

This study reports the crystallization of a new nucleotide state of the ATPase domain of a bacterial transcription activator NtrC1 from the hyperthermophilic bacterium Aquifex aeolicus. Wild-type NtrC1 ATPase domain was crystallized in the presence of the ATP analog ADP–BeFx–Mg and the crystals diffracted anisotropically to at best 3.2, 5.2 and 3.2 Å resolution in the a*, b* and c* directions, respectively.
Keywords: enhancer-binding protein, σ54 transcription activator, AAA+ ATPases, NtrC1, Aquifex aeolicus
Abstract
One way that bacteria regulate the transcription of specific genes to adapt to environmental challenges is to use different σ factors that direct the RNA polymerase holoenzyme to distinct promoters. Unlike σ70 RNA polymerase (RNAP), σ54 RNAP is unable to initiate transcription without an activator: enhancer-binding protein (EBP). All EBPs contain one ATPase domain that belongs to the family of ATPases associated with various cellular activities (AAA+ ATPases). AAA+ ATPases use the energy of ATP hydrolysis to remodel different target macromolecules to perform distinct functions. These mechanochemical enzymes are known to form ring-shaped oligomers whose conformations strongly depend upon nucleotide status. Here, the crystallization of the AAA+ ATPase domain of an EBP from Aquifex aeolicus, NtrC1, in the presence of the non-hydrolyzable ATP analog ADP–BeFx is reported. X-ray diffraction data were collected from two crystals from two different protein fractions of the NtrC1 ATPase domain. Previously, this domain was co-crystallized with ADP and ATP, but the latter crystals were grown from the Walker B substitution variant E239A. Therefore, the new data sets are the first for a wild-type EBP ATPase domain co-crystallized with an ATP analog and they reveal a new crystal form. The resulting structure(s) will shed light on the mechanism of EBP-type transcription activators.
1. Introduction
ATPases associated with various cellular activities (AAA+ ATPases) use the energy of ATP hydrolysis to remodel different target macromolecules to perform various functions (White & Lauring, 2007 ▶; Neuwald et al., 1999 ▶; Tucker & Sallai, 2007 ▶; Snider & Houry, 2008 ▶; Erzberger & Berger, 2006 ▶). EBPs (enhancer-binding proteins) belong to a subgroup of the AAA+ ATPases family that modifies σ54 RNA polymerase (RNAP) to assist transcription initiation via remodeling the σ54 factor (Wedel & Kustu, 1995 ▶; Popham et al., 1989 ▶; Morett & Segovia, 1993 ▶).
Most often the AAA+ ATPase in an EBP is flanked by a regulatory domain at the N-terminus of the protein and a DNA-binding domain at the C-terminus (Studholme & Dixon, 2003 ▶; Buck et al., 2006 ▶; Morett & Segovia, 1993 ▶). Like other AAA+ ATPases, EBPs self-assemble to form oligomeric rings (Rippe et al., 1998 ▶; De Carlo et al., 2006 ▶; Doucleff et al., 2005 ▶; Rappas et al., 2005 ▶; Schumacher et al., 2004 ▶; Wedel et al., 1990 ▶; Wyman et al., 1997 ▶; Flashner et al., 1995 ▶; Porter et al., 1993 ▶). The function of the N-terminal regulator domain is to regulate assembly of the functional EBP oligomer in response to a particular signal. Such regulation allows bacteria to activate the transcription of a particular gene under specific conditions. For example, the receiver domain of the NtrC protein is phosphorylated by NtrB kinase under nitrogen limitation. Phosphorylated NtrC assembles into an active hexamer to promote the transcription of glutamine synthetase and other genes in the NtrC region (Wedel & Kustu, 1995 ▶; De Carlo et al., 2006 ▶; Magasanik, 1988 ▶; Weiss & Magasanik, 1988 ▶). The C-terminal DNA-binding domain of an EBP brings the activator into close proximity of the specific promoter from which the σ54 RNAP will start transcription (Wedel et al., 1990 ▶; Ronson et al., 1987 ▶; Nixon et al., 1986 ▶; Cannon et al., 2000 ▶; Buck et al., 1987 ▶; Su et al., 1990 ▶). This domain possesses a helix–turn–helix fold found in many DNA-binding proteins (Ray et al., 2002 ▶; Pelton et al., 1999 ▶; Vidangos et al., 2013 ▶).
