

Abstract
Ankylosing spondylitis (AS) is a chronic inflammatory disease with complex genetic traits. Multiple sequence variations have been associated with AS, but explained only a proportion of heritability. The studies herein aimed to explore potential associations between genomic copy number variation (CNV) and AS of Han Chinese. Five AS patients were examined with the high-density comparative genomic hybridization (CGH) microarrays in the first screen test for AS associated CNVs. A total of 533 AS patients and 792 unrelated controls were examined in confirmation studies with the AccuCopy assays. A significant association was observed between the CNV of the HLA-DQA1 and AS. Comparing with controls, AS patients showed an aberrant copy number (CN), and significantly increased number of patients had more than 2 copies of the HLA-DQA1. Therefore, CNV of the HLA-DQA1 may play an important role in susceptibility to AS in Han Chinese population.
Introduction
Ankylosing spondylitis (AS) is an immune-mediated complex disease with inflammation of the spine and extra-spinal sites such as the peripheral joints, entheses, eyes, and elsewhere. Although, etiopathogenesis is not fully understood, genetic predisposition is strongly associated with AS. A spectrum of sequence variants including HLA-B27, ERAP1, RUNX3, IL23R, IL12B, PTGER4, CARD9, STAT3, TNFRSF1A, LTBR, TBKBP1, TRADD, ANTXR2 and IL1R2 [1–3] have been associated with heritability of AS. Genetic contribution of whole risk alleles based on genome-wide association studies (GWAS) to AS was estimated as 25.39%, and the majority of which comes from HLA-B27 [1,2].
Recently, CNV in the human genome is increasingly recognized to also play important roles in trait heritability. CNV is another class of genetic variation that alters quantity of gene instead of DNA sequence. Common CNVs include deletion, duplication and insertion that occur in about 15% of the human genome [4]. Specific CNVs may explain a proportion of disease risk in addition to sequence variations, which has been reported in a number of human complex diseases, such as systemic lupus erythematosus (SLE) [5], type 1 diabetes (T1D) [6], human immunodeficiency virus (HIV) acquisition and progression [7] and Alzheimer disease [8].
Like many other human complex diseases, sequence variations of genetic risk may explain only a proportion of trait heritability to AS. The aim of the studies herein was to explore specific CNVs that potentially contribute to genetic susceptibility to AS.
Results
Characteristics of Study Subjects
The median and interquartile (IQR) of age of all AS cases and controls were 32 years (IQR=16) and 60.5 years (IQR=17), respectively. In the first and second cohorts, they were 33 (IQR=18) and 32 (IQR=16) in cases, 64 (IQR=11) and 58 (IQR=17) in controls, respectively.
Male to female ratios were 3.6/1 (77.3% vs. 19.5%) in cases and 0.81/1 (44.7% vs. 55.2%) in controls. In the first and second cohorts, they were 137/43 and 243/61 male/female cases, and 126/99 and 228/338 male/female controls, respectively.
HLA-B27 allele was observed in 90.6% of AS cases. There is no significant association between HLA-B27 positive and age, gender, or CNVs of the tested genomic region.
Analysis of CNV and AS
The CNV studies using CGH microarrays showed aberrant CN at 6p21.32 in majority of AS patients. In particular, aberrant CN of a 42-kb fragment at the region containing the HLA-DRB5 showed in 4 of 5 AS patients, and a 12-kb fragment of the region containing HLA-DQA1 showed in 3 of 5 AS patients. The CGH result for CNV of the DRB5 and DQA1 regions was exampled in Figure 1.
Figure 1.
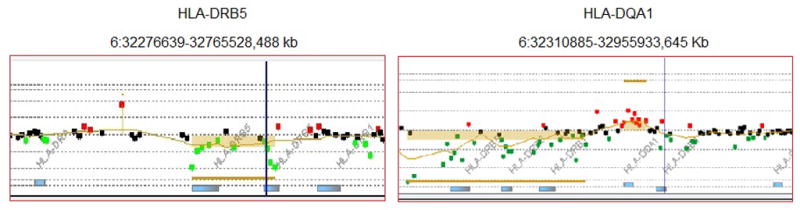
CGH result for CNVs of the DRB5 and DQA1 regions. Red dots indicate increase hybridization signal, green dots represent decreased hybridization signal, and copy number alterations are called and marked with colored bars.
