, Helmholz Center Munich [21, 22]. All procedures were performed with approval from the Government of Upper Bavaria (reference no. 55.2-1-54-2531-70-07) and adhered to the Principles of Laboratory Animal Care (NIH publication no. 85-23, revised 1985, http://grants1.nih.gov/grants/olaw/references/phspol.htm).
Male wild-type and db/db mice were fed a high-fat diet postweaning (S0372-E010, ssniff Spezialdiäten, Soest, Germany). At 8 weeks, mice were fasted for 4 h and weighed, and blood glucose (Contour, Bayer Vital, Leverkusen, Germany) and HbA1c (A1cNow+, Bayer Vital) concentrations were measured. Inclusion criteria for diabetic mice were weight greater than 44 g and blood glucose above 19.43 mmol/l. Mice that met these criteria were matched into two cohorts: one cohort was treated with metformin (dbM, 300 mg kg−1 day−1; Sigma Aldrich, Munich, Germany), while the other cohort and wild-type mice were given placebo (n = 10 in each group). Metformin was dissolved in 0.5% placebo solution (95% hydroxyethylcellulose 250G Pharm, Fagron, Barsbüttel, Germany; 5% Solutol HS15, BASF, Ludwigshafen, Germany) and applied daily by gavage. At 10 weeks, mice were fasted for 4 h, weighed, had blood glucose and HbA1c measurements taken and were killed with an isoflurane overdose 18 h after treatment. The eyes were enucleated, and retinas were either dissected, frozen in liquid nitrogen and stored at −80°C until further processing, or the anterior chamber was removed and the posterior eyecup fixed in 4% paraformaldehyde in 0.1 mol/l phosphate buffer (PB; pH 7.4), and processed for immunofluorescence [23]. Figure 1 shows body weight, blood glucose and HbA1c levels.
Fig. 1.
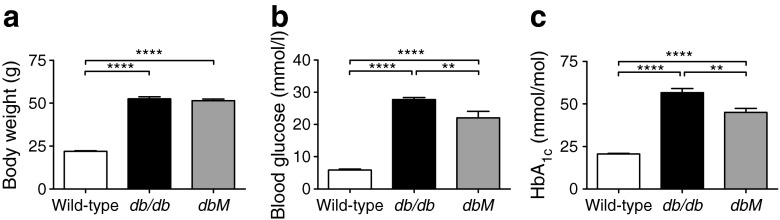
(a) Body weight, (b) blood glucose and (c) HbA1c measurements for wild-type, db/db and dbM mice. db/db and dbM mice had significantly increased body weight compared with wild-type mice (wild-type 21.88 ± 0.371 g; db/db 52.36 ± 1.207 g; dbM 51.44 ± 1.013 g; p < 0.0001). Blood glucose was significantly increased in db/db and dbM mice (wild-type 5.899 ± 0.296 mmol/l; db/db 27.76 ± 0.625 mmol/l; dbM 22.05 ± 2.009 mmol/l; p < 0.0001), as were HbA1c values (wild-type 20.06 ± 0.400 mmol/mol; db/db 56.57 ± 2.543 mmol/mol; dbM 45.00 ± 2.400 mmol/mol; p < 0.0001). Diabetic db/db and dbM mice had similar body weight (p > 0.999), but dbM mice had significantly decreased blood glucose (p < 0.001) and HbA1c values (p < 0.001). **p < 0.001, ****p < 0.0001. To convert values for HbA1c in mmol/mol into %, multiply by 0.09148 and add 2.152, or use the conversion calculator at www.hba1c.nu/eng/
Proteomic sample preparation
Membrane proteins were extracted as previously described [24]. Frozen retinal samples (n = 9 per group, in pools of three) were mechanically homogenised in a high-salt buffer (2 mol/l NaCl; 10 mmol/l HEPES NaOH, pH 7.4; 1 mmol/l EDTA), lightly centrifuged (61 g, 5 min, 4°C) to remove non-grinded tissue and then strongly centrifuged (17,500 g, 30 min, 4°C). The remaining pellet was repeatedly homogenised in a high pH buffer (0.1 mol/l Na2CO3, pH 11.3; 1 mmol/l EDTA), incubated on ice and centrifuged (17,500 g, 30 min, 4°C), with the supernatant fraction removed following each round. Following homogenisation, membrane protein pellets were solubilised in 36 μl of 50 mmol/l ammonium bicarbonate and 4 μl 2% RapiGest (Waters, Milford, MA, USA), reduced with 2 μl of 100 mmol/l dithiothreitol and incubated at 60°C for 10 min. Samples were alkylated with 2 μl of 300 mmol/l iodoacetamide at room temperature in the dark and digested with 4 μl trypsin (0.5 mg/ml, Promega, Madison, WI, USA) at 37°C overnight. The reaction was stopped with concentrated HCl. This was followed by centrifugation, with the soluble phase collected for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis.
Mass spectrometry
LC-MS/MS analysis was performed on an UltiMate 3000 nano rapid separation LC system (Dionex, Idstein, Germany) coupled to a linear trap quadrupole Velos mass spectrometer (Thermo Fisher Scientific, Schwerte, Germany) [25]. Samples were automatically loaded into a HPLC system (Dionex) at a 6 μl/min flow rate, in 0.5% trifluoracetic acid in HPLC-grade water onto an Acclaim PepMap100 column (75 μm inner diameter ×2 cm, C18, 3 μm, 100 Å, Dionex). After 5 min, peptides were eluted and separated on an Acclaim PepMap RSLC column (75 μm × 25 cm, C18, 2 μm, 100 Å, Dionex) by a linear gradient from 2% to 35% of buffer B (80% acetonitrile, 0.08% formic acid in HPLC-grade water) in buffer A (2% acetonitrile, 0.1% formic acid in HPLC-grade water) at a 300 nl/min flow rate over 150 min. Remaining peptides were eluted by a short gradient from 35% to 100% buffer B in 5 min. The eluted peptides were analysed online in the linear trap quadrupole Velos. From the high-resolution MS prescan with a mass range from 300 to 1,500 Da, the ten most intense peptide ions were automatically selected for fragment analysis in the linear ion trap if they exceeded an intensity of 200 counts and if they were at least doubly charged. Target peptides selected for MS/MS were dynamically excluded for 20 s.
Label-free analysis
Acquired spectra (Thermo raw files) were analysed using Progenesis LC-MS software (Version 4.0, Nonlinear Dynamics, Newcastle upon Tyne, UK) [24]. The profile data of the MS scans were transformed into peak lists with respective intensities, abundances and m/z values. MS/MS spectra were treated similarly. Using one sample as the reference, the retention times of the other samples were manually and automatically aligned to a maximal overlay of the two-dimensional features. Features with one charge or more than eight charges were excluded from further analyses. Samples were then allocated to their respective experimental groups (wild-type, db/db or dbM).