ATP hydrolysis by an EBP frees energy which is required for isomerization of the ‘closed’ σ54 RNAP–promoter complex (Wedel & Kustu, 1995 ▶; Ninfa et al., 1987 ▶). The functional linkage between the ATPase activity of an EBP and melting of the DNA promoter is not well understood. ATP binding is required for an EBP to interact with the σ54 factor (Chaney et al., 2001 ▶) and ATP hydrolysis is necessary for promoter melting (Weiss et al., 1991 ▶).
The AAA+ ATPase domain of the EBP itself contains two conserved stem loops, L1 and L2, that are important for the interaction with σ54 RNAP (Wang et al., 1997 ▶; Bordes et al., 2004 ▶; De Carlo et al., 2006 ▶; Schumacher et al., 2007 ▶; Dago et al., 2007 ▶; Chen et al., 2007 ▶; Zhang et al., 2009 ▶; González et al., 1998 ▶). Loop L1 bears a GAFTGA sequence motif that is strictly conserved among EBPs. Based on structural and functional studies it is hypothesized that progression through the catalytic cycle causes conformational changes of the GAFTGA-bearing loops within the ATPase oligomer that allow σ54 to bind to the activator (Wang & Hoover, 1997 ▶; Wang et al., 1997 ▶, 2003 ▶; González et al., 1998 ▶; Zhang et al., 2009 ▶; Schumacher et al., 2007 ▶; Bordes et al., 2004 ▶; Dago et al., 2007 ▶; Chen et al., 2007 ▶, 2010 ▶; Chaney et al., 2001 ▶). Consequently σ54 is thought to be remodeled by some other structural change within the EBP and such remodeling allows the σ54 factor to melt the promoter (Hong et al., 2009 ▶; Bose et al., 2008 ▶).
Nucleotide-binding pockets in the AAA+ ATPase ensembles are located at the intersubunit interfaces. As a consequence the overall ring assembly is highly sensitive to the presence of nucleotide. For the NtrC1 ATPase domain two oligomeric states have been established, thought to be inactive dimer and active heptameric rings with either ADP or ATP occupying every intersubunit interface (Chen et al., 2010 ▶; Lee et al., 2003 ▶). The most recent ATP-bound structure was obtained by co-crystallization of an NtrC1 variant that was unable to hydrolyze ATP but was able to bind nucleotide and to respond with conformational changes and binding to σ54 similar to that of the wild-type protein (Chen et al., 2010 ▶). The differences between those highly symmetric ring structures gave rise to a model of rigid-body roll in which the L1/L2 loops undergo a highly cooperative concerted movement relative to the rest of the ATPase domain. However, biochemical evidence suggests that heterogeneity exists in the nucleotide occupancies of an optimally functioning EBP (Chen et al., 2010 ▶; Schumacher et al., 2006 ▶). To date, there is no known structure of a wild-type NtrC1 ATPase in complex with ATP. Therefore, we co-crystallized the ATPase domain with the ground-state ATP analog ADP–BeFx–Mg. Data sets were collected for two crystals obtained from two different fractions of the NtrC1 ATPase: the so-called ‘S’ and ‘Q’ fractions. Previously we showed that the Q fraction contains nucleotides that are tightly bound to a fraction of the NtrC1 ATPase domain, while the S fraction is nucleotide free (Chen et al., 2009 ▶). The crystals of the S fraction showed mild anisotropy, diffracting to about 3.6 Å resolution. The other crystal (Q crystal) was strongly anisotropic, diffracting to 3.2, 5.2 and 3.2 Å resolution in the a*, b* and c* directions, respectively.