Specific probes were designed for validation assays with the AccuCopy™ technology. Two cohorts were examined in validation assays. The 1st cohort (N=430: 205 cases, 225 controls) showed that CN distributions of HLA-DQA1 alleles among the AS patients and controls are significantly different by Chi-squared test for trend proportions with p-values of 0.0033, but no significant association for HLA-DRB5 was observed (p = 0.78). In particular, higher CN (≥2) of the HLA-DQA1 allele were presented more in AS patients compared to the controls (AS: CN<2 = 4.39%, CN=2 = 88.8% and CN>2 = 6.83% vs. control: CN<2 = 12.9%, CN=2 = 84.4% and CN>2 = 2.67%) (Table 1). In the 2nd cohort (N=895: 328 cases and 567 controls), similar trend in proportions of the CN of HLA-DQA1 was observed (Table 1). In combined analysis of both cohorts, Chi-squared test for trend proportions showed a significant difference between cases and controls (p = 0.00016) (Table 1). A low CN (<2) of the HLA-DQA1 appeared less in AS (p = 0.0016, OR = 0.42, 95% CI = 0.24–0.72). In contrast, A high CN (>2) may be a potential risk for AS (p = 0.047, OR =1.64, 95% CI = 1.0–2.72).
Table 1.
Copy numbers at HLA-DRB5 and HLA-DQA1 of the HLA region in two cohorts
| Cohort 1 | Cohort 2 | Combined cohorts | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CN | <2 | =2 | >2 | p* | CN | <2 | =2 | >2 | p* | CN | <2 | =2 | >2 | p* | |
| DRB5 | Case 205 | 186 | 13 | 6 | 0.7791 | Case 328 | 304 | 18 | 6 | 0.3787 | Case 533 | 490 | 31 | 12 | 0.447 |
| Control 225 | 203 | 14 | 8 | Control 567 | 518 | 33 | 16 | Control 792 | 721 | 47 | 24 | ||||
| DQA1 | Case 205 | 9 | 182 | 14 | 0.0033 | Case 328 | 8 | 299 | 21 | 0.023 | Case 533 | 17 | 481 | 35 | 0.00016 |
| Control 225 | 29 | 190 | 6 | Control 567 | 30 | 512 | 25 | Control 792 | 59 | 702 | 31 | ||||
p-values were based on Chi-squared test for trend proportions; significant association is set at p < 0.05.
Such differences are displayed on the histograms, in which the distribution of CN of the HLA-DQA1 allele in AS patients is switched to the right side (CN>2) of that of controls, while the HLA-DRB5 showed overlapped distribution of CN in cases and controls (Figure 2).
Figure 2.
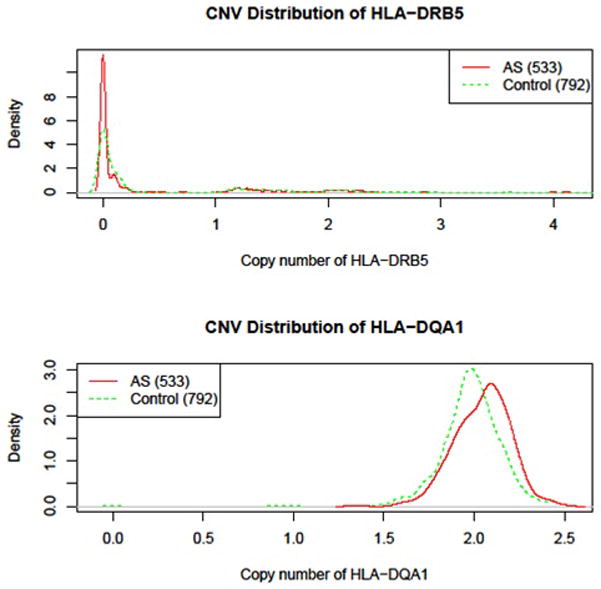
Copy number density of DRB5 and DQA1 in AS cases (red) and controls (green) in the AccuCopy assay. X-axis represents copy number status, Y-axis represents density values of CN distribution. The area under the curve is the proportion of such CNV.
In considering the heterogeneity of gender in study population, we further tested the associations between the CNV and AS using mixed model for logistic regression analysis and linear mixed model with gender as covariates. The results showed that AS association with the CN of the HLA-DQA1 allele became stronger (logistic regression analysis: p = 6.99 × 10−8, OR = 8.72, 95% CI =3.97–19.17; linear mixed model: p < 10−8, Beta = 1.5, 95% CI = 1.3–1.74). The association between AS and the CN of the HLA-DRB5 remained insignificant. In addition, we performed analysis on male and female subgroups for CN of HLA-DQA1 association with AS. The results showed that in male and female subgroups, the achieved p values were 4.75 × 10−5 (OR = 6.55, 95% CI = 2.65–16.19) and 1.88 × 10−4 (OR =19.77, 95% CI =4.13–94.6), respectively.
