

Abstract
Context
Kynurenic acid, a metabolite of the kynurenine pathway of tryptophan degradation, is an antagonist at N-methyl-d-aspartate and α7 nicotinic acetylcholine receptors and modulates glutamate, dopamine, and acetylcholine signaling. Cortical kynurenic acid concentrations are elevated in the brain and cerebrospinal fluid of schizophrenia patients. The proximal cause may be an impairment of kynurenine 3-monooxygenase (KMO), a rate-limiting enzyme at the branching point of the kynurenine pathway.
Objectives
To examine KMO messenger RNA expression and KMO enzyme activity in postmortem tissue from the frontal eye field (FEF; Brodmann area 6) obtained from schizophrenia individuals compared with healthy control individuals and to explore the relationship between KMO single-nucleotide polymorphisms and schizophrenia oculomotor endophenotypes.
Design
Case-control postmortem and clinical study.
Setting
Maryland Brain Collection, outpatient clinics.
Participants
Postmortem specimens from schizophrenia patients (n=32) and control donors (n=32) and a clinical sample of schizophrenia patients (n=248) and healthy controls (n=228).
Main Outcome Measures
Comparison of quantitative KMO messenger RNA expression and KMO enzyme activity in postmortem FEF tissue between schizophrenia patients and controls and association of KMO single-nucleotide polymorphisms with messenger RNA expression in postmortem FEF and schizophrenia and oculomotor endophenotypes (ie, smooth pursuit eye movements and oculomotor delayed response).
Results
In postmortem tissue, we found a significant and correlated reduction in KMO gene expression and KMO enzyme activity in the FEF in schizophrenia patients. In the clinical sample, KMO rs2275163 was not associated with a diagnosis of schizophrenia but showed modest effects on predictive pursuit and visuospatial working memory endophenotypes.
Conclusion
Our results provide converging lines of evidence implicating reduced KMO activity in the etiopathophysiology of schizophrenia and related neurocognitive deficits.
Impairment of the kynurenine pathway (KP) of tryptophan metabolism has been suggested to play a role in the pathophysiology of schizophrenia and related cognitive deficits.1-5 The KP generates 3 neuroactive metabolites with purported links to neuropsychiatric diseases.6-8 These compounds—kynurenic acid (KYNA), 3-hydroxykynurenine, and quinolinic acid—are downstream products of the regulatory enzymes tryptophan 2,3-dioxygenase (TDO), indoleamine 2,3-dioxygenase, and kynurenine 3-monooxygenase (KMO) (Figure 1). The levels of KYNA, an endogenous antagonist of the glycine coagonist (glycineB) site of the glutamatergic N-methyl-d-aspartate receptor (NMDAR) and the α7 nicotinic acetylcholine receptor (α7nAChR),6,9,10 are elevated in the prefrontal cortex and cerebrospinal fluid of schizophrenia patients.1,11-13 This unique receptor affinity profile of KYNA is particularly interesting when viewed against accumulated evidence implicating NMDAR hypofunction in the pathophysiology of schizophrenia14-19 and α7nAChR hypofunction in schizophrenia-related cognitive deficits.20,21 In rats, endogenous KYNA controls extracellular levels of cortical acetylcholine, dopamine, and glutamate22-24 and the firing rate and burst activity of midbrain dopaminergic neurons.25 Remarkably, experimentally increased brain KYNA levels in rats induce deficits in visuospatial working memory, contextual learning, sensory gating, and prepulse inhibition of the acoustic startle reflex.26-29 These neurocognitive deficits are of particular relevance to schizophrenia because they are highly frequent in schizophrenia individuals and their unaffected first-degree relatives and are considered endophenotypes.30,31 Taken together, these findings suggest that cognitive deficits in schizophrenia could be causally related to elevated cortical KYNA concentrations.5
Figure 1.
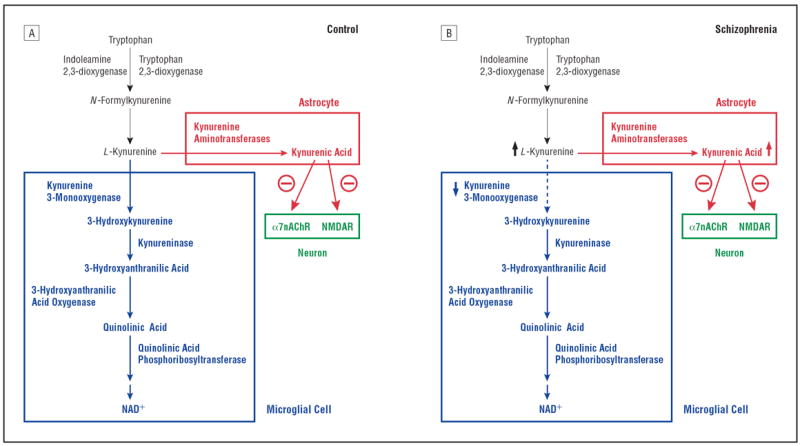
The kynurenine pathway of tryptophan degradation. A, Metabolism is initiated by the oxidative ring opening of tryptophan by indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase. In the brain, the pivotal metabolite kynurenine is enzymatically converted to 3-hydroxykynurenine and kynurenic acid in microglial cells and astrocytes, respectively. B, In schizophrenia, a persistent reduction of microglial kynurenine 3-monooxygenase activity would result in increased kynurenic acid formation in, and release from, astrocytes. This could cause increased inhibition of neuronal α7 nicotinic receptors (α7nAChRs) and N-methyl-d-aspartate receptors (NMDARs) (modified from Wonodi and Schwarcz5). NAD+ indicates nicotinamide adenine dinucleotide.
Recent genetic studies provide support for a role for abnormal KP metabolism in the etiopathophysiology of schizophrenia. These studies include the demonstration of upregulated brain TDO2 (OMIM 191070) messenger RNA (mRNA) (encoding the TDO enzyme) in postmortem tissue from schizophrenia patients32,33 and in the brains of newborn mice experimentally infected with influenza virus (a relevant animal model of schizophrenia34,35), possibly signifying an enhanced ability of the tissue to generate KYNA downstream (Figure 1). Moreover, an association between polymorphisms in the KMO gene (OMIM 603538) (which encodes the KMO enzyme) and schizophrenia was demonstrated in a Japanese cohort.36 This finding was not replicated in an independent sample drawn from the same population,36 nor was an association with schizophrenia demonstrated in a recent report on a European sample.37 These findings are of particular interest because the KMO gene maps to chromosome 1q42-q44, a region that has shown linkage in schizophrenia samples.38
Difficulties replicating genetic findings are not uncommon in schizophrenia, likely due to the polygenic and heterogeneous nature of this complex disorder. This phenotypic and genetic heterogeneity has hindered the search for schizophrenia liability genes using approaches from classical genetics, yielding associations with candidate loci and genes that are rarely replicated.39 As an alternative to using schizophrenia as an end point, one popular approach is to focus on genes that are directly or indirectly related to neurochemical pathways associated with pathogenesis, thus reducing neurochemical and genetic heterogeneity.3,40 Another approach aims to decrease phenotypic and genetic heterogeneity by focusing on disease-associated heritable quantitative traits (ie, endophenotypes).41,42 Using the latter approach, we have studied abnormal smooth pursuit eye movements (SPEMs), an established schizophrenia endophenotype, 43-45 and found that the predictive smooth pursuit component (hereafter referred to as predictive pursuit) is highly heritable.31,46,47 A phylogenetically recent function fully preserved only in primates, SPEM (or eye tracking) is a highly developed behavioral response subserved by a known neuronal network48 and is an important component of the oculomotor response to smoothly moving objects in visual space. In combination with saccadic eye movements, SPEMs capture and maintain the image of a moving object on the fovea as the eyes track it in space and time. In the course of our studies, we have used predictive pursuit to parse differences in small gene effects between schizophrenia groups and healthy control individuals that otherwise would have gone unnoticed using traditional global SPEM measures, clinical diagnosis, or behavioral symptoms.49,50
In this study, we integrated these 2 alternative approaches and investigated the KP and the predictive pursuit endophenotype. Initially, we compared KMO gene expression and KMO enzyme activity in postmortem brain tissue, focusing on the frontal eye field (FEF; Brodmann area 6). On the basis of neuroimaging data from monkeys and humans, this cortical area is a key region associated with SPEMs, and FEF deficits have been consistently found in schizophrenia patients.51-56 Comparisons between postmortem FEF specimens from schizophrenia patients and control samples revealed a significant and correlated downregulation of KMO gene expression and KMO enzyme activity in the tissues from the former compared with the latter. Subsequently, we examined the association of 2 KMO single-nucleotide polymorphisms (SNPs) with gene expression in the FEF tissues and 2 schizophrenia oculomotor endophenotypes in a clinical sample.