2. Materials and methods
2.1. Expression and purification of the NtrC1 ATPase domain
The NtrC1 ATPase domain (GenBank AAC07159.1; amino acids 137–384) was overexpressed in Escherichia coli strain Rosetta2(DE3) (Novagen) as described by Chen et al. (2007 ▶). Two different fractions of the NtrC1 AAA+ ATPase domain were purified: Q and S fractions, as described by Chen et al. (2009 ▶). Briefly, for purification of the S fraction 40–50 g of cell paste were suspended in 200 ml of ice-cold lysis buffer. The cells, submerged in ice water, were sonicated for ten 30 s bursts at full power (large horn, Artek Sonic Dismembrator 150). Subsequently, the lysate was cleared by centrifugation for 30 min at 100 000g (31 krev min−1 in a Beckman Ti70 rotor, r max of 91.9 mm) and heated to 343 K in the presence of 5 mM TCEP for 30 min. The solution was mixed two to three times during the heating. The cleared heated lysate was subjected to ammonium sulfate precipitation at 80% saturation for 1 to 2 h at room temperature or overnight at 277 K. The precipitate was captured by centrifugation for 30 min at 31 krev min−1, redissolved in buffer consisting of 20 mM Tris–HCl pH 7.9, 5%(w/v) glycerol and then dialyzed against the same buffer overnight at room temperature.
The dialyzed protein was applied onto a HiTrap S column (GE Healthcare) and the NtrC1 protein was eluted with a KCl gradient in the loading buffer, eluting from the column at between 0.15 and 0.30 M KCl. The final preparation of protein was dialyzed against 20 mM Tris–HCl pH 7.9, 200 mM KCl, 5%(w/v) glycerol supplemented with 5 mM TCEP and frozen at 193 K until use. The Q fraction was purified by substituting the S column in that protocol with a HiTrap Q column (GE Healthcare) at 277 K. The protein yield was typically 1–2% of the cell mass with a final purity of greater than 99% as determined by SDS–PAGE. To confirm the ability of the ATPase to respond to binding of ATP analog by assembling to a higher order oligomer that was capable of binding to σ54 protein, size-exclusion chromatography was performed using a Superdex 200 column (GE Healthcare) as described by Chen et al. (2007 ▶, 2010 ▶).
2.2. Preparation of the ATP analog
The ATP metal fluoride analog ADP–BeFx–Mg was prepared from 0.1 M ADP, 1 M BeCl2, 1 M NaF and 1 M MgCl2. ADP (Sigma–Aldrich) was dissolved in 20 mM Tris–HCl pH 7.9, 200 mM KCl buffer to create a 0.1 M stock solution for which the pH was brought to between 6 and 7 by the addition of 10 M and 1 M KOH. Aliquots of 0.1 M ADP were frozen and stored at 193 K. Dissolving BeCl2 in too little water can cause the water to boil, so we poured the final volume of water on crystals of the salt in a fume hood, swirling to evenly distribute the released heat throughout the solution. NaF is not soluble at such concentrations, but makes a fine slurry that was readily suspended and pipetted into solutions as needed. Stocks of the metal fluorides with concentrations higher than 1 mM were prepared with the same ratio of the components: 1:1:8:1 ADP:BeCl2:NaF:MgCl2. The concentrations of MgCl2 and NaF were kept at a 5 mM excess over the amount of BeCl2 to assure excess Mg2+ ion and optimal free F− ions to favor formation of the ternary complex ion BeF3 − over the other possible complex metal ions (Mesmer & Baes, 1969 ▶; Bigay et al., 1987 ▶).