To examine independency of the association of the HLA-DQA1 from HLA-B27, we performed analysis of HLA-B27 negative cases and controls on available data. The high CN of the HLA-DQA1 (>2) was observed in 5 of 28 B27 negative AS patients (17.9%) and 1 of 43 B27 negative controls (2.3%) (p = 0.022, OR = 9.13, 95% CI = 1.0–82.9).
Discussion
In studies of genetic predisposition to AS, sequence variations have been extensively investigated, and from which multiple genetic susceptibility loci for AS have been identified [14–16]. However, sequence variations may not fully cover the genetic contribution to AS. CNV is another significant source of human genetic variation and attributing cause for disease and population diversity. Recent studies indicated that CNVs of the HLA region are abundant in human genome [17,18]. Our studies demonstrated that aberrant CN of the HLA-DQA1 is strongly associated with AS, which provided first evidence that CNV may also play important roles in heritability of AS.
The HLA-DQA1 is a HLA class II gene encoding an alpha chain of HLA-DQ molecule, along with a beta chain (DQB) to form a heterodimer anchored in membrane of antigen presenting cells (APC). Like other HLA class II molecules, HLA-DQ plays central role in immune response to foreign antigens by presenting specific antigenic peptides to T cells. Genetic variations including sequence and copy number of HLA genes contribute to enhance the recognition repertoire of the immune system, as well as to wide range of disease susceptibility. Specific alleles and gene-dosage of the HLA-DQA1 have been associated with celiac disease [19–21] and type 1 diabetes [21–24]. Although HLA class I region, especially HLA-B27 has been clearly defined as the major genetic factor to AS, the CNV of the HLA-DQA1 identified herein suggested that the HLA class II gene may also play important roles in disease pathogenesis.
Although, the results are appealing and the statistical p-values of which greatly exceed random expectation, there are still several limitations presented in the studies. Only five patients were examined in genome-wide CNV screen with CGH arrays, which would potentially miss other important genomic regions in association with AS, as well as it might result in false positive at discovery phase. A typical example is that deletion of the HLA-DRB5 appeared more in AS patients in the CNV screen with CGH arrays. However, it was not confirmed in the validation studies, which suggests that interpretation of small number of cases on the CGH arrays should be cautious. The findings observed in the studies are limited in AS patients of Chinese Han population. Further confirmation studies, especially in other ethnic populations are necessary. In addition, biological functions of the CNV of the HLA-DQA1 have not been studied. Whether it impacts on presenting specific antigenic peptides to T cells, and induces or accelerates an immune response would be interesting to know.
Nonetheless, this is the first report in studies of CNVs in AS. The results suggest that the CN of the HLA-DQA1 allele is strongly associated with susceptibility to AS in Chinese Han population. Further replication and functional studies of this finding will be necessary.
Materials and Methods
Subjects
A total of 533 AS patients and 792 unrelated controls of Chinese Han were examined in the studies. AS patients and controls were enrolled from Shanghai and Taizhou cities in China. First 205 patients were enrolled from year 2010 to 2011, and were studied earlier as the 1st cohort. The remaining 328 patients were enrolled from year 2012 to 2013, and were examined later as 2nd cohort. All patients met the modified New York diagnostic criteria for AS [9]. Controls were selected on the base of without history of immune-mediated diseases. All participants signed informed consent. The studies were approved by Ethic Committee of both Fudan University and the University of Texas Medical School at Houston.
Genome-Wide Copy Number Variation Analysis
Five AS patients were examined by Agilent SurePrint G3 Human CGH Microarrays (1x1M) following the manufacturer’s protocol. Commercial genomic DNA (Promega, Madison, WI, USA) was used as the internal controls. Briefly, genomic DNA of each subject was treated with restriction digestion, and then was labeled with ULS-Cy5 (for patients) and ULS-Cy3 (for sex-matched controls). The labeled products of one patient and control were mixed and hybridized to the array for 40 hours at 60°C. Then, the array was washed and scanned on an Agilent Microarray Scanner. The data was extracted by Agilent Feature Extraction 10.7.3.1 and analyzed by Agilent Workbench 7.0. ADM-2 was used as statistical algorithm with p-value threshold of 0.05. Copy number (CN) gains or losses of at least five consecutive oligomers on the array were selected for further analysis. The genome-wide common CNVs in human populations were adopted from the public data via Database of Genomic Variants (http://projects.tcag.ca/variation/) [10] and our private Chinese CNV data [11]. Genes present in these common aberrations regions (refer to as copy number variation regions, CNVRs) were identified using human genome browser at UCSC [12].