METHODS
BRAIN TISSUE FOR GENE EXPRESSION AND BIOCHEMICAL STUDIES
Human postmortem brains (n=64 total), stored at −80°C, were obtained from the Maryland Brain Collection (http://www.mprc.umaryland.edu/mbc.asp). The brains were slowly warmed to −20°C, and the FEF was removed via dissection with a Stryker bone saw from the location denoted by Rosano and colleagues.57
Specimens were obtained from 32 schizophrenia donors (15 had taken first-generation antipsychotic medications, 5 had taken second-generation antipsychotics, 2 had taken combined first-generation antipsychotics and second-generation antipsychotics, 5 had not taken antipsychotics for 6 months or more before death, and 5 were of unknown antipsychotics status at the time of death). Normal control specimens (n=32) were from individuals with no history of psychosis, mood disorders, drug dependence, psychiatric treatment, or hospitalization, as determined by medical examiner documentation and telephone screening of the next of kin. Specimens were selected as case-control pairs and were matched on sex, age, postmortem interval, pH, hemisphere, and freezer storage time (Table 1). Clinical information was obtained through family interviews, including the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-IV). Senior psychiatrists performed best-estimate diagnoses using the Diagnostic Evaluation After Death58 in conjunction with review of past medical and psychiatric records. Postmortem clinical diagnoses were established according to the method developed by Roberts and colleagues.59 All specimens were obtained with informed consent of the legal next of kin with approval from the office of the chief medical examiner of the state of Maryland.
Table 1.
Postmortem Frontal Eye Field Tissue Specimensa
| Study Group | No. of Specimens | Ethnicity of Specimens (EA/AA) No.b | Sex (F/M), No. | Mean (SD)
|
|||
|---|---|---|---|---|---|---|---|
| Age, y | PMI, h | pH | RIN | ||||
| Healthy control individuals | 32 | 20/12 | 5/27 | 46.9 (13.3) | 14.5 (5.8) | 6.63 (0.2) | 6.6 (0.8) |
| Schizophrenia patients | 32 | 21/11 | 5/27 | 44.7 (12.7) | 14.0 (6.9) | 6.70 (0.2) | 7.1 (0.6) |
Abbreviations: AA, African American/mixed ethnicity; EA, European American; PMI, postmortem interval; RIN, RNA integrity number (scale, 1-10).
None of the characteristics differed significantly between the schizophrenia and control groups.
Designates postmortem specimens from donors from the control group and from schizophrenia patients.
ASSOCIATION AND PHENOTYPIC STUDY PARTICIPANTS
Schizophrenia individuals (n=248) were recruited from outpatient research programs at the Maryland Psychiatric Research Center and Baltimore area mental health centers (Table 2). Enrollment was based on consensus diagnosis of schizophrenia derived from all available information, including past medical and psychiatric histories and the SCID-IV (patient version).60 We attempted to match healthy controls (n=228) with the demographic characteristics of our outpatient population via 2 methods. Using public databases, we conducted random telephone screenings to recruit controls by matching them to the age (SD, 3 years), sex, ethnicity, and zip code of recruited patients. The zip code was determined by the residence at the time of the patient’s first psychotic episode. Alternately, if zip codes were unavailable, we used targeted local community media advertisements based on patients’ county of residence. The design was intended to re-create, in the controls, the environment in which patients resided during the premorbid period that led to the first psychotic break. Thus, comparison subjects were drawn from similar geographic areas as the patient group. Because many neurophysiologic measures may be affected by age, particularly in individuals 60 years or older,61 endophenotypic studies were restricted to individuals between ages 18 and 58 years. Healthy controls did not meet DSM-IV criteria for Axis I disorders (confirmed by SCID-IV, nonpatient version) and had no family history of psychosis based on the Family History Research Diagnostic Criteria interview that extended through 3 generations. Respondents were excluded if they had general medical or neurologic conditions that could affect measurement of oculomotor movements, reported substance dependence within 6 months before study enrollment or current substance abuse, or had mental retardation.
Table 2.
Demographic and Endophenotypic Measures of Study Participants
| Demographics | Mean (SD)a
|
P Value | |
|---|---|---|---|
| Healthy Control Participantsb | Schizophrenia Patientsc | ||
| Age, y | 43.3 (14.6) | 45.2 (12.4) | .14 |
| Female sex, % | 47.8 | 26.2 | <.001 |
| Ethnicity (EA/AA), % | 57.9/42.1 | 50.0/50.0 | .19 |
| Predictive pursuit gain | 0.57 (0.22) (n = 144) | 0.48 (0.21) (n = 162) | <.001 |
| Visuospatial working memory, absolute error in degreesd | 1.57 (0.58) (n = 73) | 2.64 (1.83) (n = 82) | <.001 |
Abbreviations: AA, African American/mixed ethnicity; EA, European American.
Data are presented as mean (SD) unless otherwise indicated.
n = 228 unless otherwise indicated.
n = 248 unless otherwise indicated.
On the basis of the Oculomotor Delayed Response Task.
The study sample of 476 individuals included 256 self-identified European Americans and 220 who self-identified as African American/of mixed ethnicity. Completed endophenotypic measurements were available in subgroups as predictive pursuit subcomponent of SPEM (n=306) and oculomotor delayed response (ODR; also known as memory-guided saccade, a visuospatial working memory measure) (n=156).62 All participants gave written informed consent in accordance with the University of Maryland Institutional Review Board guidelines. In schizophrenia patients, the evaluation of the capacity to sign consent was performed to assess their understanding of the study before signing consent.
LABORATORY PROCEDURES
RNA Isolation and Reverse Transcription
We performed RNA isolation and reverse transcription, as previously described.50 Briefly, tissues were homogenized and total RNA extracted using TRIZOL Reagent (Life Technologies Corporation, Carlsbad, California), purified using Qiagen RNeasy mini spin columns (Qiagen, Hilden, Germany), and treated with DNase. The quality of the RNA was confirmed by high-resolution capillary electrophoresis (Agilent Bioanalyzer 2100; Agilent Technologies, Santa Clara, California) and assessed by RNA integrity number (RIN), obtained from the entire Agilent electrophoretic trace with RIN software algorithm (on a scale of 1 to 10, with 1 being the lowest and 10 the highest RNA quality).63 The RNA samples that showed clearly defined, sharp 18S and 28S ribosomal peaks, 28S/18S ratios greater than 1.7, and an RIN of 6.0 or higher were included. Total RNA (1 μg) was used for 20 μL of reverse transcriptase reaction to synthesize complementary DNA (cDNA) (iScript cDNA Synthesis Kit; Bio-Rad Laboratories Inc, Hercules, California).