Since the composition of the mixture is not strictly known, we use the notation ADP–BeFx throughout the text for the description of the ATP analog in solution. Prior to use, an aliquot of ADP was thawed and warmed to room temperature. A higher pH of the ADP stock or a low temperature of the solutions caused increased precipitation while forming the metal fluoride ATP analog. The identity of the precipitate was not known to us, but it was always present in variable amounts. The buffer composition and the order of mixing were also crucial for preparing the metal fluoride analog. Firstly, ADP stock was added to buffer solution (pH 7.9) being stirred at room temperature. Secondly, quickly following the nucleotide, the required amounts of freshly prepared mixtures of BeCl2 and then NaF were added. Some precipitation was sometimes observed during this mixing and the pH of the solution dropped by two or three units upon the addition of BeCl2 and rose back upon the addition of NaF. The last component added was MgCl2 and this was followed by more obvious precipitation. The solution was clarified by passing it through a 0.2 µm cellulose acetate filter. In parallel studies with AlCl3 in lieu of BeCl2, it was always noted that the aluminium fluoride analog ADP–AlFx–Mg gave more precipitation than did the beryllium-based analog. In some cases stock solutions with up to a 40 mM concentration of ADP–BeFx–Mg were prepared and these were stable for several hours.
2.3. Crystallization of the NtrC1 protein in the presence of the ATP analog
Crystallization of the NtrC1 protein (S fraction) was screened under oil using the Hampton batch screen sets I and II (Hampton Research, Aliso Viejo, California, USA). One condition with 25%(v/v) ethylene glycol as the precipitant gave diffracting crystals. Crystal growth was refined by varying the concentration of the precipitant and varying the amount of nucleotide analog in the protein solution in the hanging-drop diffusion evaporation method, mixing 2 µl protein solution and 2 µl precipitant solution. The best conditions were 20% ethylene glycol with 300–500 µM ADP–BeFx–Mg in the initial protein solution [20 g l−1 protein in 20 mM Tris–HCl pH 7.9, 200 mM KCl, 5%(w/v) glycerol, 5 mM TCEP]. The crystals obtained were mainly polycrystallites of multiple thin plates, sometimes apparently joined into bulk crystals (Fig. 1a ▶). Sporadically some drops produced visibly clear and massive crystals which were extremely sensitive to a mechanical stress and produced highly anisotropic diffraction with diffuse spots. In an attempt to improve the crystal quality we reproduced the growth conditions for the Q fraction of the NtrC1, with only slight improvement of the diffraction quality.
Figure 1.

Crystals of the AAA+ ATPase domain of the NtrC1 activator grown in the presence of ADP–BeFx–Mg. (a) Representative example of NtrC1 crystals grown in the presence of the ATP analog and ethylene glycol. (b) Example of the darkening observed upon exposure of some of the NtrC1 crystals to the X-ray beam. Arrows show the tracks of several exposures at different regions of the crystal cooled in a 0.4 mm loop.
2.4. Preliminary X-ray analysis of the NtrC1 crystals and data collection
The S-fraction crystals were flash-cooled directly from the crystallization drops into liquid nitrogen and were sent to beamline 8.3.1 at the Advanced Light Source, Berkeley, California, USA for remote data collection using an ADSC Q-315 charge-coupled device (CCD) detector. One data set turned out to be useful although it suffered from the problem of ice rings which reduced the completeness of the data set (Table 1 ▶, Fig. 2 ▶ a).
Table 1. Data-collection statistics for the NtrC1 crystals.
Ranges in parentheses are for the highest resolution shells. Where there are two values in parentheses, these are for the lowest and the highest resolution shells.
| S crystal | Q crystal | |
|---|---|---|
| Space group | P1 | P1 |
| Unit-cell parameters | ||
| a (Å) | 119.3 | 120.0 |
| b (Å) | 130.0 | 130.8 |
| c (Å) | 206.4 | 208.2 |
| α (°) | 90.0 | 90.0 |
| β (°) | 89.7 | 90.0 |
| γ (°) | 89.9 | 89.9 |
| Resolution (Å) | 37.9–3.60 (3.64–3.60) | 44.3–2.87 (2.97–2.87)† |
| R merge ‡ | 0.084 (0.038, 0.363) | 0.118 (0.032, 0.511) |
| 〈I/σ(I)〉 | 5.0 (13.4, 1.2) | 3.0 (12.1, 0.8)† |
| Completeness (%) | 76.4 (84.8, 78.0) | 88.89 (90.3, 47.0)† |
| Multiplicity | 1.66 (1.66, 1.50) | 1.76 (1.81, 1.62) |
| No. of reflections | 109669 | 256906† |
Because of the anisotropy on the b* axis the data were truncated at 3.2, 5.2 and 3.2 Å resolution in the a*, b* and c* directions to provide an elliptical set of reflections at an F/σ(F) of 3.0. This data set retained 109 010 unique reflections with completenesses of 91% (4.3–10.0 Å), 88% (6.7–6.2 Å), 60% (6.2–4.5 Å), 34% (4.5–3.7 Å), 26% (3.7–3.5 Å), 22% (3.5−3.4 Å) and 10% (3.4–3.2 Å).