All MS/MS spectra were exported in Mascot generic format (mgf) and used for peptide identification with Mascot (version 2.4; Matrix Science, Boston, MA, USA) using the Ensembl mouse protein database (www.ensembl.org; Mus musculus; release 66 NCBIM37, 56,416 sequences, ftp://ftp.ensembl.org/pub/current_fasta/mus_musculus/). Search variables were: 10 ppm peptide mass tolerance; 0.6 Da MS/MS tolerance; one missed cleavage allowed; methionine oxidation, asparagine and glutamine deamidation as variable modifications; and carbamidomethylation as a fixed modification. The Mascot-integrated decoy database search calculated a false discovery rate of less than 1% when searching was performed on the concatenated mgf files with a percolator ion score cut-off of 15, and the significance threshold p adjusted to reach the maximum false discovery rate. Identifications were re-imported into Progenesis. All unique peptides were included for quantification. Total cumulative protein abundance was calculated by summing the intensities of the individual peptides allocated to the respective protein; when two peptides are identified as belonging to protein X, if the intensity of peptide A is 1,000 and peptide B is 2,000, the total abundance of protein X is 3,000 [24]. No minimal thresholds were set for the method of peak picking or selection of data to use for quantifications. This study used proteins identified and quantified by at least two peptides for further analysis.
Protein network and pathway enrichment analysis
Protein interaction maps were created by uploading gene identifiers and fold changes (db/wild-type) for significantly different proteins into the Genomatix Pathway System (GePS, version 2.3.0, www.genomatix.de, accessed 27 March 2013, Genomatix, Munich, Germany). Interaction maps were based on Mus musculus and ‘function word’ as co-citation level for literature mining. The network was extended by ten genes via frequent co-citations for Ctbp2 (protein name RIBEYE), Slc17a7 (vesicular glutamate transporter 1 [VGLUT1]), Ndufa4 (NDUA4). Ndufa9 (NDUA9), Ndufa11 (NDUAB), Ndufa12 (NDUAC), Ndufa13 (NDUAD), Ndufb6 (NDUB6) and Ndufb10 (NDUBA), and connected by the shortest path. Candidates without interactions were removed. Based on db/wild-type fold-change, upregulated proteins were coloured green and downregulated proteins were coloured red. Grey colouring represented network-extended proteins.
Pathway enrichment analyses were performed for down- or upregulated proteins using Ingenuity Pathway Analysis (IPA; www.ingenuity.com, accessed 13 December 2012, Ingenuity Systems, CA, USA). Protein lists were uploaded with Ensembl mouse accession numbers and core analysis was performed to identify the biological functions most significantly represented in the dataset. The analysis used the Ingenuity Knowledge Base (Ingenuity Systems) as the reference set, and considered molecules and/or relationships with Mus musculus as the species in experimentally observed results. p values were calculated using Fisher’s exact test to determine the probability that a function or disease assigned to the dataset would occur by chance.
Metformin quantification
Retinal metformin levels were determined using HPLC-MS/MS quantification (unpublished data, S Zukunft, C Muschet, J Adamski). Frozen retinal samples of placebo- and metformin-treated db/db mice were transferred into dry-ice-cooled 1.4 mm Precellys tubes (Peqlab Biotechnologie, Erlangen, Germany) and homogenised in HPLC-grade methanol (20 μl/mg tissue; AppliChem, Darmstadt, Germany). The homogenates were diluted 1:100 in HPLC-grade methanol containing 5 ng/ml deuterium-labelled metformin (metformin-d6), which served as an internal standard. MS/MS measurements were performed on a QTRAP 5500 System (AB Sciex, Darmstadt, Germany) coupled to an 1260 Infinity Series HPLC (Agilent, Böblingen, Germany) and an HTC-xt PAL autosampler (CTC Analytics, Zwingen, Switzerland), controlled by Analyst 1.6.1 software (AB Sciex). Reverse-phase LC was performed applying a methanolic isocratic mobile phase. Metformin was identified by multiple reaction monitoring using 130.04/60.00 Da as Q1/Q3 mass pairs for metformin and 136.09/60.00 Da for metformin-d6. Quantification was assessed by comparing sample peak areas with those of a methanol-dissolved metformin standard curve.
Relative qRT-PCR quantification
Total RNA was isolated from wild-type and db/db retinas (n = 3 per group) [26]. Samples were homogenised in 750 μl TRIzol (Invitrogen, Karlsruhe, Germany) and incubated for 5 min. Chloroform (150 μl) was added, followed by incubation for 3 min and centrifuged (17,500 g, 15 min, 4°C). The supernatant fraction was transferred to a new tube with 375 μl isopropanol, incubated for 10 min and centrifuged (17,500 g, 10 min, 4°C). The pellet was washed with 75% ethanol and treated with the RQ1 RNase-Free DNase kit (Promega) to eliminate contaminating genomic DNA. Total RNA was quantified using a NanoDrop photometer (Thermo Fisher Scientific) and cDNA generated using the Fermentas RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. To improve the yield, oligo(dT)20-primers and random hexamer primers were used for synthesis. Slc17a7 gene expression was assessed relative to the Hprt1 housekeeping gene (primer sequences in Electronic Supplementary Material [ESM] Table 1). Real-time PCR was performed using a LightCycler 480 (Roche Diagnostics, Mannheim, Germany) using the KAPA SYBR FAST qPCR MasterMix for Roche LightCycler 480 Kit (Peqlab Biotechnologie).
Immunofluorescence and photography
VGLUT1 expression was examined with immunofluorescence (n = 6) [23]. Vertical cryosections of 12 μm were blocked with 10% normal goat serum, 1% BSA and 0.5% Triton-X 100 in PB for 1 h, and incubated in anti-VGLUT1 antibody (AB#5905, Millipore, Schwabach, Germany) diluted 1:5,000 in buffer (3% normal goat serum, 1% BSA and 0.5% Triton-X 100 in PB) overnight at room temperature. After rinsing, sections were incubated with goat anti-guinea pig Alexa Fluor 488 secondary antibody (Invitrogen) diluted 1:1,000 in buffer for 90 min, washed in PB and coverslipped. All sections were photographed with constant variables on an AxioImager Z1 with ApoTome attachment (Zeiss, Göttingen, Germany). Mean intensities of VGLUT1 labelling in the outer plexiform layer (OPL) and inner plexiform layer (IPL) were measured using the Definiens Enterprise Image Intelligence Suite (Definiens, Munich, Germany).
Statistical analysis
All values are expressed as mean ± SEM. Body weight, blood glucose and HbA1c were analysed with one-way ANOVA and Bonferroni post hoc test using an alpha of 0.05. All other data were analysed using Student’s t test. Results were considered significant at p < 0.05.