TaqMan Gene Expression Assays and Real-Time Quantitative Polymerase Chain Reaction
Commercially available assays were used for KMO (Hs00175738_m1) and GAPDH (Hs99999905_m1) (Gene Expression Assays; Life Technologies Corporation). Expression levels of KMO mRNA transcripts were measured with real-time quantitative polymerase chain reaction (PCR) using relative quantitation by comparative CT assay in an ABI 7900HT Fast System with standard 96-well reaction plates normalized to GAPDH (Life Technologies Corporation). The PCR cycle parameters were 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, and 59°C or 60°C for 1 minute. The PCR data were acquired with the Sequence Detection Software, version 2.3 (Life Technologies Corporation), and quantified by relative quantitation analysis. Amplification efficiency was quantified by a standard curve method with serial dilutions of pooled cDNA from the FEF of 4 control samples. For all experiments, the R2 values of the curves, the slopes, and amplification efficiencies were 0.96 to 0.99, −3.48 to −3.78, and 91% to 95%, respectively. Negative controls (ie, those having no-template cDNA) resulted in no detectable signal. All assays were performed in single wells with the target gene and endogenous control genes as triplicates on the same 96-well plate.
KMO Activity
On the day of the assay, tissues were thawed and homogenized (1:5, wt/vol) in ultrapure water. After further dilution (1:5, vol/vol) in 100 mM Tris hydrochloride buffer (pH 8.1) containing 10 mM potassium chloride and 1 mM editic acid, 80 μL of the tissue preparation was incubated for 40 minutes at 37°C in a solution containing 1 mM nicotinamide adenine dinucleotide phosphate, 3 mM glucose-6-phosphate, 1 U/mL glucose-6-phosphate dehydrogenase, 100 μM l-kynurenine, 10 mM potassium chloride, and 1 mM editic acid, in a total volume of 200 μL. Blanks were obtained by including the specific enzyme inhibitor Ro 61-8048 (100 μM; provided by W. Fröstl, MD, Novartis AG, Basel, Switzerland64) in the incubation solution. After centrifugation (16 000g for 15 minutes), 20 μL of the supernatant was applied to a 3-μm high-performance liquid chromatography column (HR-80; 80×4.6 mm; ESA Biosciences Inc, Chelmsford, Massachusetts), using a mobile phase consisting of 1.5% acetonitrile, 0.9% triethylamine, 0.59% phosphoric acid, 0.27 mM editic acid, and 8.9 mM sodium heptane sulfonic acid and a flow rate of 1.0 mL/min. In the eluate, the reaction product, 3-hydroxykynurenine, was detected electrochemically using an HTEC 500 detector (Eicom Corporation, San Diego, California; oxidation potential, +0.5 V). The retention time of 3-hydroxykynurenine was approximately 11 minutes. The protein content of the samples was measured spectrophotometrically using the method developed by Lowry et al.65
Genotyping
The SNP genotyping was performed on genomic DNA isolated with the QIAamp DNA Maxi Kit (Qiagen) from postmortem tissue for the gene expression association studies and from leukocytes obtained from the clinical sample for the endophenotype association studies. Context sequences and assay identification numbers of genotyped KMO SNPs are given in Table 3. For each PCR reaction, 2 ng of genomic DNA was used in a 5-μL reaction mixture (1.3 mM magnesium chloride, 200 μM deoxyribonucleotide mix, 0.25 μM of each primer, 5% dimethyl sulfoxide, and 1.5 U Taq polymerase). The PCR program included denaturation of DNA (5 minutes at 94°C), 30 cycles of 30 seconds at 94°C, 30 seconds at 62°C, and 30 seconds at 72°C. This sequence was followed by a final extension of 10 minutes at 72°C. All PCR reactions were performed using the Bio-rad Multiplatform Thermocycler, which has 4 blocks of 384 wells (Bio-Rad Laboratories Inc).
Table 3.
Effect of KMO Genotype on Endophenotypes in Healthy Control Individuals and Schizophrenia Patients Combined
| Variable |
KMO rs2275163 Genotype, Mean (SD)
|
||
|---|---|---|---|
| TT | CT | CC | |
| Predictive pursuit gaina | 0.51 (0.15) (n = 24) | 0.53 (0.15) (n = 105) | 0.47 (0.13) (n = 157) |
| Visuospatial working memory, absolute error in degreesa,b | 1.99 (2.46) (n = 12) | 1.77 (0.78) (n = 60) | 2.46 (1.65) (n = 84) |
Significant main effects of genotype corrected P < .05.
Based on the Oculomotor Delayed Response Task.
SNP Selection
We selected 2 KMO SNPs for analysis: 1 that results in a nonsynonymous change in KMO gene sequence (KMO rs1053230) and 1 (KMO rs2275163) that was previously reported to be associated with schizophrenia.36 Both SNPs have minor allele frequencies of 0.15 or more in European American and African American/mixed ethnicity populations (http://hapmap.ncbi.nlm.nih.gov/)66 (Table 4).
Table 4.
SNP Genotyping
| Gene Symbol | Chromosome | ABI Assay Identification No. | SNP | Function | RefSNP Alleles | MA | Context Sequence |
|---|---|---|---|---|---|---|---|
| KMO | 1q42-q44 | C_8856260_10 | rs1053230 | Nonsyn | A/G | A | CTACATGTCACCACGATCTTTCCTC[C/T]GCTTGAGAAGACCATGGAACTGGAT |
| KMO | 1q42-q44 | C_16183814_10 | rs2275163a | Intron | C/T | T | CAGAAACCTACATTAGAGCAAAAGT[C/T]TAAGTGGATATTGTGCTGTGAGCAG |
Abbreviations: ABI, Applied Biosystems Inc; MA, minor allele; Nonsyn, nonsynonymous; RefSNP Alleles, National Center for Biotechnology Information reference SNP alleles; SNP, single-nucleotide polymorphism.
Haplotype-tagged SNP is based on a minor allele frequency of ≥0.15.
PHENOTYPIC MEASURES
Oculomotor Measures
Oculomotor measures were scored by investigators masked to the identity and group membership of study participants. Self-reported smokers were requested to refrain from smoking for at least 1 hour before testing.
Smooth Pursuit Eye Movement Task
In the smooth pursuit task, a target starts from a center fixation and moves back and forth across the screen. Participants were instructed to follow the moving target (velocity of 18.7° per second; amplitude, greater or less than 12°) across the computer monitor with their eyes. After 4 to 6 half-cycles, the target was unpredictably masked (ie, made invisible) for 500 milliseconds. Of the 25 trials presented, 15 had the masking appear at some point during a half-cycle and the rest at the change in ramp direction. Participants were instructed to follow the moving target even when it became briefly invisible. Pursuit gain was defined as the ratio of pursuit eye velocity to target velocity. Pursuit gain measures from the masked period from these 2 types of trials were averaged to obtain a measure of predictive pursuit gain. The scoring algorithm of the measures has been fully described elsewhere.46
Oculomotor Delayed Response Task
In this task, participants fixated on a centrally located set of crosshairs. A target was flashed for 250 milliseconds between 2.5° and 10.0° to the left or right of the central fixation point. Participants continued to fixate until the fixation cross was turned off 10 seconds after the target was briefly shown. With this offset of the center crosshairs, participants were instructed to move their eyes to the location where the target had flashed. Feedback was provided by a small circle that appeared after 1.5 seconds, indicating where the target had flashed at the beginning of the trial. Absolute error in degrees was the primary dependent measure.