R
merge = 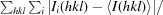
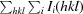 .
.
Figure 2.

Exemplary diffraction patterns and anisotropy of data from the Q crystal. (a), (b) Diffraction from the Q crystal. The black circle locates 3.7 Å resolution diffraction. (c) Plot of F/σ versus resolution along the a*, b* and c* axes of for the Q crystal obtained via the UCLA Diffraction Anisotropy Server (Strong et al., 2006 ▶).
The Q-fraction NtrC1 crystals were flash-cooled in a stream of nitrogen gas after a brief incubation in freshly prepared mother liquor supplemented with nucleotide and 20% ethylene glycol. X-ray diffraction data for Q-fraction crystals were collected on beamline X-29 of the National Synchrotron Light Source, Upton, New York, USA using an ADSC Q315 CCD detector (Figs. 2a and 2b ▶). Collected data were processed with the CrystalClear software (Rigaku Americas, The Woodlands, Texas, USA) and the statistics are given in Table 1 ▶. The data were processed in space group P1, but phenix.xtriage (Adams et al., 2010 ▶) indicated a possibility of higher symmetry for the space groups P2 or P222. This break in higher symmetry turned out to be due to minor but real differences in protomers and nucleotide occupancies among otherwise very similar hexamer rings. Based on our gel-filtration and X-ray solution scattering studies (not shown), we surmise that the crystal might contain hexameric NtrC1 assemblies. This is supported by the self-rotation function, which reveals a strong symmetry peak between pentamer and hexamer, and notably no peak expected for heptamers (Fig. 3 ▶). We thus suggest that the P1 unit cell contains four hexameric complexes and a Matthews coefficient of 4.54 Å3 Da−1 (Matthews, 1968 ▶). This implies an unusually high solvent content (∼73%) that is consistent with the observed fragility and poor diffraction of the crystals.
Figure 3.

Selected stereoprojections of the self-rotation function for Q-crystal diffraction. The chosen κ angle maps reveal the major peak (see arrows) at ω = 94.0°, ϕ = 88.4°, κ = 66.1°. Note that this peak lies between sixfold and fivefold symmetries (60 and 72°, respectively), but is far removed from sevenfold symmetry (51.4°). The axis corresponds to a pseudo-sixfold symmetry axis in the new hexamer ring structure found by molecular replacement, the details of which will be published elsewhere (Sysoeva et al., 2013 ▶). Calculations were made using POLARRFN from the CCP4 suite (Winn et al., 2011 ▶).
3. Results and discussion
Single crystals grew as thin uneven plates and rhombs of up to 0.5 mm in the longest dimension and of ∼10–30 µm in thickness (Fig. 1 ▶). The results of our crystallization attempts revealed that crystal formation and growth were highly sensitive to nucleotide concentration and protein preparations. Several protein preparations obtained via the same purification procedures did not form crystals under identical precipitation conditions. All crystals showed some anisotropy in the diffraction pattern (some were very anisotropic) and they were all highly susceptible to radiation damage. In some crystals we observed that radiation damage caused a darkening of the crystal area exposed to the X-rays within the first few seconds of exposure (Fig. 1b ▶). To the best of our knowledge, none of the components were suspected to decompose with gas release as observed for solutions containing cacodylate or other components. It is not clear what is causing the crystals to become more opaque, but it may result from radiation damage to the buffer or the protein component. This effect was not pronounced for all screened crystals and was not investigated further. The best crystal that we obtained diffracted to ∼5.2 Å resolution in the worst direction and to ∼3.2 Å resolution in the other two directions. To deal with the Q data set with high anisotropy we truncated the data set using the UCLA Diffraction Anisotropy Server (Strong et al., 2006 ▶). From our prior small-angle X-ray scattering studies it is apparent that NtrC1 ATPase undergoes large-scale conformational changes upon binding ATP and ATP analogs; therefore, we expect that the new data sets will shed light on some of the observed structural changes upon interaction with the nucleotide, despite the low resolution of the data sets.