Results
Identification of proteins and protein networks in diabetic retina
A total of 844 proteins were identified in the membrane-enriched fractions of retinal tissue from wild-type, db/db and dbM mice, of which 513 proteins were identified and quantified by more than one peptide and used for further analysis (ESM Tables 2 and 3). Analysis of the subcellular location of the identified proteins was completed using the UniProtKB/Swiss-Prot database. Overall, 64% of identified proteins were annotated with ‘membrane’ or variations thereof (ESM Fig. 1), which is comparable with other membrane proteome studies [24].
A total of 98 proteins were differentially abundant (p < 0.05) between wild-type and db/db retinas, of which 80 were downregulated and 18 upregulated (ESM Table 4). A protein interaction network map was generated using GePS to identify functional clusters affected by diabetes (Fig. 2). Nine candidates were chosen to extend the network: Ndufa4, Ndufa9, Ndufa11, Ndufa12, Ndufa13, Ndufb6 and Ndufb10, which are subunits of NADH dehydrogenase (ubiquinone) I, also known as complex I, from the electron transport chain; and Ctbp2 (RIBEYE) and Slc17a7 (VGLUT1), which are known to have important roles in retinal synaptic transmission. Extending by the most frequently co-cited genes and then removing results without connections produced a loosely interconnected network of 41 proteins from the dataset, with 12 additional proteins. Two proteins added to the network formed many connections with other proteins from the dataset and with other added proteins: Ins2, the mouse orthologue of insulin (INS), and Trp53, the mouse orthologue of tumour protein 53 (TP53).
Fig. 2.
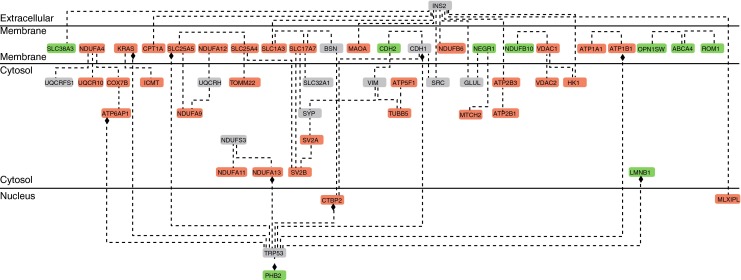
Interaction map of differentially abundant retinal proteins in diabetes. A total network was generated with GePS using functional word as connector. Proteins abbreviations are based on official gene symbols (NCBI Entrez Gene). Upregulated proteins are presented in green, downregulated proteins in red and network-extended proteins in grey. The network indicates a moderate degree of functional interaction among proteins affected by diabetes
Pathway enrichment analysis with IPA was completed to determine the biological processes most affected in diabetic retinas. The 80 proteins with decreased abundances in diabetes were significantly associated with 367 biological functions (ESM Table 5). Table 1 lists the top five molecular and cellular functions. Cell-to-cell signalling was the highest ranking category with 11 annotated functions, of which synaptic transmission was the most significantly annotated (p = 7.93 × 10−6). The synaptic transmission annotation contained eight proteins, which included several proteins identified in the GePS protein interaction network. Other highly ranking categories include molecular transport and amino acid metabolism. IPA identified 210 significantly enriched biological functions associated with the 18 upregulated proteins (ESM Table 6). Cell death and survival was the highest ranking molecular function, with four proteins and four functions (Table 2), of which recovery of retinal rods was the most significantly annotated function (p = 1.17 × 10−6). These biological functions were not enriched when the entire set of 513 detected and quantified proteins underwent pathway analysis (ESM Table 7).
Table 1.
Top five molecular and cellular functions associated with proteins downregulated in diabetic retinas
| Category and annotations | p value | Molecules | No. molecules |
|---|---|---|---|
| Cell-to-cell signalling and interaction | |||
| Synaptic transmission | 7.93 × 10−6 | DPP6, GLRB, SLC12A5, SLC17A7, SLC1A3, SV2A, SV2B, VDAC1 | 8 |
| Long-term potentiation | 4.10 × 10−3 | DPP6, KRAS, SLC17A7, SYNGR1, VDAC1 | 5 |
| Long-term potentiation of synapse | 4.59 × 10−3 | DPP6, SLC17A7, SYNGR1, VDAC1 | 4 |
| Accumulation of 5-hydroxytryptamine | 4.81 × 10−3 | MAOA | 1 |
| Sequestration of neurotransmitter | 4.81 × 10−3 | SLC17A7 | 1 |
| Release of l-glutamic acid | 6.87 × 10−3 | SLC17A7, SLC1A3 | 2 |
| Paired-pulse facilitation of synapse | 1.81 × 10−2 | SYNGR1, VDAC1 | 2 |
| Aggregation of neurons | 2.38 × 10−2 | ATP1B1 | 1 |
| Catabolism of dopamine | 2.38 × 10−2 | MAOA | 1 |
| Plasticity of synapse | 4.23 × 10−2 | KRAS, SYNGR1 | 2 |
| Formation of adherens junctions | 4.25 × 10−2 | KRAS | 1 |
| Cellular assembly and organisation | |||
| Activation of permeability transition pores | 2.28 × 10−5 | SLC25A4, SLC25A5 | 2 |
| Fragmentation of microtubules | 4.81 × 10−3 | TUBB | 1 |
| Organisation of axon terminals | 4.81 × 10−3 | MAOA | 1 |
| Expansion of thalamocortical axons | 9.59 × 10−3 | MAOA | 1 |
| Quantity of thalamocortical axons | 9.59 × 10−3 | MAOA | 1 |
| Segregation of sensory projections | 9.59 × 10−3 | MAOA | 1 |
| Morphology of Golgi apparatus | 1.44 × 10−2 | TMED10 | 1 |
| Organisation of Golgi apparatus | 2.85 × 10−2 | TMED10 | 1 |
| Formation of adherens junctions | 4.25 × 10−2 | KRAS | 1 |
| Morphology of mitochondria | 4.35 × 10−2 | NDUFA13, SLC25A4 | 2 |
| Amino acid metabolism | |||
| Import of l-glutamic acid | 6.81 × 10−5 | SLC17A7, SLC1A3 | 2 |
| Synthesis of GABA | 4.81 × 10−3 | SLC1A3 | 1 |
| Synthesis of l-glutamic acid | 4.81 × 10−3 | SLC1A3 | 1 |
| Release of l-glutamic acid | 6.87 × 10−3 | SLC17A7, SLC1A3 | 2 |
| Metabolism of l-glutamic acid | 1.91 × 10−2 | SLC1A3 | 1 |
| Molecular transport | |||
| Import of l-glutamic acid | 6.81 × 10−5 | SLC17A7, SLC1A3 | 2 |