AccuCopy™ Technology for CNVs Validation
Two sets of primers were designed to examine CNVs of two regions (HLA-DRB5 and HLA-DQA1) at 6p21.32 selected from the results of the CGH arrays. The primer sequences for these two regions are: 5′-GAGCGGGTGCGGTTCCTGC-3′/5′-ACAGCGACGTGGGGGAGTAC-3′ and 5′-GGTCACAGTGTTTTCCAAGTCTCCC-3′/5′-TATGACTGCAAGGTGGAGCACTG-3′. For the HLA-DRB5 and HLA-DQA1 regions, the probes cover human chromosomes (Chr; genome assembly of UCSC hg19) chr6:32489824-32489901 and chr6:32609759-32609998, respectively.
A total of 205 AS cases and 225 controls were examined as the 1st cohort, and then 328 cases and 567 controls were examined as the 2nd cohort for validation of specific CNVs identified from the array-based CGH studies with the AccuCopy™ assay following manufacturer’s protocol [13]. Briefly, genomic DNA of each subject was mixed with fluorescence-labeled specific primers, PCR Master mix and a competitive DNA with known copy number for a multiple competitive real-time PCR reaction. The PCR products were diluted, then were loaded on an ABI 3730XL sequencer for quantification analysis. Raw data were analyzed by Gene Mapper 4.0. The HG19 was used for the genome build for the genomic coordinates. The peak ratio between sample DNA and corresponding competitive DNA (S/C) was calculated and then normalized to the median of four preset two-copy reference genes respectively. Two normalized S/C ratios were further normalized to the median value in all samples for each reference gene and then averaged. The copy number of each target fragment was determined by the average S/C ratio times two. Cases and controls were examined and read at the same time to minimize non-random errors.
HLA-B sequence typing
The HLA-B genotyping was performed with sequence-based typing (SBT) method using SeCore Kits (Life Technologies, USA). Briefly, the allele-specific polymerase chain reactions (PCR) were performed using primers supplied in the SeCore kits, and then were followed by sequencing exon 2 and 3 of the HLA-B gene. The HLA SBT uTYPE 6.0 program (Life Technologies) was used in sequencing analysis and assigning HLA-B alleles.
Association analysis
Median and inter-quartile range (IQR) were used to describe the distribution of the epidemiological variables in the studies. The distributions of copy numbers between patients and controls after copy number assignment according to the pre-defined threshold were compared using Chi-squared test for trend in proportions with R. Logistic regression models were constructed to determine the odds ratio (OR) and 95% confidence intervals (CI) in the condition of adjusting for gender using SPSS (version 17.0, SPSS Inc., 2008). Thresholds for deletions and duplications were empirically set at below 1.75 and above 2.35, respectively, in above CNV validation assays according to the manufacture’s instruction [13]. All samples were tested in duplicates.
Acknowledgments
The studies were supported by research grants from the National Basic Research Program (2012CB944600), International S&T Cooperation Program of China (2013DFA30870), the Science and Technology Committee of Shanghai Municipality (11410701800, 11DJ1400102), Ministry of Science and Technology (2011BAI09B00), Ministry of Health (201002007) and the US NIH NIAID UO1, 1U01AI09090.
Footnotes
Disclosure statement: The authors have declared no conflicts of interest.