Statistical Analysis
Postmortem Brain: KMO mRNA Expression, KMO Activity, and SNP Analyses
Measurements of KMO mRNA expression and KMO enzyme activity were performed masked to diagnostic groups. The KMO mRNA expression data were approximately normally distributed (skewness, 0.10; median, 0.83; standard error of skewness, 0.3) and were compared between diagnosis groups using 1-way analysis of variance (ANOVA). Comparisons via ANOVA were performed for KMO activity. Pearson correlational analysis examined the correlation between KMO mRNA expression and KMO activity. In an exploratory framework, we examined the relationship between KMO SNPs and KMO mRNA expression in the FEF tissue samples. Because the sample size was small, we collapsed all minor allele carriers into 1 group (ie, CT/TT for rs2275163 and AG/AA for rs1053230) and compared them with participants with the homozygous major allele (ie, CC for rs2275163 and GG for rs1053230) genotype to assess the effects of genotype on KMO mRNA expression using ANOVA.
Clinical Sample: Endophenotype and Single-Nucleotide Polymorphism Association Analyses
Before analysis, the distribution of genotypes was evaluated for their fit to Hardy-Weinberg equilibrium expectations using the χ2 test. On the basis of the results of the exploratory analyses of KMO SNPs on KMO mRNA expression in the postmortem brain samples, we used multiple logistic regression to assess the association of the KMO rs2275163 SNP genotype with schizophrenia endophenotypes. Cognizant of ethnic differences in allelic frequencies, we determined the minor allele frequency (MAF) in both groups in our sample. Consequently, MAFs were 0.23 and 0.14 in the European American and African American/mixed ethnicity groups, respectively. The MAFs based on case-control status in each group were 0.21 and 0.18 in the European American cases and controls, respectively, and 0.10 and 0.08 in the African American/mixed ethnicity cases and controls, respectively. Thus, the MAFs were in the same direction in cases and controls in both ethnic groups. Analysis of variance was used to compare mean endophenotype scores (ie, predictive pursuit and ODR-visuospatial working memory) between different genotype classes. A subgroup analysis was conducted limited to SNP-endophenotype comparisons between healthy controls and schizophrenia patients who displayed poor predictive pursuit. For these analyses, we considered schizophrenia patients who had mean predictive pursuit values below 0.4025 to have poor predictive pursuit.67 This exploratory analysis was motivated by an attempt to apply this endophenotype in an extreme trait design framework. Smooth pursuit shares a similar neurocognitive construct to working memory,68 which involves higher-order mnemonic processing of transient information in service of a response.69 The FEF neurons controlling predictive pursuit maintain firing when the target sensory information is removed, and their firing rates directly correlate with predictive eye velocity.70 During predictive pursuit, the patient relies on an internal representation of the target velocity information, as occurs during target masking.71 Indeed, preliminary analyses showed that schizophrenia patients who displayed poor eye tracking performed significantly worse than schizophrenia patients who displayed good eye tracking on 4 of 5 neuropsychological domains of cognitive function (data not shown, available on request from the authors). We adjusted the P values for multiple correlated tests using methods described by Conneely and Boehnke,72 using their software program (http://csg.sph.umich.edu/boehnke/p_act.php).
RESULTS
KMO mRNA EXPRESSION
Expression of KMO mRNA in the FEF was significantly reduced (−33%) in schizophrenia compared with control samples (F1,63=8.59; P=.005; Figure 2A). The mean (SD) KMO mRNA expression values observed in FEF tissue from patients who had not been taking medications for 6 months or more before death (0.78 [0.47]; n=5) were not different from those in patients who had been taking medications at the time of death (0.94 [0.36]; n=27).
Figure 2.

KMO messenger RNA (mRNA) expression and kynurenine 3-monooxygenase (KMO) enzyme activity. A, Mean KMO mRNA expression and KMO enzyme activity in frontal eye field (FEF) tissues obtained post mortem from schizophrenia (SZ) patients and healthy control individuals. Both measures are significantly reduced in the FEF of SZ patients: *P<.05, †P=.005 (analysis of variance). Error bars indicate SD; RQ, relative quantitation. B, Scatterplot of Pearson correlation (r= 0.66) between KMO gene expression (RQ values, normalized to GAPDH) and KMO enzyme activity in FEF tissue from 30 healthy controls and 30 SZ patients.
KMO ENZYME ACTIVITY
KMO activity was also significantly reduced (−30%) in schizophrenia compared with control FEFs (F1,61=5.64; P=.02; Figure 2A). The KMO gene expression and enzyme activity in the FEF were significantly correlated (Pearson correlation: combined group r=0.66, P < .001; within groups: schizophrenia, r=0.43; P < .01; and controls, r=0.63; P < .001; Figure 2B). The mean (SD) KMO activity in samples from patients who had not been taking medications for 6 months or more before death (15.70 [8.44] pmol/h per milligram of protein; n=5) was not different from that in patients who had been taking medications (15.62 [8.35] pmol/h per milligram of protein; n=27).
EFFECTS OF KMO SNPs ON KMO mRNA EXPRESSION IN FEF
KMO mRNA levels were slightly higher in schizophrenia samples carrying the rs2275163 minor allele (TT/CT) compared with the major allele homozygotes (ie, CC) (nominal P = .05; effect size=0.61; Figure 3), although this difference did not achieve our threshold for statistical significance. No significant differences were found in expression levels between samples with and without the minor allele at rs1053230 (P = .50; effect size=0.30). No significant genotype effect on KMO activity was found.
Figure 3.

Mean effects of kynurenine 3-monooxygenase (KMO) rs2275163 genotype groups on KMO gene expression (ie, relative quantitation values normalized to GAPDH) in postmortem frontal eye field (FEF) samples. KMO gene expression is compared between carriers of the minor allele (TT and CT) (black bars) and carriers that are homozygous for the major allele (CC) (gray bars) in control and schizophrenia FEF tissue specimens: *P<.05, nominal (analysis of variance). Error bars indicate SD. TT and CT: control specimens: n=18, schizophrenia specimens: n=17; CC: control specimens: n=14, schizophrenia specimens: n=15.
ENDOPHENOTYPE AND SNP ASSOCIATION STUDIES IN THE CLINICAL SAMPLE
Results of SNP genotyping were available for 286 of the 306 study participants who completed predictive pursuit testing and for all 156 study participants who completed ODR testing to measure visuospatial working memory (Table 2 and Figure 4). The KMO rs2275163 genotype was significantly associated with predictive pursuit in the combined group of patients and controls (F2,279=4.32; corrected P < .05). Post hoc comparisons showed lower predictive pursuit in individuals with the CC genotype (mean [SD], 0.47 [0.13]; n=157) compared with those with the CT genotype (0.53 [0.15]; n=105; P < .001; effect size=0.46) and no difference regarding those with TT genotypes (0.51 [0.15]; n=24; effect size=0.31; Figure 4A). The SNP rs2275163 also was associated with ODR performance (F2,149=3.78; corrected P < .05), with post hoc comparisons revealing more visuospatial errors in participants with the CC genotype (2.46° [1.65°]; n=84) than in those with the CT genotype (1.77° [0.78°]; n=60; P < .009; effect size=0.47; Figure 4B) but not different from those with TT genotypes (1.99° [2.46°]; n=12; effect size=0.28).