| Transport of anions | 1.39 × 10−4 | GLRB, SLC12A5, SLC4A3, VDAC1 | 4 |
| Transport of ions | 5.13 × 10−4 | ATP2B1, CAMK2D, CAMK2G, GLRB, SLC12A5, SLC4A3, VDAC1 | 7 |
| Transport of molecules | 1.51 × 10−3 | ATP2B1, CAMK2D, CAMK2G, GLRB, SLC12A5, SLC17A7, SLC1A3, SLC4A3, TMED10, VDAC1 | 10 |
| Transport of Ca2+ | 3.14 × 10−3 | ATP2B1, CAMK2D, CAMK2G | 3 |
| Accumulation of 5-hydroxytryptamine | 4.81 × 10−3 | MAOA | 1 |
| Exchange of ADP | 4.81 × 10−3 | SLC25A4 | 1 |
| Exchange of ATP | 4.81 × 10−3 | SLC25A4 | 1 |
| Release of lactic acid | 4.81 × 10−3 | SLC1A3 | 1 |
| Translocation of ADP | 4.81 × 10−3 | VDAC1 | 1 |
| Release of l-glutamic acid | 6.87 × 10−3 | SLC17A7, SLC1A3 | 2 |
| Transport of chloride | 6.87 × 10−3 | GLRB, SLC12A5 | 2 |
| Redistribution of cholesterol | 1.91 × 10−2 | ATP1A1 | 1 |
| Transport of cations | 2.47 × 10−2 | ATP2B1, CAMK2D, CAMK2G, SLC12A5 | 4 |
| Small-molecule biochemistry | |||
| Import of l-glutamic acid | 6.81 × 10−5 | SLC17A7, SLC1A3 | 2 |
| Accumulation of 5-hydroxytryptamine | 4.81 × 10−3 | MAOA | 1 |
| Exchange of ADP | 4.81 × 10−3 | SLC25A4 | 1 |
| Exchange of ATP | 4.81 × 10−3 | SLC25A4 | 1 |
| Oxidation of lactic acid | 4.81 × 10−3 | MLXIPL | 1 |
| Release of lactic acid | 4.81 × 10−3 | SLC1A3 | 1 |
| Sequestration of neurotransmitters | 4.81 × 10−3 | SLC17A7 | 1 |
| Synthesis of GABA | 4.81 × 10−3 | SLC1A3 | 1 |
| Synthesis of l-glutamic acid | 4.81 × 10−3 | SLC1A3 | 1 |
| Translocation of ADP | 4.81 × 10−3 | VDAC1 | 1 |
| Release of l-glutamic acid | 6.87 × 10−3 | SLC17A7, SLC1A3 | 2 |
| Metabolism of l-glutamic acid | 1.91 × 10−2 | SLC1A3 | 1 |
| Redistribution of cholesterol | 1.91 × 10−2 | ATP1A1 | 1 |
| Catabolism of dopamine | 2.38 × 10−2 | MAOA | 1 |
| Synthesis of glucocorticoids | 2.85 × 10−2 | ATP1A1 | 1 |
Protein abbreviations are based on official gene symbols (NCBI Entrez Gene)
Table 2.
Top five molecular and cellular functions associated with proteins upregulated in diabetic retina
| Category and annotations | p value | Molecules | No. molecules |
|---|---|---|---|
| Cell death and survival | |||
| Recovery of retinal rods | 1.17 × 10−6 | ABCA4, RGS9BP | 2 |
| Delay in recovery of retinal rods | 1.11 × 10−3 | ABCA4 | 1 |
| Apoptosis of endothelial cell lines | 4.45 × 10−3 | CDH2 | 1 |
| Apoptosis of photoreceptors | 1.22 × 10−3 | ROM1 | 1 |
| Cellular compromise | |||
| Degeneration of photoreceptors | 1.07 × 10−3 | ABCA4, ROM1 | 2 |
| Neurodegeneration of photoreceptors | 6.67 × 10−3 | ROM1 | 1 |
| Degeneration of retinal rods | 1.33 × 10−2 | ROM1 | 1 |
| Amino acid metabolism | |||
| Transport of l-histidine | 1.11 × 10−3 | SLC38A3 | 1 |
| Transport of l-glutamine | 2.23 × 10−3 | SLC38A3 | 1 |
| Carbohydrate metabolism | |||
| Quantity of N-retinylidene-phosphatidylethanolamine | 1.11 × 10−3 | ABCA4 | 1 |
| Cell morphology | |||
| Size of rod outer segment disks | 1.11 × 10−3 | ROM1 | 1 |
| Abnormal morphology of photoreceptor outer segments | 1.12 × 10−3 | ABCA4, ROM1 | 2 |
| Blebbing of nucleus | 2.23 × 10−3 | LMNB1 | 1 |
| Length of rod outer segments | 4.45 × 10−3 | ROM1 | 1 |
| Abnormal morphology of rod outer segments | 1.55 × 10−2 | ROM1 | 1 |
| Innervation of muscle | 2.43 × 10−2 | LMNB1 | 1 |
| Abnormal morphology of photoreceptors | 3.73 × 10−2 | ABCA4 | 1 |
| Extension of axons | 4.16 × 10−2 | CDH2 | 1 |
Protein abbreviations are based on official gene symbols (NCBI Entrez Gene)
Effect of metformin on diabetic retinas
In order to confirm that metformin crossed the blood–retinal barrier, a direct measurement of metformin levels was conducted on retinal tissue from 4 h drug-treated db/db mice. A total of 8.52 ± 1.28 nmol/g of metformin was detected in retinas from drug-treated mice, while none was detected in the placebo-treated group.
Retinal membrane proteins from metformin-treated db/db mice were analysed to examine whether drug treatment ameliorated diabetes-induced changes. A total of 63 proteins were differentially abundant between the db/db and dbM samples (p < 0.05, ESM Table 8). Combining these results with the analysis of wild-type vs db/db samples, 43 of the identified proteins occurred in both datasets (Fig. 3); 55 were not significantly altered, and a further 20 proteins were not detected in the wild-type vs db/db dataset. Six of the 43 normalised proteins were complex I subunits: NDUA4, NDUA9, NDUAC, NDUAD, NDUB6 and NDUBA. The abundance of these proteins was similar in wild-type and dbM mice, indicating normalisation through metformin treatment (Fig. 4). Of the 55 proteins from the wild-type vs db/db dataset that did not exhibit a significant change following metformin treatment, some protein abundances e.g. VGLUT1, synaptogyrin 1 (SNG1) and sodium-coupled neutral amino acid transporter 3 (S38A3) were different from wild-type levels following treatment (Fig. 5a–c), while others e.g. opsin 1 blue -sensitive (OPSB), NDUAB and plasma membrane calcium ATPase (ATP2B3) showed large variations in sample protein levels (Fig. 5d–f). However, protein abundances demonstrated a trend towards normalisation. Some of the 20 proteins significantly altered in db/db vs dbM mice, but not in wild-type vs db/db mice, could have resulted from variations in sample abundance (Fig. 6a, b). Nevertheless, abundances of other proteins e.g. opsin 2 rhodopsin (OPSD), retinol dehydrogenase 12 (RDH12), cyclic nucleotide gated channel alpha 1 (CNGA1) and 5′-nucleotidase (5NTD) were dramatically altered following metformin treatment (Fig. 6c–f).