References
- 1.Reveille JD, Brown MA. Epidemiology of ankylosing spondylitis: IGAS 2009. J Rheumatol. 2010;37:2624–2625. doi: 10.3899/jrheum.100891. [DOI] [PubMed] [Google Scholar]
- 2.Thomas GP, Brown MA. Genomics of ankylosing spondylitis. Discov Med. 2010;10:263–271. [PubMed] [Google Scholar]
- 3.Brown MA. Genomewide screens in ankylosing spondylitis. Adv Exp Med Biol. 2009;649:148–158. doi: 10.1007/978-1-4419-0298-6_11. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Yang T, Wang L, Yan H, Zhang Y, Guo Y, et al. Whole genome distribution and ethnic differentiation of copy number variation in Caucasian and Asian populations. PLoS One. 2009;4:e7958. doi: 10.1371/journal.pone.0007958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grayson BL, Smith ME, Thomas JW, Wang L, Dexheimer P, Jeffrey J, et al. Genome-wide analysis of copy number variation in type 1 diabetes. PLoS One. 2010;5:e15393. doi: 10.1371/journal.pone.0015393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shostakovich-Koretskaya L, Catano G, Chykarenko ZA, He W, Gornalusse G, Mummidi S, et al. Combinatorial content of CCL3L and CCL4L gene copy numbers influence HIV-AIDS susceptibility in Ukrainian children. AIDS. 2009;23:679–688. doi: 10.1097/QAD.0b013e3283270b3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swaminathan S, Kim S, Shen L, Risacher SL, Foroud T, Pankratz N, et al. Genomic Copy Number Analysis in Alzheimer’s Disease and Mild Cognitive Impairment: An ADNI Study. Int J Alzheimers Dis. 2011;2011:729478. doi: 10.4061/2011/729478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mau W, Zeidler H, Mau R, Majewski A, Freyschmidt J, Stangel W, et al. Evaluation of early diagnostic criteria for ankylosing spondylitis in a 10 year follow-up. Z Rheumatol. 1990;49:82–87. [PubMed] [Google Scholar]
- 10.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 11.Lou H, Li S, Yang Y, Kang L, Zhang X, Jin W, et al. A map of copy number variations in Chinese populations. PLoS One. 2011;6:e27341. doi: 10.1371/journal.pone.0027341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Feuk L, Duggan GE, Khaja R, Scherer SW. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 13.Du R, Lu C, Jiang Z, Li S, Ma R, An H, et al. Efficient typing of copy number variations in a segmental duplication-mediated rearrangement hotspot using multiplex competitive amplification. J Hum Genet. 2012;57:545–551. doi: 10.1038/jhg.2012.66. [DOI] [PubMed] [Google Scholar]
- 14.Lin Z, Bei JX, Shen M, Li Q, Liao Z, Zhang Y, et al. A genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitis. Nat Genet. 2012;44:73–77. doi: 10.1038/ng.1005. [DOI] [PubMed] [Google Scholar]
- 15.Australo-Anglo-American Spondyloarthritis Consortium (TASC) Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. 2010;42:123–127. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MA, Pile KD, Kennedy LG, Campbell D, Andrew L, March R, et al. A genome-wide screen for susceptibility loci in ankylosing spondylitis. Arthritis Rheum. 1998;41:588–595. doi: 10.1002/1529-0131(199804)41:4<588::AID-ART5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 17.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 19.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169:345–350. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray JA, Moore SB, Van Dyke CT, Lahr BD, Dierkhising RA, Zinsmeister AR, et al. HLA DQ gene dosage and risk and severity of celiac disease. Clin Gastroenterol Hepatol. 2007;5:1406–1412. doi: 10.1016/j.cgh.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petronzelli F, Multari G, Ferrante P, Bonamico M, Rabuffo G, Campea L. Different dose effect of HLA-DQ alpha beta heterodimers in insulin-dependent diabetes mellitus and celiac disease susceptibility. Hum Immunol. 1993;36:156–162. doi: 10.1016/0198-8859(93)90119-l. [DOI] [PubMed] [Google Scholar]
- 22.Todd JA, Fukui Y, Kitagawa T, Sasazuki T. The A3 allele of the HLA-DQA1 locus is associated with susceptibility to type 1 diabetes in Japanese. Proc Natl Acad Sci U S A. 1990;87:1094–1098. doi: 10.1073/pnas.87.3.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen L, Wong FS, Tang J, Chen NY, Altieri M, David C, et al. In vivo evidence for the contribution of human histocompatibility leukocyte antigen (HLA)-DQ molecules to the development of diabetes. J Exp Med. 2000;191:97–104. doi: 10.1084/jem.191.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Britten AC, Mijovic CH, Barnett AH, Kelly MA. Differential expression of HLA-DQ alleles in peripheral blood mononuclear cells: alleles associated with susceptibility to and protection from autoimmune type 1 diabetes. Int J Immunogenet. 2009;36:47–57. doi: 10.1111/j.1744-313X.2008.00823.x. [DOI] [PubMed] [Google Scholar]