

Abstract
Background
Drug-induced arrhythmia is the most common cause of drug development failure and withdrawal from market. This study tested whether human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) combined with a low-impedance microelectrode array (MEA) system could improve upon industry-standard, preclinical cardiotoxicity screening methods, identify the effects of well-characterized drugs, and elucidate underlying risk factors for drug-induced arrhythmia. Human iPSC-CMs may be advantageous over immortalized cell lines because they possess similar functional characteristics as primary human cardiomyocytes and can be generated in unlimited quantities.
Methods and Results
Pharmacological responses of beating embryoid bodies (EBs) exposed to a comprehensive panel of drugs at 65 to 95 days post-induction were determined. Responses of hiPSC-CMs to drugs were qualitatively and quantitatively consistent with the reported drug effects in literature. Torsadogenic hERG blockers such as sotalol and quinidine produced statistically and physiologically significant effects, consistent with patch-clamp studies on human embryonic stem cell-derived cardiomyocytes (hESC-CMs). False negative and false positive hERG blockers were identified accurately. Consistent with published studies using animal models, early afterdepolarizations (EADs) and ectopic beats were observed in 33% and 40% of embryoid bodies treated with sotalol and quinidine, respectively, compared to negligible EADs and ectopic beats in untreated controls.
Conclusions
We found that drug-induced arrhythmias can be recapitulated in hiPSC-CMs and documented with MEA. Our data indicate that the MEA/hiPSC-CM assay is a sensitive, robust, and efficient platform for testing drug effectiveness and for arrhythmia screening. We believe that this system holds great potential for reducing drug development costs and may provide significant advantages over current industry standard assays that use immortalized cell lines or animal models.
Keywords: stem cell, cardiomyocyte, pharmacology, arrhythmia, genomics, pharmacogenomics
Introduction
Cardiac toxicity is a major cause of drug attrition during preclinical development 1, 2. In addition, the risk of drug-induced arrhythmia is the most common cause of restriction or withdrawal of drugs from the market 3. Between 1990 and 2001, eight non-cardiovascular drugs were withdrawn at an estimated cost of $12 billion due to problems such as delayed cardiac repolarization, prolonged QT interval, and Torsade de Pointes (TdP) 4, 5. TdP is a rare polymorphic ventricular tachyarrhythmia with a characteristic twist of the QRS complex around the isoelectric baseline that often resolves spontaneously but can also result in true ventricular tachycardia, ventricular fibrillation, and sudden cardiac death. QT prolongation, early afterdepolarizations (EADs), ectopic beats, and increased spatial dispersion of repolarization are risk factors associated with TdP6. EADs are considered the underlying mechanism that generates TdP, as they can progress to triggered activity, lead to ectopic beats, and contribute to the dispersion of ventricular repolarization. Importantly, drugs that do not induce EADs, ectopic beats, or increased dispersion of ventricular repolarization have a very low incidence of TdP 3, 7, 8.
One of the problems in drug development is that primary human cardiomyocytes are very difficult to obtain and survive only transiently in culture. The combination of stably-transfected cell lines and automated, high-throughput patch-clamping is a standard assay for cardiac safety screening because the majority of clinical cases of drug-induced cardiotoxicity have been associated with blockage of the hKv11.1 potassium channel, also known as hERG 8-10. The limitations of this approach are well-understood because the QT interval is the net result of the activities of several interdependent ion channels 11. To address this issue, animal models such as rabbit or dog Purkinje fibers are used in later stages of the drug screening process to study the action potential (AP) via conventional patch-clamping. However, the lack of sensitivity of these assays to torsadogenic drugs is well-known 12-14. Furthermore, the vulnerability of the cardiomyocyte calcium current to “run-down” due to dialysis of the cytoplasm with conventional whole-cell patch-clamping limits the duration of drug tests 15-18.
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) have great potential in drug screening because they express human ion channels similar to primary human CMs 19, 20. As undifferentiated hiPSCs can, in theory, be cultured indefinitely, the supply of human CMs is likewise unlimited 21-23. Significantly, patient-specific hiPSC-CMs can be produced to study drug effects in the context of a particular risk factor or disease mutation 24-30. Microelectrode arrays (MEAs) have been used previously in many pharmacological studies using cardiac tissue, including hiPSC-CMs 25, 26, 28, 31. MEAs can easily extract basic electrophysiological parameters such as beat frequency and field potential duration (FPD) that correlate closely with cardiomyocyte action potential durations (APD) obtained via patch-clamping, as well as with heart rates and QT intervals obtained via clinical EKGs. Furthermore, the noninvasive nature of MEAs allows the study of drug effects on beating rate and rhythm. While many academic labs, pharmaceutical companies, and contract research organizations have begun to adopt MEA assays for hiPSC-CM research, no published study has compared results from this approach to clinical data in order to address whether the MEA/iPSC-CM system is predictive of drug toxicity effects against industry-standard technologies such as patch-clamping with recombinant cell lines or animal models. Additionally, previous drug screening studies using MEAs to study cardiomyocytes derived from PSCs have utilized hESC-CMs but not patient-specific hiPSC-CMs 1, 32.