Fig. 3.
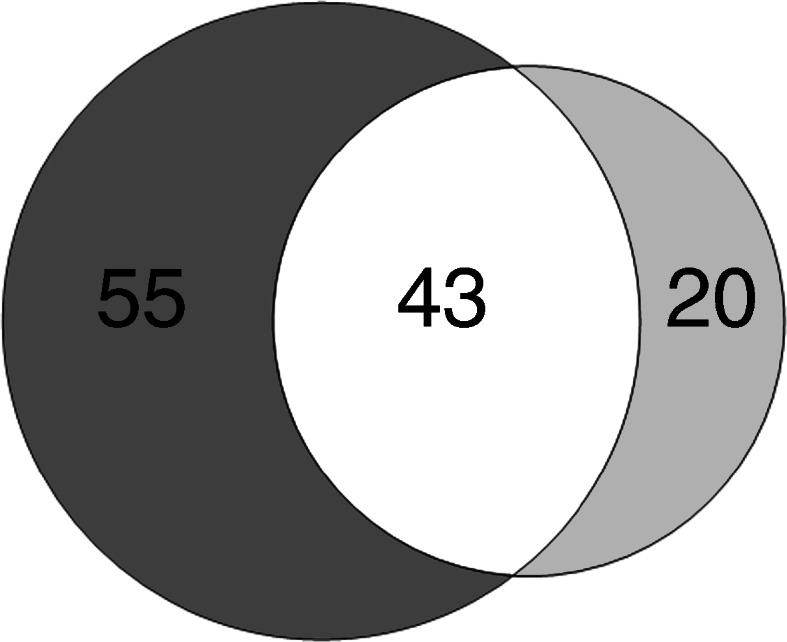
Venn diagram showing the number of proteins identified in retinas from wild-type vs db/db (dark grey) and db/db vs dbM (light grey) mice. A total of 98 proteins were identified as being differentially abundant in diabetic vs non-diabetic mice. Metformin treatment produced 63 differentially abundant proteins from untreated diabetic mice. A total of 43 proteins were identified as occurring in both datasets (overlap; white)
Fig. 4.

Complex I proteins normalised by metformin treatment as measured by MS. NDUA4 (a), NDUA9 (b), NDUAC (c), NDUAD (d) and NDUB6 (e) abundances were significantly decreased in db/db mice compared with wild-type and dbM mice, while NDUBA (f) was increased. Levels of these proteins were comparable in dbM and wild-type mice. *p < 0.05. Protein abundances are arbitrary units
Fig. 5.

Selected non-significantly different proteins between db/db and dbM treatment groups, as measured by MS. While all of the proteins presented were significantly altered from wild-type levels, they were not significantly affected (p > 0.05) by metformin treatment. (a) VGLUT1, (b) SNG1, (c) S38A3, (d) OPSB, (e) NDUAB and (f) ATP2B3 protein abundance. *p < 0.05. Protein abundances are arbitrary units
Fig. 6.

Selected significantly different proteins between db/db and dbM treatment groups. Mass spectrometric measurements showed variations in the individual wild-type sample abundances of (a) monocarboxylate transporter 1(MOT1) and (b) signal-regulatory protein alpha (SIRPA), demonstrating that some proteins may not have been identified as significantly altered between wild-type and db/db measurements. However, metformin treatment did change the abundances of non-altered proteins, such as (c) OPSD, (d) RDH12, (e) CNGA1 and (f) 5NTD. *p < 0.05. Protein abundances are arbitrary units
Slc17a7/VGLUT1 alterations in diabetic retinas
VGLUT1, encoded by Slc17a7, was selected for validation because it was differentially abundant between wild-type and db/db samples, not altered by metformin treatment and identified by GePS and IPA as having significant protein connections and functions. Using quantitative PCR, gene expression of Slc17a7 in diabetic retinas was found to be 41% of wild-type expression (Fig. 7). Because VGLUT1 is expressed at the glutamatergic synapses of photoreceptors and bipolar cells in the OPL and IPL [27], immunofluorescence was used to determine if VGLUT1 downregulation occurred in a specific location. Strong punctate labelling with VGLUT1 antibody was observed in the OPL and IPL in wild-type animals (Fig. 8a). In comparison, expression of VGLUT1 appeared to be decreased in both corresponding layers of the db/db retina (Fig. 8b). Quantification of VGLUT1 immunoreactivity found a significant decrease in db/db mice in the OPL (p < 0.01, Fig. 8c) and an even greater reduction in the IPL, compared with wild-type (p < 0.001, Fig. 8d). Taken together, these data indicate decreases in retinal VGLUT1 protein and gene expression, and anatomic localisation in diabetes.
Fig. 7.

Changes in Slc17a7 gene expression in a mouse model of diabetes. The mRNA level of Slc17a7 was decreased in diabetic retinas (db/db) compared with non-diabetic wild-type animals. Data are expressed as mean ± SD
Fig. 8.

Altered retinal VGLUT1 immunoreactivity in diabetes. Retinas from (a) wild-type and (b) diabetic mice were assessed for VGLUT1 immunolabelling. The intensity of VGLUT1 labelling was assessed in (c) the OPL and (d) the IPL. In wild-type retinas, VGLUT1 labelling was strongly present in the OPL with punctate labelling in the IPL. The intensity of VGLUT1 labelling appeared decreased in diabetic retinas in both layers. Quantification of fluorescence intensity indicated significant decreases in both the OPL (**p < 0.01) and the IPL (***p < 0.001). Scale bar: 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer
Discussion
We used label-free quantitative MS to examine the retinal membrane proteome at an early stage of diabetes. We identified pathways most affected by disease and investigated the effect of metformin on the membrane proteome of the diabetic retina. Our study revealed 98 differentially abundant proteins between wild-type and diabetic mice. Bioinformatics analysis indicated that these proteins were associated with cell signalling, particularly synaptic transmission. A total of 63 differentially abundant retinal membrane proteins were detected in diabetic mice treated with metformin compared with untreated diabetic retinas. Of these, 43 were found to overlap with the wild-type vs diabetes dataset, leaving 55 proteins not significantly affected by metformin treatment. Finally, qPCR and immunofluorescence analyses indicated a significant decrease in VGLUT1 protein and gene expression levels in the diabetic retina.