There are two known structures of the wild-type ATPase domain in dimeric and heptameric forms (Lee et al., 2003 ▶). A structure of a heptamer of the Walker B mutant E239A of NtrC1 has also been established (Chen et al., 2010 ▶). These NtrC1 structures and other EBP crystal structures (PspF, ZraR and NtrC4; Rappas et al., 2005 ▶; Sallai & Tucker, 2005 ▶; Batchelor et al., 2009 ▶) were candidate targets for molecular replacement to solve the phase problem. A successful solution was obtained using the structural model of a single protomer of the ATP-bound Walker B mutant of NtrC1 (PDB entry 3m0e; Chen et al., 2010 ▶) and the two new structures will be described elsewhere (Sysoeva et al., 2013 ▶). This is the first report of co-crystallization of an EBP with ADP–BeFx–Mg. The resulting structures provide a third, novel state for EBP ATPases. Combined, the NtrC1 structures present a unique opportunity to glimpse a single σ54-dependent ATPase in dimeric and higher order oligomeric ring forms to advance our understanding of the EBP functioning cycle.
Acknowledgments
These studies were facilitated by Hemant Yennawar (Biochemistry and Molecular Biology Department, Penn State University) and Mark Signs of the Shared Fermentation Facility of the Huck Institutes of Life Sciences at Penn State University, to whom we express our deep gratitude. The work was funded by an NIH grant to BTN. Use of the Advanced Light Source and the National Synchrotron Light Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science under contract Nos. DE-AC02-05CH11231 and DE-AC02-98CH10866.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Batchelor, J. D., Sterling, H. J., Hong, E., Williams, E. R. & Wemmer, D. E. (2009). J. Mol. Biol. 393, 634–643. [DOI] [PMC free article] [PubMed]
- Bigay, J., Deterre, P., Pfister, C. & Chabre, M. (1987). EMBO J. 6, 2907–2913. [DOI] [PMC free article] [PubMed]
- Bordes, P., Wigneshweraraj, S. R., Chaney, M., Dago, A. E., Morett, E. & Buck, M. (2004). Mol. Microbiol. 54, 489–506. [DOI] [PubMed]
- Bose, D., Pape, T., Burrows, P. C., Rappas, M., Wigneshweraraj, S. R., Buck, M. & Zhang, X. (2008). Mol. Cell, 32, 337–346. [DOI] [PMC free article] [PubMed]
- Buck, M., Bose, D., Burrows, P., Cannon, W., Joly, N., Pape, T., Rappas, M., Schumacher, J., Wigneshweraraj, S. & Zhang, X. (2006). Biochem. Soc. Trans. 34, 1067–1071. [DOI] [PubMed]
- Buck, M., Cannon, W. & Woodcock, J. (1987). Mol. Microbiol. 1, 243–249. [DOI] [PubMed]
- Cannon, W. V., Gallegos, M. T. & Buck, M. (2000). Nature Struct. Biol. 7, 594–601. [DOI] [PubMed]
- Chaney, M., Grande, R., Wigneshweraraj, S. R., Cannon, W., Casaz, P., Gallegos, M. T., Schumacher, J., Jones, S., Elderkin, S., Dago, A. E., Morett, E. & Buck, M. (2001). Genes Dev. 15, 2282–2294. [DOI] [PMC free article] [PubMed]