Here we report and quantify drug-induced EADs, ectopic cardiomyocyte contraction, and the pharmacology of a comprehensive panel of drugs from various classes using a MEA/hiPSC-CM platform. This also represents the first study to employ this platform for the purpose of recapitulating drug-induced arrhythmias in vitro. Our study demonstrates a novel means of studying cardiomyocyte pharmacology and toxicology by utilizing the MEA/hiPSC-CM technology to evaluate more than 10 known arrhythmia-regulating compounds through a broad set of cellular, genetic, and physiological assays.
Methods
An expanded Methods section is available in the Supplemental Material.
Derivation of hiPSCs
All the protocols for this study were approved by the Stanford University Institutional Review Board. Briefly, dermal fibroblasts obtained from a healthy 14-year old male subject via skin biopsy were reprogrammed and characterized as described previously, using a lentiviral vector. Stem cells were cultured on feeder-free Matrigel-coated tissue culture dishes (BD Biosciences, San Jose, CA) with mTESR-1 hESC Growth Medium (STEMCELL Technologies, Vancouver, Canada)
Differentiation of hiPSC-CMs from embryoid bodies
Colonies fulfilling established “stemness” criteria were differentiated into cardiomyocytes using standard 3D differentiation protocols and maintained in a 5% CO2/air environment 33. Briefly, on day 0 hiPSC colonies were dissociated with Accutase (Sigma) into small clumps of 10-20 cells. Cells were resuspended in 2 ml of basic media containing StemPro34 (Invitrogen), 2 mM glutamine (Invitrogen), 0.4 mM monothioglycerol (Sigma), 50 μg/ml ascorbic acid (Sigma), and 0.5 ng/ml BMP4 (R&D Systems, Minneapolis, MN) to form EBs. For the first 1-4 days, cells were treated with 10 ng/ml BMP4, 5 ng/ml human bFGF (R&D Systems), and 3 ng/ml activin A (R&D Systems) in the basic media. On days 4-8, EBs were re-fed with basic media containing human 50 ng/ml DKK1 (R&D Systems) and 10 ng/ml human VEGF (R&D Systems). After day 8, cells were cultured in basic media containing 5 ng/ml human bFGF and 10 ng/ml human VEGF. To further corroborate data obtained from EB differentiation, hiPSC-CMs were also created through a monolayer differentiation technique (See Supplementary Methods).
Gene expression and immunocytochemistry
RNA was extracted with the miRNeasy kit (Qiagen). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and Real-time PCR was done with Taqman assays (Applied Biosystems) using StepOne (Applied Biosystems). The Hierarchical clustering was performed and the heatmap was generated by TM4 (www.tm4.org). Immunostaining was performed according to previously established protocols 22. Imaging was performed using a DMIL-LED inverted tissue culture microscope (Leica Microsystems, Buffalo Grove, IL) or an A1-R Resonant Confocal System running NIS Elements software (Nikon, Tokyo, Japan).
Microelectrode array recordings
Beating EBs were plated on 0.1-0.2% gelatin-coated AL-MED P5004A MEA probes (Alpha Med Scientific, Osaka, Japan) (Supplemental Figure 1A). Prior to MEA recordings, AVI videos were recorded on the DMIL-LED microscope. Signals were acquired at 20 kHz using a MED64 amplifier (Alpha Med Scientific) and digitized via an Optiplex 390 PC (Dell, Round Rock, TX) with PCI-6071 A/D cards (National Instruments, Austin, TX) running Mobius QT software (Witwerx, Inc., Tustin CA) (Supplemental Figure 1B). Data were analyzed by Mobius QT software (Witwerx, Inc., Tustin CA), Igor Pro (Wave Metrics), Microcal Origin 6.1 (OringinLab), or Prism 5 (GraghPad). Steady state parameters were averaged and FPD was normalized to beat rate using the Bazett correction formula (FPDc).
Statistical Methods
Data are presented as mean ± SEM. All statistical analyses were performed using the software Sigma plot and Sigma stat. In our study, before performing the Student's t-test between 2 groups of data, we ran the Shapiro-Wilk test, and the data set with a p-value larger than 0.05 was considered normally distributed. Afterwards, we examined the two groups of data using the Equal Variance Test, and the two groups of data with the p-value larger than 0.05 were considered to have similar variance. Only data that passed both of the two tests were then further analyzed with Student's test, and the two-tailed p-values smaller than 0.05 were considered to be statistically significant between 2 groups. Responses to drugs were normalized and compared to baseline via t-tests, with significant differences defined by p<0.05.