Proteomic studies of diabetes and diabetic complications can provide valuable information on the pathogenesis of the disease by identifying and quantifying biomarkers. The earliest proteomic diabetic retinopathy studies were conducted in 2007. Comparing healthy and diabetic rat retinas, two-dimensional electrophoresis was used to separate proteins, which were subsequently identified using matrix-assisted laser desorption/ionisation time of flight (MALDI-TOF) and MS/MS. Alterations in a number of proteins, including succinyl CoA ligase, phosducin, glutamine synthetase and αA-crystallin, were described [28, 29]. Later studies identified upregulation of α-, β-, and γ-crystallin isoforms, but reported conflicting results as to whether insulin treatment normalised these alterations [30, 31]. The identification of changes to crystallin isoforms is notable as these proteins belong to the heat-shock protein family and are involved in apoptosis regulation and vascular remodelling [32, 33]. Despite these promising results, relatively few proteomic studies have since been conducted on the diabetic vitreous or retina [28–31, 34–38], and none has concentrated specifically on the membrane proteome.
We have focused on the membrane proteome for a number of reasons. Membrane-bound proteins include signal transducers, transporters and receptors, which are likely to be among the first proteins affected by systemic hyperglycaemia and, being on the cell surface, are possible therapeutic targets. A previous proteomic diabetic retinopathy study commented on the relatively low abundance of membrane proteins compared with cytosolic proteins in their results [34], but improvements in sample preparation methods have now made it possible to analyse this specific subset of proteins [24]. Finally, immunohistochemical studies examining retinas from other diabetic animal models have reported defects in cell membrane proteins such as occludin [39], connexin-26 [23] and connexin-43 [40]. These proteins were not differentially abundant in our study; however, a number of proteins in our dataset were associated with cell signalling, particularly in synaptic transmission.
Decreased abundances of synaptic proteins may have relevance regarding the neuronal dysfunction observed in diabetic retinopathy. A number of electroretinogram studies in humans and animal models have reported decreases in inner retinal waveform components [12, 23, 41], indicating that postreceptoral neurons are particularly susceptible in diabetic retinopathy. The cause of these defects is unknown, but the downregulation of synaptic transmission proteins may indicate alterations in the steps required to transmit synaptic signals. VGLUT1 is the major transporter responsible for loading the excitatory neurotransmitter glutamate into synaptic vesicles, and is expressed at ribbon synapses in photoreceptors in the OPL and ‘ON’ bipolar cells in the IPL [27]. In our study, VGLUT1 was decreased in the diabetic retina at the mRNA and protein levels, and was not normalised by metformin treatment. As the electroretinogram is a serial waveform corresponding to the activity of different neuronal populations [42], a decrease in VGLUT1 might result in altered rates and amounts of glutamate loading, affecting the magnitude of signal transmission from photoreceptors to bipolar cells and between postreceptoral neurons.
While metformin has been detected in the central nervous system [43], this is the first study to demonstrate its presence in the retina. Metformin was detected at comparable levels with those found in different brain regions of metformin-treated rats [43], indicating that retinal alterations following drug treatment could be caused by a direct effect. We observed the downregulation of complex I subunits in the db/db retina and subsequent normalisation by metformin. Decreases in the ratio of free cytosolic NAD+ to NADH in diabetic retinas have been linked to altered aldose reductase activity [44, 45]; however, as complex I catalyses the transfer of electrons from NADH to coenzyme Q, reduced abundance of complex I subunits could lessen the dehydrogenase ability, resulting in increased free NADH. In contrast to our results, previous work using rat livers found that metformin directly inhibited complex I [46]. The difference in metformin response between these studies could be attributed to experimental design. First, the rats received metformin for 5 days, and were then culled 4 h after treatment [46]; animals in our study were treated for 14 days and culled 18 h later. Therefore, we primarily investigated effects caused by subchronic metformin treatment. Second, disparities in our studies could be due to differences between liver and retinal tissue. That metformin may have varied effects in diverse tissues over time is supported by a recent study in which skeletal muscle samples from type 2 diabetic patients, who had taken their last dose of metformin 12 h before biopsy collection, were found to have normal complex I function [47]. Nonetheless, the effect of metformin within the retina should be examined further.
That metformin treatment did not reverse all diabetic alterations was not unexpected. Previous studies have found that chronic insulin treatment only partially normalised changes in the retinal proteome and transcriptome [31, 48]. In addition, because HbA1c and glycaemic levels were not fully normalised with metformin in this study, longer and/or more vigorous treatment may produce additional differences in retinal proteins. Although glycaemic regulation substantially decreases the risk of developing diabetic complications, the reduction of ophthalmic complications is closely linked to hypertension control, particularly via the renin–angiotensin system [49, 50]. A study examining the effect of angiotensin-1 receptor antagonism on the whole retinal proteome of diabetic mice found that 72% of the proteins altered in diabetes were normalised by treatment, and that these were associated with metabolic and apoptotic abnormalities [34]. The authors did not report whether their animals also received treatment for hyperglycaemia. It would therefore be interesting to examine how the combination of anti-hyperglycaemic and anti-hypertensive treatment affects the diabetic retinal proteome.
In summary, we identified 98 differentially abundant retinal membrane proteins in a mouse model of type 2 diabetes using label-free MS. Network and pathway enrichment analysis indicated defects in proteins involved with electron transport and cell signalling. Metformin treatment partially normalised these changes. Our findings may have relevance to the neuronal dysfunction observed in diabetic retinopathy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 163 kb)
(XLSX 204 kb)
(XLSX 4048 kb)
(PDF 302 kb)
(XLSX 61 kb)
(XLSX 94 kb)
(XLSX 56 kb)
(PDF 415 kb)
(PDF 146 kb)
Acknowledgements
A. Feuchtinger (Institute of Pathology, Helmholtz Zentrum München, Germany) aided the analysis of VGLUT1 immunofluorescence. C. Bobe, N. Senninger and M. Priller (Research Unit Protein Science, Helmholtz Zentrum München, Germany) and A. Seelig and G. Zieglmeier (Institute of Experimental Genetics, Helmholtz Zentrum München, Germany) gave technical assistance. We further thank the animal caretaker crew of the German Mouse Clinic, Neuherberg, Germany.
Funding
This project was funded by the German Center for Diabetes Research (DZD) and the Ministry of Education and Research of the Federal Republic of Germany (BMBF) under project no. 0315494A (SysMBo – Systems Biology of Metabotypes). Funding to the German Mouse Clinic: NGFN-Plus grant no. 01GS0850 and Infrafrontier grant 01KX1012. AL is supported by BMBF grant no. 0315505A (SysTec-Verbund IMAGING).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AL, MFS, JA, MHdA, SN, SMH and MU conceived and designed the experiments. AL, MFS, SZ, CM and JM acquired, analysed and interpreted data. JA, MHdA, SN, SMH and MU contributed reagents, materials or analysis tools. AL, MFS, SZ, CM, JM, JA, MHdA, SN, SMH and MU wrote the paper. All authors have given approval to the final version of the manuscript.