Figure 4.

Results of single-nucleotide polymorphism (SNP) genotyping. A, Effect of kynurenine 3-monooxygenase (KMO) SNP rs2275163 CC genotype on predictive pursuit function in the combined clinical sample of schizophrenia patients and healthy control individuals. Participants with the CC genotype had significantly worse predictive pursuit function compared with participants with CT or TT genotypes: *P<.003 (analysis of variance post hoc test). B, Effect of KMO SNP rs2275163 CC genotype on visuospatial working memory in the combined clinical sample of schizophrenia patients and healthy control participants. Participants with the CC genotype made significantly more visuospatial (spatial) working memory errors compared with participants with the CT or TT genotypes: *P<.009 (analysis of variance post hoc test).
No association was found between rs2275163 and the diagnosis of schizophrenia (P = .62). Subanalyses based on poor and good predictive pursuit in patients revealed that schizophrenia patients who had poor predictive pursuit (n=53) were less likely to be minor allele carriers than were healthy controls (n=126) (25.0% vs 50.0%; P = .03). In contrast, the proportion of schizophrenia patients who had good predictive pursuit and were minor allele carriers was virtually identical to the proportion of controls with those qualities (49% vs 50%). Age did not differ significantly (P = .70) between healthy controls (43.0 [14.0] years) and schizophrenia individuals who had poor eye tracking (44.3 [11.3] years).
NEUROLEPTIC EXPOSURE
Neuroleptic medication dosing in schizophrenia patients was converted to chlorpromazine equivalent doses, as previously described.73-75 The mean chlorpromazine equivalents for schizophrenia with good eye-tracking performance (760.3 [448.2] mg; n=114) were not significantly different (P = .14) from schizophrenia with poor eye-tracking performance (819.6 [410.2] mg; n=53).
COMMENT
The present study provides the first demonstration, to our knowledge, of reduced KMO gene expression in postmortem cortical samples from schizophrenia patients. Moreover, the correlated reduction of KMO gene expression and KMO activity in FEF samples from schizophrenia patients shown here complements the recent report by some of us76 of significantly decreased KMO activity in the prefrontal cortex of patients and preliminary observation77 of a similar reduction in enzyme activity in the basal ganglia. For several reasons, the differences between patients and controls reported here are unlikely to be artifacts of antemortem antipsychotic medication exposure. First, similar KMO gene expression and KMO enzyme activity were observed in the FEF of schizophrenia patients who had not been taking medications for 6 or more months before death. Second, long-term (ie, 28-day) treatment of rats with risperidone does not affect cortical KMO gene expression (I. Wonodi, K.V. Sathyasaikumar, and R. Schwarcz, unpublished data, available on request from the authors) or KMO enzyme activity.78 Moreover, in postmortem brain studies of schizophrenia patients, total RNA quality rather than exposure to antipsychotic drugs was identified as the most critical factor affecting gene expression,79 and the RNA quality of the tissues used was high (RIN ≥ 6.0; Table 1).62 Finally, although an effect of excessive nicotine exposure due to the high prevalence of smoking in schizophrenia patients80-82 cannot be discounted categorically, it is notable that no effect on KMO gene expression was found in a study83 in which differential expression of more than 200 genes was observed in schizophrenia smokers compared with schizophrenia nonsmokers.
In functional terms, our data suggest that KP flux toward 3-hydroxykynurenine and quinolinic acid might be compromised in several brain regions and possibly throughout the brain of schizophrenia individuals. This implies that the substrate of KMO (ie, kynurenine) might eventually accumulate in hyperphysiologic concentrations, resulting in a shift toward enhanced KYNA formation (Figure 1). Notably, elevations in brain kynurenine and secondary increases in KYNA formation may be accentuated further by increased synthesis of the metabolite in the brain and/or increased kynurenine influx from the circulation secondary to peripheral KP activation.3,32-34,84 Of particular relevance to schizophrenia, redirected cortical KP metabolism toward increased KYNA production is positioned to play a critical role in NMDAR and α7nAChR hypofunction, which are presumed to be causally related to cognitive deficits in the disease (Figure 1).14-21 We are currently in the process of examining KYNA and several other KP metabolites and enzymes as part of a comprehensive follow-up study.
Our SNP association analyses revealed that the KMO CC genotype was associated with neurocognitive endophenotypic deficits (ie, in predictive pursuit and visuospatial working memory) in the clinical sample and revealed a trend toward reduced KMO mRNA expression in FEF tissue from schizophrenia patients in the postmortem sample. Although no significant difference was observed in CC genotype frequencies between healthy controls and the total sample of schizophrenia patients, patients with good predictive pursuit had similar proportions of the CC genotype as controls. Differences only emerged when CC genotype frequencies were compared between healthy controls and schizophrenia patients who displayed poor eye tracking. These results merit further exploration. For example, in a genetic background that includes KMO risk variants, additional environmental and/or genetic “hits” could lead to increased cortical KYNA levels and KYNA-related cognitive impairments. We also note that in a previous study36 that examined a Japanese sample, the CC genotype was nominally less frequent in schizophrenia.
The mechanism by which KMO rs2275163, which maps to intron 9 of the KMO gene, might affect KMO gene expression is unclear. Because it is neither a nonsynonymous coding nor a promoter SNP,85 our findings might indicate susceptibility mutation(s) in linkage disequilibrium with KMO rs2275163 or multiple functional rare variants in synthetic association with the KMO CC genotype.86,87 Extensive molecular characterization, including gene resequencing, is currently underway and may identify the putative causative variant(s) captured by the SNP association in this study. Alternatively, KMO rs2275163 might influence KMO function by posttranscriptional and posttranslational modifications possibly involving micro-RNAs (miRNAs). The latter provides a particularly plausible mechanism because 50% of miRNA genes are located in intronic regions of protein-coding genes and control gene expression post-transcriptionally by regulating mRNA translation or stability via interactions with the 3′ UTRs of the mRNAs.88 So far, however, no miRNAs from the KMO genomic region have been deposited in the miRBase database for chromosome 1 (www.mirbase.org).89
In summary, the present study provides the first evidence of a significantly correlated reduction in KMO gene expression and KMO enzyme activity in postmortem tissue from schizophrenia patients obtained from a cortical region associated with SPEMs and KMO variation affecting predictive pursuit eye movements in a clinical schizophrenia sample. Although the results from our clinical population could provide a plausible explanation for the failure of Aoyama and colleagues36 to replicate a KMO SNP association with schizophrenia, our results also could be explained by ascertainment strategies and/or cryptic population stratification across samples. More generally, our data support the concept that schizophrenia-related endophenotypes could be used to dissect the effects of susceptibility genes of small effect.30,90-93 It follows that extreme-trait designed studies87 focusing on subgroups identified by endophenotypic markers might help resolve inconsistencies across genetic data sets. Therefore, future studies probing the complex interactions between molecular genetic and specific neurochemical perturbations using endophenotypes may advance our understanding of the etiopathophysiology of schizophrenia. This, in turn, could facilitate the development of novel, targeted treatments for cognitive deficits in schizophrenia and possibly in other neuropsychiatric diseases.