- Chen, B., Doucleff, M., Wemmer, D. E., De Carlo, S., Huang, H. H., Nogales, E., Hoover, T. R., Kondrashkina, E., Guo, L. & Nixon, B. T. (2007). Structure, 15, 429–440. [DOI] [PMC free article] [PubMed]
- Chen, B., Sysoeva, T. A., Chowdhury, S., Guo, L., De Carlo, S., Hanson, J. A., Yang, H. & Nixon, B. T. (2010). Structure, 18, 1420–1430. [DOI] [PMC free article] [PubMed]
- Chen, B., Sysoeva, T. A., Chowdhury, S., Guo, L. & Nixon, B. T. (2009). FEBS J. 276, 807–815. [DOI] [PMC free article] [PubMed]
- Dago, A. E., Wigneshweraraj, S. R., Buck, M. & Morett, E. (2007). J. Biol. Chem. 282, 1087–1097. [DOI] [PubMed]
- De Carlo, S., Chen, B., Hoover, T. R., Kondrashkina, E., Nogales, E. & Nixon, B. T. (2006). Genes Dev. 20, 1485–1495. [DOI] [PMC free article] [PubMed]
- Doucleff, M., Chen, B., Maris, A. E., Wemmer, D. E., Kondrashkina, E. & Nixon, B. T. (2005). J. Mol. Biol. 353, 242–255. [DOI] [PubMed]
- Erzberger, J. P. & Berger, J. M. (2006). Annu. Rev. Biophys. Biomol. Struct. 35, 93–114. [DOI] [PubMed]
- Flashner, Y., Weiss, D. S., Keener, J. & Kustu, S. (1995). J. Mol. Biol. 249, 700–713. [DOI] [PubMed]
- González, V., Olvera, L., Soberón, X. & Morett, E. (1998). Mol. Microbiol. 28, 55–67. [DOI] [PubMed]
- Hong, E., Doucleff, M. & Wemmer, D. E. (2009). J. Mol. Biol. 390, 70–82. [DOI] [PMC free article] [PubMed]
- Lee, S.-Y., De La Torre, A., Yan, D., Kustu, S., Nixon, B. T. & Wemmer, D. E. (2003). Genes Dev. 17, 2552–2563. [DOI] [PMC free article] [PubMed]
- Magasanik, B. (1988). Trends Biochem. Sci. 13, 475–479. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mesmer, R. E. & Baes, C. F. Jr (1969). Inorg. Chem. 8, 618–626.
- Morett, E. & Segovia, L. (1993). J. Bacteriol. 175, 6067–6074. [DOI] [PMC free article] [PubMed]
- Neuwald, A. F., Aravind, L., Spouge, J. L. & Koonin, E. V. (1999). Genome Res. 9, 27–43. [PubMed]
- Ninfa, A. J., Reitzer, L. J. & Magasanik, B. (1987). Cell, 50, 1039–1046. [DOI] [PubMed]
- Nixon, B. T., Ronson, C. W. & Ausubel, F. M. (1986). Proc. Natl Acad. Sci. USA, 83, 7850–7854. [DOI] [PMC free article] [PubMed]
- Pelton, J. G., Kustu, S. & Wemmer, D. E. (1999). J. Mol. Biol. 292, 1095–1110. [DOI] [PubMed]
- Popham, D. L., Szeto, D., Keener, J. & Kustu, S. (1989). Science, 243, 629–635. [DOI] [PubMed]
- Porter, S. C., North, A. K., Wedel, A. B. & Kustu, S. (1993). Genes Dev. 7, 2258–2273. [DOI] [PubMed]
- Rappas, M., Schumacher, J., Beuron, F., Niwa, H., Bordes, P., Wigneshweraraj, S., Keetch, C. A., Robinson, C. V., Buck, M. & Zhang, X. (2005). Science, 307, 1972–1975. [DOI] [PMC free article] [PubMed]