Abbreviations
- 5NTD
5′-Nucleotidase
- ATP2B3
Plasma membrane calcium ATPase 3
- CNGA1
Cyclic nucleotide gated channel alpha 1
- dbM
db/db Mice treated with metformin
- GePS
Genomatix Pathway System
- IPA
Ingenuity Pathway Analysis
- IPL
Inner plexiform layer
- LC
Liquid chromatography
- LC-MS/MS
Liquid chromatography tandem mass spectrometry
- MS
Mass spectrometry
- OPL
Outer plexiform layer
- OPSB
Opsin 1 blue-sensitive
- OPSD
Opsin 2 rhodopsin
- PB
Phosphate buffer
- RDH12
Retinol dehydrogenase 12
- SNG1
Synaptogyrin 1
- S38A3
Sodium-coupled neutral amino acid transporter 3
- VGLUT1
Vesicular glutamate transporter 1
References
- 1.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998;21:1414–1431. doi: 10.2337/diacare.21.9.1414. [DOI] [PubMed] [Google Scholar]
- 2.Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81:4059–4067. doi: 10.1210/jc.81.11.4059. [DOI] [PubMed] [Google Scholar]
- 3.UKPDS Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. doi: 10.1016/S0140-6736(98)07037-8. [DOI] [PubMed] [Google Scholar]
- 4.Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16:434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 5.Perneger TV, Brancati FL, Whelton PK, Klag MJ. End-stage renal disease attributable to diabetes mellitus. Ann Intern Med. 1994;121:912–918. doi: 10.7326/0003-4819-121-12-199412150-00002. [DOI] [PubMed] [Google Scholar]
- 6.Fong DS, Aiello L, Gardner TW, et al. Diabetic retinopathy. Diabetes Care. 2003;26:226–229. doi: 10.2337/diacare.26.1.226. [DOI] [PubMed] [Google Scholar]
- 7.Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366–378. doi: 10.1001/archopht.1961.00960010368014. [DOI] [PubMed] [Google Scholar]
- 8.Di Leo MA, Caputo S, Falsini B, et al. Nonselective loss of contrast sensitivity in visual system testing in early type I diabetes. Diabetes Care. 1992;15:620–625. doi: 10.2337/diacare.15.5.620. [DOI] [PubMed] [Google Scholar]
- 9.Hardy KJ, Lipton J, Scase MO, Foster DH, Scarpello JH. Detection of colour vision abnormalities in uncomplicated type 1 diabetic patients with angiographically normal retinas. Br J Ophthalmol. 1992;76:461–464. doi: 10.1136/bjo.76.8.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizutani M, Gerhardinger C, Lorenzi M. Muller cell changes in human diabetic retinopathy. Diabetes. 1998;47:445–449. doi: 10.2337/diabetes.47.3.445. [DOI] [PubMed] [Google Scholar]
- 11.Bearse MA, Jr, Han Y, Schneck ME, Adams AJ. Retinal function in normal and diabetic eyes mapped with the slow flash multifocal electroretinogram. Invest Ophthalmol Vis Sci. 2004;45:296–304. doi: 10.1167/iovs.03-0424. [DOI] [PubMed] [Google Scholar]
- 12.Phipps JA, Fletcher EL, Vingrys AJ. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest Ophthalmol Vis Sci. 2004;45:4592–4600. doi: 10.1167/iovs.04-0842. [DOI] [PubMed] [Google Scholar]
- 13.Tyrberg M, Lindblad U, Melander A, Lovestam-Adrian M, Ponjavic V, Andreasson S. Electrophysiological studies in newly onset type 2 diabetes without visible vascular retinopathy. Doc Ophthalmol. 2011;123:193–198. doi: 10.1007/s10633-011-9298-6. [DOI] [PubMed] [Google Scholar]
- 14.Ottlecz A, Garcia CA, Eichberg J, Fox DA. Alterations in retinal Na+, K(+)-ATPase in diabetes: streptozotocin-induced and Zucker diabetic fatty rats. Curr Eye Res. 1993;12:1111–1121. doi: 10.3109/02713689309033509. [DOI] [PubMed] [Google Scholar]
- 15.Phipps JA, Yee P, Fletcher EL, Vingrys AJ. Rod photoreceptor dysfunction in diabetes: activation, deactivation, and dark adaptation. Invest Ophthalmol Vis Sci. 2006;47:3187–3194. doi: 10.1167/iovs.05-1493. [DOI] [PubMed] [Google Scholar]
- 16.Yee P, Weymouth AE, Fletcher EL, Vingrys AJ. A role for omega-3 polyunsaturated fatty acid supplements in diabetic neuropathy. Invest Ophthalmol Vis Sc. 2009;51:1755–1764. doi: 10.1167/iovs.09-3792. [DOI] [PubMed] [Google Scholar]
- 17.Kodama H, Fujita M, Yamaguchi I. Development of hyperglycaemia and insulin resistance in conscious genetically diabetic (C57BL/KsJ-db/db) mice. Diabetologia. 1994;37:739–744. doi: 10.1007/BF00404329. [DOI] [PubMed] [Google Scholar]
- 18.Barile GR, Pachydaki SI, Tari SR, et al. The RAGE axis in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:2916–2924. doi: 10.1167/iovs.04-1409. [DOI] [PubMed] [Google Scholar]
- 19.Cheung AK, Fung MK, Lo AC, et al. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes. 2005;54:3119–3125. doi: 10.2337/diabetes.54.11.3119. [DOI] [PubMed] [Google Scholar]
- 20.Xiao C, He M, Nan Y, et al. Physiological effects of superoxide dismutase on altered visual function of retinal ganglion cells in db/db mice. PloS One. 2012;7:e30343. doi: 10.1371/journal.pone.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuchs H, Gailus-Durner V, Neschen S, et al. Innovations in phenotyping of mouse models in the German Mouse Clinic. Mamm Genome. 2012;23:611–622. doi: 10.1007/s00335-012-9415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gailus-Durner V, Fuchs H, Becker L, et al. Introducing the German Mouse Clinic: open access platform for standardized phenotyping. Nat Methods. 2005;2:403–404. doi: 10.1038/nmeth0605-403. [DOI] [PubMed] [Google Scholar]
- 23.Ly A, Yee P, Vessey KA, Phipps JA, Jobling AI, Fletcher EL. Early inner retinal astrocyte dysfunction during diabetes and development of hypoxia, retinal stress, and neuronal functional loss. Invest Ophthalmol Vis Sci. 2011;52:9316–9326. doi: 10.1167/iovs.11-7879. [DOI] [PubMed] [Google Scholar]
- 24.Hauck SM, Dietter J, Kramer RL, et al. Deciphering membrane-associated molecular processes in target tissue of autoimmune uveitis by label-free quantitative mass spectrometry. Mol Cell Proteomics: MCP. 2010;9:2292–2305. doi: 10.1074/mcp.M110.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogt A, Fuerholzner B, Kinkl N, Boldt K, Ueffing M. Isotope coded protein labeling coupled immunoprecipitation (ICPL-IP): a novel approach for quantitative protein complex analysis from native tissue. Mol Cell Proteomics. 2013;12:1395–1406. doi: 10.1074/mcp.O112.023648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTech. 1993;15(532–534):536–537. [PubMed] [Google Scholar]