Acknowledgments
Funding/Support: This study was supported by grants MH-K12RR023250, MH49826, MH67014, MH075101, MH079172, and MH68580 from the National Institute of Mental Health; the National Alliance for Research on Schizophrenia andDepression; and the VA Capitol Health Care Network Mental Illness Research, Education, and Clinical Center, respectively; the University of Maryland General Clinical Research Center grant M01-RR16500; the National Center for Research Resources; and the National Institutes of Health.
Additional Contributions: We thank our patients and their families who participated in our studies and the families of the deceased who altruistically donated postmortem tissue samples to benefit science. Also, we thank Joy K. Roche, MS, Stephanie Tucker, PhD, and the staff of the Maryland Brain Collection for providing the postmortem brain specimens and Robert Conley, MD, William T. Carpenter, MD, and Carol Tamminga, MD, for postmortem diagnosis and Jing Yin, DDS, Shan Li, MS, and Li Tang, BS, for exceptional technical assistance.
Footnotes
Previous Presentation: This study was previously presented at the Society for Neuroscience Annual Meeting; October 21, 2009; Chicago, Illinois.
Financial Disclosure: None reported.
References
- 1.Schwarcz R, Rassoulpour A, Wu H-Q, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001;50(7):521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- 2.Erhardt S, Schwieler L, Nilsson L, Linderholm K, Engberg G. The kynurenic acid hypothesis of schizophrenia. Physiol Behav. 2007;92(1-2):203–209. doi: 10.1016/j.physbeh.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 3.Müller N, Schwarz M. Schizophrenia as an inflammation-mediated dysbalance of glutamatergic neurotransmission. Neurotox Res. 2006;10(2):131–148. doi: 10.1007/BF03033242. [DOI] [PubMed] [Google Scholar]
- 4.Miller CL, Llenos IC, Cwik M, Walkup J, Weis S. Alterations in kynurenine precursor and product levels in schizophrenia and bipolar disorder. Neurochem Int. 2008;52(6):1297–1303. doi: 10.1016/j.neuint.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Wonodi I, Schwarcz R. Cortical kynurenine pathway metabolism: a novel target for cognitive enhancement in schizophrenia. Schizophr Bull. 2010;36(2):211–218. doi: 10.1093/schbul/sbq002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45(3):309–379. [PubMed] [Google Scholar]
- 7.Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303(1):1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- 8.Pérez-De La Cruz V, Königsberg M, Santamaría A. Kynurenine pathway and disease: an overview. CNS Neurol Disord Drug Targets. 2007;6(6):398–410. doi: 10.2174/187152707783399229. [DOI] [PubMed] [Google Scholar]
- 9.Hilmas C, Pereira EFR, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21(19):7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perkins MN, Stone TW. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982;247(1):184–187. doi: 10.1016/0006-8993(82)91048-4. [DOI] [PubMed] [Google Scholar]
- 11.Erhardt S, Blennow K, Nordin C, Skogh E, Lindström LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001;313(1-2):96–98. doi: 10.1016/s0304-3940(01)02242-x. [DOI] [PubMed] [Google Scholar]
- 12.Nilsson LK, Linderholm KR, Engberg G, Paulson L, Blennow K, Lindström LH, Nordin C, Karanti A, Persson P, Erhardt S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr Res. 2005;80(2-3):315–322. doi: 10.1016/j.schres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Linderholm KR, Skogh E, Olsson SK, Dahl ML, Holtze M, Engberg G, Samuelsson M, Erhardt S. Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr Bull. doi: 10.1093/schbul/sbq086. published online ahead of print August 20, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmüller B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980;20(3):379–382. doi: 10.1016/0304-3940(80)90178-0. [DOI] [PubMed] [Google Scholar]
- 15.Lodge D, Anis NA, Burton NR. Effects of optical isomers of ketamine on excitation of cat and rat spinal neurones by amino acids and acetylcholine. Neurosci Lett. 1982;29(3):281–286. doi: 10.1016/0304-3940(82)90330-5. [DOI] [PubMed] [Google Scholar]
- 16.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 17.Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE, Coyle JT. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry. 1995;52(10):829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- 18.Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harv Rev Psychiatry. 1996;3(5):241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- 19.Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- 20.Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995;38(1):22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- 21.Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R. Association of promoter variants in the α7 nicotine acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry. 2002;59(12):1085–1096. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- 22.Wu HQ, Pellicciari R, Schwarcz R. Bidirectional regulation of extracellular dopamine by endogenous kynurenic acid in the rat medial prefrontal cortex. Abstr Soc Neurosci. 2006;32:624.3. [Google Scholar]
- 23.Wu H-Q, Pereira EFR, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R. The astrocyte-derived α7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci. 2010;40(1-2):204–210. doi: 10.1007/s12031-009-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zmarowski A, Wu H-Q, Brooks JM, Potter MC, Pellicciari R, Schwarcz R, Bruno JP. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur J Neurosci. 2009;29(3):529–538. doi: 10.1111/j.1460-9568.2008.06594.x. [DOI] [PubMed] [Google Scholar]
- 25.Erhardt S, Engberg G. Increased phasic activity of dopaminergic neurones in the rat ventral tegmental area following pharmacologically elevated levels of endogenous kynurenic acid. Acta Physiol Scand. 2002;175(1):45–53. doi: 10.1046/j.1365-201X.2002.00962.x. [DOI] [PubMed] [Google Scholar]
- 26.Chess AC, Simoni MK, Alling TE, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull. 2007;33(3):797–804. doi: 10.1093/schbul/sbl033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chess AC, Landers AM, Bucci DJ. L-kynurenine treatment alters contextual fear conditioning and context discrimination but not cue-specific fear conditioning. Behav Brain Res. 2009;201(2):325–331. doi: 10.1016/j.bbr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Shepard PD, Joy B, Clerkin L, Schwarcz R. Micromolar brain levels of kynurenic acid are associated with a disruption of auditory sensory gating in the rat. Neuropsychopharmacology. 2003;28(8):1454–1462. doi: 10.1038/sj.npp.1300188. [DOI] [PubMed] [Google Scholar]