- Ray, P., Smith, K. J., Parslow, R. A., Dixon, R. & Hyde, E. I. (2002). Nucleic Acids Res. 30, 3972–3980. [DOI] [PMC free article] [PubMed]
- Rippe, K., Mücke, N. & Schulz, A. (1998). J. Mol. Biol. 278, 915–933. [DOI] [PubMed]
- Ronson, C. W., Nixon, B. T. & Ausubel, F. M. (1987). Cell, 49, 579–581. [DOI] [PubMed]
- Sallai, L. & Tucker, P. A. (2005). J. Struct. Biol. 151, 160–170. [DOI] [PubMed]
- Schumacher, J., Joly, N., Rappas, M., Bradley, D., Wigneshweraraj, S. R., Zhang, X. & Buck, M. (2007). J. Biol. Chem. 282, 9825–9833. [DOI] [PubMed]
- Schumacher, J., Joly, N., Rappas, M., Zhang, X. & Buck, M. (2006). J. Struct. Biol. 156, 190–199. [DOI] [PubMed]
- Schumacher, J., Zhang, X., Jones, S., Bordes, P. & Buck, M. (2004). J. Mol. Biol. 338, 863–875. [DOI] [PubMed]
- Snider, J. & Houry, W. A. (2008). Biochem. Soc. Trans. 36, 72–77. [DOI] [PubMed]
- Strong, M., Sawaya, M. R., Wang, S., Phillips, M., Cascio, D. & Eisenberg, D. (2006). Proc. Natl Acad. Sci. USA, 103, 8060–8065. [DOI] [PMC free article] [PubMed]
- Studholme, D. J. & Dixon, R. (2003). J. Bacteriol. 185, 1757–1767. [DOI] [PMC free article] [PubMed]
- Su, W., Porter, S., Kustu, S. & Echols, H. (1990). Proc. Natl Acad. Sci. USA, 87, 5504–5508. [DOI] [PMC free article] [PubMed]
- Sysoeva, T. A., Chowdhury, S., Guo, L. & Nixon, B. T. (2013). Genes Dev. 27, 2500–2511. [DOI] [PMC free article] [PubMed]
- Tucker, P. A. & Sallai, L. (2007). Curr. Opin. Struct. Biol. 17, 641–652. [DOI] [PubMed]
- Vidangos, N., Maris, A. E., Young, A., Hong, E., Pelton, J. G., Batchelor, J. D. & Wemmer, D. E. (2013). Biopolymers, 99, 1082–1096. [DOI] [PMC free article] [PubMed]
- Wang, Y.-K. & Hoover, T. R. (1997). J. Bacteriol. 179, 5812–5819. [DOI] [PMC free article] [PubMed]
- Wang, Y.-K., Lee, J. H., Brewer, J. M. & Hoover, T. R. (1997). Mol. Microbiol. 26, 373–386. [DOI] [PubMed]
- Wang, Y.-K., Park, S., Nixon, B. T. & Hoover, T. R. (2003). J. Bacteriol. 185, 6215–6219. [DOI] [PMC free article] [PubMed]
- Wedel, A. & Kustu, S. (1995). Genes Dev. 9, 2042–2052. [DOI] [PubMed]
- Wedel, A., Weiss, D. S., Popham, D., Dröge, P. & Kustu, S. (1990). Science, 248, 486–490. [DOI] [PubMed]
- Weiss, D. S., Batut, J., Klose, K. E., Keener, J. & Kustu, S. (1991). Cell, 67, 155–167. [DOI] [PubMed]
- Weiss, V. & Magasanik, B. (1988). Proc. Natl Acad. Sci. USA, 85, 8919–8923. [DOI] [PMC free article] [PubMed]
- White, S. R. & Lauring, B. (2007). Traffic, 8, 1657–1667. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wyman, C., Rombel, I., North, A. K., Bustamante, C. & Kustu, S. (1997). Science, 275, 1658–1661. [DOI] [PubMed]
- Zhang, N., Joly, N., Burrows, P. C., Jovanovic, M., Wigneshweraraj, S. R. & Buck, M. (2009). Nucleic Acids Res. 37, 5981–5992. [DOI] [PMC free article] [PubMed]