- 27.Sherry DM, Wang MM, Bates J, Frishman LJ. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. J Comp Neurol. 2003;465:480–498. doi: 10.1002/cne.10838. [DOI] [PubMed] [Google Scholar]
- 28.Quin G, Len AC, Billson FA, Gillies MC. Proteome map of normal rat retina and comparison with the proteome of diabetic rat retina: new insight in the pathogenesis of diabetic retinopathy. Proteomics. 2007;7:2636–2650. doi: 10.1002/pmic.200600486. [DOI] [PubMed] [Google Scholar]
- 29.Wang YD, Wu JD, Jiang ZL, et al. Comparative proteome analysis of neural retinas from type 2 diabetic rats by two-dimensional electrophoresis. Curr Eye Res. 2007;32:891–901. doi: 10.1080/02713680701593702. [DOI] [PubMed] [Google Scholar]
- 30.Fort PE, Freeman WM, Losiewicz MK, Singh RS, Gardner TW. The retinal proteome in experimental diabetic retinopathy: up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics: MCP. 2009;8:767–779. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.VanGuilder HD, Bixler GV, Kutzler L, et al. Multi-modal proteomic analysis of retinal protein expression alterations in a rat model of diabetic retinopathy. PloS One. 2011;6:e16271. doi: 10.1371/journal.pone.0016271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Gehlbach P, Gongora C, et al. A potential role for beta- and gamma-crystallins in the vascular remodeling of the eye. Dev Dyn. 2005;234:36–47. doi: 10.1002/dvdy.20494. [DOI] [PubMed] [Google Scholar]
- 33.Mao YW, Liu JP, Xiang H, Li DW. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- 34.Gao BB, Phipps JA, Bursell D, Clermont AC, Feener EP. Angiotensin AT1 receptor antagonism ameliorates murine retinal proteome changes induced by diabetes. J Proteome Res. 2009;8:5541–5549. doi: 10.1021/pr9006415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao BB, Chen X, Timothy N, Aiello LP, Feener EP. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res. 2008;7:2516–2525. doi: 10.1021/pr800112g. [DOI] [PubMed] [Google Scholar]
- 36.Decanini A, Karunadharma PR, Nordgaard CL, Feng X, Olsen TW, Ferrington DA. Human retinal pigment epithelium proteome changes in early diabetes. Diabetologia. 2008;51:1051–1061. doi: 10.1007/s00125-008-0991-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simo R, Higuera M, Garcia-Ramirez M, Canals F, Garcia-Arumi J, Hernandez C. Elevation of apolipoprotein A-I and apolipoprotein H levels in the vitreous fluid and overexpression in the retina of diabetic patients. Arch Ophthalmol. 2008;126:1076–1081. doi: 10.1001/archopht.126.8.1076. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Ramirez M, Canals F, Hernandez C, et al. Proteomic analysis of human vitreous fluid by fluorescence-based difference gel electrophoresis (DIGE): a new strategy for identifying potential candidates in the pathogenesis of proliferative diabetic retinopathy. Diabetologia. 2007;50:1294–1303. doi: 10.1007/s00125-007-0627-y. [DOI] [PubMed] [Google Scholar]
- 39.Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–3568. [PubMed] [Google Scholar]
- 40.Bobbie MW, Roy S, Trudeau K, Munger SJ, Simon AM. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Invest Ophthalmol Vis Sci. 2010;51:3758–3763. doi: 10.1167/iovs.09-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hancock HA, Kraft TW. Oscillatory potential analysis and ERGs of normal and diabetic rats. Invest Ophthalmol Vis Sci. 2004;45:1002–1008. doi: 10.1167/iovs.03-1080. [DOI] [PubMed] [Google Scholar]
- 42.Rodieck RW. Components of the electroretinogram—a reappraisal. Vis Res. 1972;12:773–780. doi: 10.1016/0042-6989(72)90003-X. [DOI] [PubMed] [Google Scholar]
- 43.Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep: PR. 2010;62:956–965. doi: 10.1016/s1734-1140(10)70357-1. [DOI] [PubMed] [Google Scholar]
- 44.Salceda R, Vilchis C, Coffe V, Hernandez-Munoz R. Changes in the redox state in the retina and brain during the onset of diabetes in rats. Neurochem Res. 1998;23:893–897. doi: 10.1023/A:1022467230259. [DOI] [PubMed] [Google Scholar]
- 45.Obrosova IG, Stevens MJ, Lang HJ. Diabetes-induced changes in retinal NAD-redox status: pharmacological modulation and implications for pathogenesis of diabetic retinopathy. Pharmacology. 2001;62:172–180. doi: 10.1159/000056091. [DOI] [PubMed] [Google Scholar]
- 46.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. doi: 10.1042/0264-6021:3480607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsen S, Rabol R, Hansen CN, Madsbad S, Helge JW, Dela F. Metformin-treated patients with type 2 diabetes have normal mitochondrial complex I respiration. Diabetologia. 2012;55:443–449. doi: 10.1007/s00125-011-2340-0. [DOI] [PubMed] [Google Scholar]
- 48.Bixler GV, Vanguilder HD, Brucklacher RM, Kimball SR, Bronson SK, Freeman WM. Chronic insulin treatment of diabetes does not fully normalize alterations in the retinal transcriptome. BMC Med Genomics. 2011;4:40. doi: 10.1186/1755-8794-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sjolie AK, Klein R, Porta M, et al. Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect 2): a randomised placebo-controlled trial. Lancet. 2008;372:1385–1393. doi: 10.1016/S0140-6736(08)61411-7. [DOI] [PubMed] [Google Scholar]
- 50.Matthews DR, Stratton IM, Aldington SJ, Holman RR, Kohner EM. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol. 2004;122:1631–1640. doi: 10.1001/archopht.122.11.1631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 163 kb)
(XLSX 204 kb)
(XLSX 4048 kb)
(PDF 302 kb)
(XLSX 61 kb)
(XLSX 94 kb)
(XLSX 56 kb)
(PDF 415 kb)
(PDF 146 kb)