- 29.Erhardt S, Schwieler L, Emanuelsson C, Geyer M. Endogenous kynurenic acid disrupts prepulse inhibition. Biol Psychiatry. 2004;56(4):255–260. doi: 10.1016/j.biopsych.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Braff DL, Light GA, Swerdlow NR. Prepulse inhibition and P50 suppression are both deficient but not correlated in schizophrenia patients. Biol Psychiatry. 2007;61(10):1204–1207. doi: 10.1016/j.biopsych.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 31.Hong LE, Turano KA, O’Neill H, Hao L, Wonodi I, McMahon RP, Elliott A, Thaker GK. Refining the predictive pursuit endophenotype in schizophrenia. Biol Psychiatry. 2008;63(5):458–464. doi: 10.1016/j.biopsych.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller CL, Llenos IC, Dulay JR, Barillo MM, Yolken RH, Weis S. Expression of the kynurenine pathway enzyme tryptophan 2,3-dioxygenase is increased in the frontal cortex of individuals with schizophrenia. Neurobiol Dis. 2004;15(3):618–629. doi: 10.1016/j.nbd.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Miller CL, Llenos IC, Dulay JR, Weis S. Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res. 2006:1073–1074. 25–37. doi: 10.1016/j.brainres.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 34.Asp L, Holtze M, Powell SB, Karlsson H, Erhardt S. Neonatal infection with neurotropic influenza A virus induces the kynurenine pathway in early life and disrupts sensorimotor gating in adult Tap1-/- mice. Int J Neuropsychopharmacol. 2010;13(4):475–485. doi: 10.1017/S1461145709990253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoyama N, Takahashi N, Saito S, Maeno N, Ishihara R, Ji X, Miura H, Ikeda M, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, Yoshida K, Iwata N, Inada T, Ozaki N. Association study between kynurenine 3-monooxygenase gene and schizophrenia in the Japanese population. Genes Brain Behav. 2006;5(4):364–368. doi: 10.1111/j.1601-183X.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 37.Holtze M, Saetre P, Erhardt S, Schwieler L, Werge T, Hansen T, Nielsen J, Djurovic S, Melle I, Andreassen OA, Hall H, Terenius L, Agartz I, Engberg G, Jönsson EG, Schalling M. Kynurenine 3-monooxygenase (KMO) polymorphisms in schizophrenia: an association study. Schizophr Res. 2011;127(1-3):270–272. doi: 10.1016/j.schres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV, Brzustowicz LM, Kaufmann CA, Garver DL, Gurling HMD, Lindholm E, Coon H, Moises HW, Byerley W, Shaw SH, Mesen A, Sherrington R, O’Neill FA, Walsh D, Kendler KS, Ekelund J, Paunio T, Lönnqvist J, Peltonen L, O’Donovan MC, Owen MJ, Wildenauer DB, Maier W, Nestadt G, Blouin J-L, Antonarakis SE, Mowry BJ, Silverman JM, Crowe RR, Cloninger CR, Tsuang MT, Malaspina D, Harkavy-Friedman JM, Svrakic DM, Bassett AS, Holcomb J, Kalsi G, McQuillin A, Brynjolfson J, Sigmundsson T, Petursson H, Jazin E, Zoëga T, Helgason T. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet. 2003;73(1):34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thaker GK, Carpenter WT., Jr Advances in schizophrenia. Nat Med. 2001;7(6):667–671. doi: 10.1038/89040. [DOI] [PubMed] [Google Scholar]
- 40.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31(5):234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160(4):636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 42.Braff DL, Freedman R, Schork NJ, Gottesman II. Deconstructing schizophrenia: an overview of the use of endophenotypes in order to understand a complex disorder. Schizophr Bull. 2007;33(1):21–32. doi: 10.1093/schbul/sbl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holzman PS, Proctor LR, Hughes DW. Eye-tracking patterns in schizophrenia. Science. 1973;181(95):179–181. doi: 10.1126/science.181.4095.179. [DOI] [PubMed] [Google Scholar]
- 44.Sweeney JA, Luna B, Srinivasagam NM, Keshavan MS, Schooler NR, Haas GL, Carl JR. Eye tracking abnormalities in schizophrenia: evidence for dysfunction in the frontal eye fields. Biol Psychiatry. 1998;44(8):698–708. doi: 10.1016/s0006-3223(98)00035-3. [DOI] [PubMed] [Google Scholar]
- 45.Thaker GK, Ross DE, Cassady SL, Adami HM, LaPorte D, Medoff DR, Lahti A. Smooth pursuit eye movements to extraretinal motion signals: deficits in relatives of patients with schizophrenia. Arch Gen Psychiatry. 1998;55(9):830–836. doi: 10.1001/archpsyc.55.9.830. [DOI] [PubMed] [Google Scholar]
- 46.Thaker GK, Avila MT, Hong EL, Medoff DR, Ross DE, Adami HM. A model of smooth pursuit eye movement deficit associated with the schizophrenia phenotype. Psychophysiology. 2003;40(2):277–284. doi: 10.1111/1469-8986.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong LE, Mitchell BD, Avila MT, Adami H, McMahon RP, Thaker GK. Familial aggregation of eye-tracking endophenotypes in families of schizophrenic patients. Arch Gen Psychiatry. 2006;63(3):259–264. doi: 10.1001/archpsyc.63.3.259. [DOI] [PubMed] [Google Scholar]
- 48.Leigh JR, Zee DS. The Neurology of Eye Movements. Philadelphia, PA: F.A. Davis Company; 1991. [Google Scholar]
- 49.Thaker GK, Wonodi I, Avila MT, Hong LE, Stine OC. Catechol o-methyltransferase polymorphism and eye tracking in schizophrenia: a preliminary report. Am J Psychiatry. 2004;161(12):2320–2322. doi: 10.1176/appi.ajp.161.12.2320. [DOI] [PubMed] [Google Scholar]
- 50.Wonodi I, Hong LE, Stine OC, Mitchell BD, Elliott A, Roberts RC, Conley RR, McMahon RP, Thaker GK. Dopamine transporter polymorphism modulates oculomotor function and DAT1 mRNA expression in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(2):282–289. doi: 10.1002/ajmg.b.30811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gottlieb JP, MacAvoy MG, Bruce CJ. Neural responses related to smooth-pursuit eye movements and their correspondence with electrically elicited smooth eye movements in the primate frontal eye field. J Neurophysiol. 1994;72(4):1634–1653. doi: 10.1152/jn.1994.72.4.1634. [DOI] [PubMed] [Google Scholar]
- 52.Fukushima K, Yamanobe T, Shinmei Y, Fukushima J. Predictive responses of periarcuate pursuit neurons to visual target motion. Exp Brain Res. 2002;145(1):104–120. doi: 10.1007/s00221-002-1088-7. [DOI] [PubMed] [Google Scholar]
- 53.O’Driscoll GA, Wolff A-L, Benkelfat C, Florencio PS, Lal S, Evans AC. Functional neuroanatomy of smooth pursuit and predictive saccades. Neuroreport. 2000;11(6):1335–1340. doi: 10.1097/00001756-200004270-00037. [DOI] [PubMed] [Google Scholar]
- 54.Rosano C, Krisky CM, Welling JS, Eddy WF, Luna B, Thulborn KR, Sweeney JA. Pursuit and saccadic eye movement subregions in human frontal eye field: a high-resolution fMRI investigation. Cereb Cortex. 2002;12(2):107–115. doi: 10.1093/cercor/12.2.107. [DOI] [PubMed] [Google Scholar]
- 55.Hong LE, Tagamets M, Avila M, Wonodi I, Holcomb H, Thaker GK. Specific motion processing pathway deficit during eye tracking in schizophrenia: a performance-matched functional magnetic resonance imaging study. Biol Psychiatry. 2005;57(7):726–732. doi: 10.1016/j.biopsych.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 56.O’Driscoll GA, Benkelfat C, Florencio PS, Wolff A-LVG, Joober R, Lal S, Evans AC. Neural correlates of eye tracking deficits in first-degree relatives of schizophrenic patients: a positron emission tomography study. Arch Gen Psychiatry. 1999;56(12):1127–1134. doi: 10.1001/archpsyc.56.12.1127. [DOI] [PubMed] [Google Scholar]
- 57.Rosano C, Sweeney JA, Melchitzky DS, Lewis DA. The human precentral sulcus: chemoarchitecture of a region corresponding to the frontal eye fields. Brain Res. 2003;972(1-2):16–30. doi: 10.1016/s0006-8993(03)02431-4. [DOI] [PubMed] [Google Scholar]
- 58.Zalcman S, Endicott J. Diagnostic Evaluation After Death. Rockville, MD: National Institute of Mental Health; 1983. [Google Scholar]
- 59.Roberts SB, Hill CA, Dean B, Keks NA, Opeskin K, Copolov DL. Confirmation of the diagnosis of schizophrenia after death using DSM-IV: a Victorian experience. Aust N Z J Psychiatry. 1998;32(1):73–76. doi: 10.3109/00048679809062709. [DOI] [PubMed] [Google Scholar]
- 60.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. New York, NY: New York State Psychiatric Institute, Biometrics Research; 1996. [Google Scholar]
- 61.Ross RG, Olincy A, Harris JG, Radant A, Adler LE, Compagnon N, Freedman R. The effects of age on a smooth pursuit tracking task in adults with schizophrenia and normal subjects. Biol Psychiatry. 1999;46(3):383–391. doi: 10.1016/s0006-3223(98)00369-2. [DOI] [PubMed] [Google Scholar]
- 62.Park S, Holzman PS, Goldman-Rakic PS. Spatial working memory deficits in the relatives of schizophrenic patients. Arch Gen Psychiatry. 1995;52(10):821–828. doi: 10.1001/archpsyc.1995.03950220031007. [DOI] [PubMed] [Google Scholar]
- 63.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Röver S, Cesura AM, Huguenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40(26):4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- 65.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 66.Howie BN, Carlson CS, Rieder MJ, Nickerson DA. Efficient selection of tagging single-nucleotide polymorphisms in multiple populations. Hum Genet. 2006;120(1):58–68. doi: 10.1007/s00439-006-0182-5. [DOI] [PubMed] [Google Scholar]
- 67.Avila MT, Adami HM, McMahon RP, Thaker GK. Using neurophysiological markers of genetic risk to define the boundaries of the schizophrenia spectrum phenotype. Schizophr Bull. 2003;29(2):299–309. doi: 10.1093/oxfordjournals.schbul.a007006. [DOI] [PubMed] [Google Scholar]
- 68.Goldman-Rakic P. Neurobiology: space and time in the mental universe. Nature. 1997;386(6625):559–560. doi: 10.1038/386559a0. [DOI] [PubMed] [Google Scholar]
- 69.Baddeley AD. Working Memory. Oxford, England: Clarendon Press; 1986. [Google Scholar]
- 70.Tanaka M, Fukushima K. Neuronal responses related to smooth pursuit eye movements in the periarcuate cortical area of monkeys. J Neurophysiol. 1998;80(1):28–47. doi: 10.1152/jn.1998.80.1.28. [DOI] [PubMed] [Google Scholar]
- 71.Wells SG, Barnes GR. Fast, anticipatory smooth-pursuit eye movements appear to depend on a short-term store. Exp Brain Res. 1998;120(1):129–133. doi: 10.1007/s002210050385. [DOI] [PubMed] [Google Scholar]
- 72.Conneely KN, Boehnke M. So many correlated tests, so little time! rapid adjustment of P values for multiple correlated tests. Am J Hum Genet. 2007;81(6):1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Woods SW. Chlorpromazine equivalent doses for the newer atypical antipsychotics. J Clin Psychiatry. 2003;64(6):663–667. doi: 10.4088/jcp.v64n0607. [DOI] [PubMed] [Google Scholar]
- 74.Buchanan RW, Kreyenbuhl J, Kelly DL, Noel JM, Boggs DL, Fischer BA, Himelhoch S, Fang B, Peterson E, Aquino PR, Keller W Schizophrenia Patient Outcomes Research Team (PORT) The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71–93. doi: 10.1093/schbul/sbp116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho B-C. Antipsychotic dose equivalents and dose-years: a standardized method for comparing exposure to different drugs. Biol Psychiatry. 2010;67(3):255–262. doi: 10.1016/j.biopsych.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sathyasaikumar KV, Stachowski E, Wonodi I, Roberts RC, Thaker GK, Schwarcz R. Impairment of kynurenine 3-monooxygenase in the frontal cortex of individuals with schizophrenia: association with the eye tracking endophenotype. Abstr Soc Neurosci. 2009;34:747.15. [Google Scholar]
- 77.Schwarcz R, Rassoulpour A, Guidetti P, Conley R, Roberts RC. Abnormal kynurenine pathway metabolism in the striatum of individuals with schizophrenia. Schizophr Bull. 2007;33(2):312. [Google Scholar]
- 78.Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, Schwarcz R. Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr Bull. doi: 10.1093/schbul/sbq112. doi:10-1093/schbul/sbq112. published online ahead of print October 29, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry. 2006;60(6):650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 80.Dalack GW, Healy DJ, Meador-Woodruff JH. Nicotine dependence in schizophrenia: clinical phenomena and laboratory findings. Am J Psychiatry. 1998;155(11):1490–1501. doi: 10.1176/ajp.155.11.1490. [DOI] [PubMed] [Google Scholar]
- 81.Leonard S, Adler LE, Benhammou K, Berger R, Breese CR, Drebing C, Gault J, Lee MJ, Logel J, Olincy A, Ross RG, Stevens K, Sullivan B, Vianzon R, Virnich DE, Waldo M, Walton K, Freedman R. Smoking and mental illness. Pharmacol Biochem Behav. 2001;70(4):561–570. doi: 10.1016/s0091-3057(01)00677-3. [DOI] [PubMed] [Google Scholar]
- 82.Leonard S, Mexal S, Freedman R. Smoking, genetics and schizophrenia: evidence for self medication. J Dual Diagn. 2007;3(3-4):43–59. doi: 10.1300/J374v03n03_05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R, Leonard S. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Brain Res Mol Brain Res. 2005;139(2):317–332. doi: 10.1016/j.molbrainres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Däubener W, Spors B, Hucke C, Adam R, Stins M, Kim KS, Schroten H. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect Immun. 2001;69(10):6527–6531. doi: 10.1128/IAI.69.10.6527-6531.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Halford S, Freedman MS, Bellingham J, Inglis SL, Poopalasundaram S, Soni BG, Foster RG, Hunt DM. Characterization of a novel human opsin gene with wide tissue expression and identification of embedded and flanking genes on chromosome 1q43. Genomics. 2001;72(2):203–208. doi: 10.1006/geno.2001.6469. [DOI] [PubMed] [Google Scholar]
- 86.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 88.Conne B, Stutz A, Vassalli J-D. The 3′ untranslated region of messenger RNA: a molecular ‘hotspot’ for pathology? Nat Med. 2000;6(6):637–641. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]
- 89.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(database issue):D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Louchart-de la Chapelle S, Nkam I, Houy E, Belmont A, Ménard J-F, Roussignol A-C, Siwek O, Mezerai M, Guillermou M, Fouldrin G, Levillain D, Dollfus S, Campion D, Thibaut F. A concordance study of three electrophysiological measures in schizophrenia. Am J Psychiatry. 2005;162(3):466–474. doi: 10.1176/appi.ajp.162.3.466. [DOI] [PubMed] [Google Scholar]
- 91.Kumari V, Ettinger U, Crawford TJ, Zachariah E, Sharma T. Lack of association between prepulse inhibition and antisaccadic deficits in chronic schizophrenia: implications for identification of schizophrenia endophenotypes. J Psychiatr Res. 2005;39(3):227–240. doi: 10.1016/j.jpsychires.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 92.Hong LE, Summerfelt A, Wonodi I, Adami H, Buchanan RW, Thaker GK. Independent domains of inhibitory gating in schizophrenia and the effect of stimulus interval. Am J Psychiatry. 2007;164(1):61–65. doi: 10.1176/ajp.2007.164.1.61. [DOI] [PubMed] [Google Scholar]
- 93.Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7(10):818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]