Results
Expression of cardiomyocyte markers
We confirmed the presence of genes specific to the cardiomyocyte lineage encoding a variety of protein classes. Cardiac-specific structural genes encoding sarcomeric myofilament proteins included ACTC1 (alpha actin 1), MYL2 (myosin regulatory light chain 2), MYH6 (myosin heavy chain 6, alpha), and MYH7 (myosin heavy chain 7, beta) (Supplemental Figure 2). Importantly, the major ion channel genes essential for cardiomyocyte function were also expressed (Figure 1A). A complete list of all structural and functional genes evaluated in this study can be found in Supplemental Table 1. Hierarchical clustering analysis demonstrates that our hiPSC-CMs were most similar to cells obtained from human adult ventricles. Expression of sarcomeric proteins was confirmed via immunofluorescence and laser confocal microscopy. Cardiac troponin T staining of hiPSC-CMs showed a typical striated pattern intercalated with alpha-actinin along the sarcomeric Z-lines (Figure 1B). hiPSC-CMs with organized parallel myofilaments and cylindrical morphology were often observed. Moreover, hiPSC-CMs derived using the iPSC monolayer differentiation method exhibited staining profiles comparable to hiPSC-CMs derived using the embryoid body method (Supplemental Figure 3).
Figure 1. hiPSC-CM expression of cardiac ion channel genes and sarcomeric proteins.

A, Heat map showing the gene expression levels of the main adrenoreceptors and ion channels essential for cardiomyocyte function in hiPSCs and human adult heart chambers. Color scale represents delta-delta Ct values relative to the expression levels in hiPSC-CMs. Green indicates upregulation and red downregulation. Strong red corresponds to genes which are not expressed. Average linkage cluster for the samples is presented. LA refers to left atrium, RA to right atrium, LV to left ventricle, and RV to right ventricle (n=4). B, Confocal microscopy image of alpha-actinin (top) and troponin T (middle) immunostaining demonstrates the presence of cardiac specific sarcomeric proteins in a single hiPSC-CM. The inset in the merged image at the bottom shows organized horizontal myofilaments with parallel red striations indicating the sarcomeric Z-lines.
Characterization of cardiomyocyte embryoid bodies
Video contractility analyses determined that the EBs were composed of almost 90% beating hiPSC-CMs (Supplemental Movie 1). The average contractile EB area was 88.4 ± 5.4% (n = 4), consistent with the percentage of TNNT (+) cells (81.34 ± 0.91%) in dissociated EBs (Figure 2). Ventricular-like, atrial-like, and nodal-like CM subtypes were identified via their AP parameters (Table 1 and Supplemental Figure 4). Cells with ventricular-like APs were the most frequently encountered, comprising 67% of all tested hiPSC-CMs, followed by atrial-like cells (28%) and nodal-like cells (5%).
Figure 2. Video contractility analyses demonstrate high percentage of hiPSC-CMs in embryoid bodies.
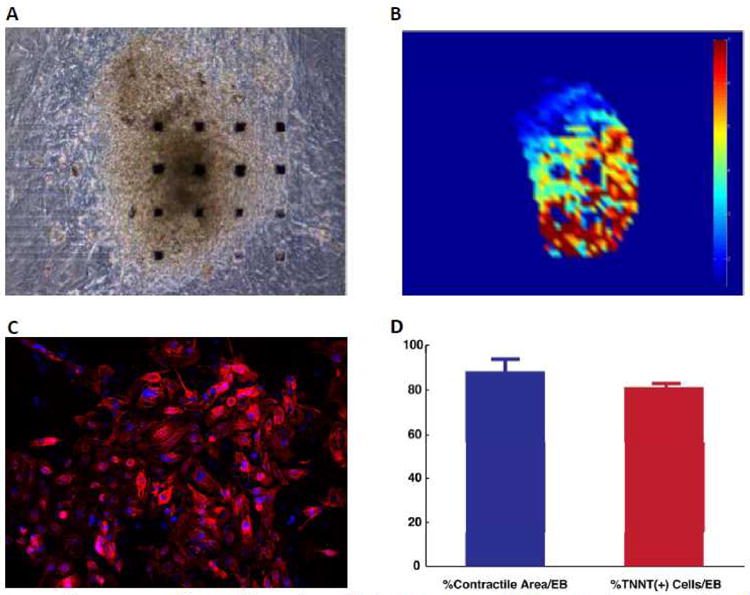
A, Embryoid body plated on a gelatin-coated MEA probe grid. B, Heat map showing the levels of contractility within the EB at a given point in time. C, Ratio of troponin T positive cells (in red) to nuclei (stained for DAPI in blue) was used to determine the percentage of cardiomyocytes in dissociated EBs. D, Graph comparing the contractile area of the EBs studied to the percentage of cells stained positively for TNNT2 (n=10).
Table 1. Baseline electrophysiology in single hiPSC-CMs obtained via patch-clamping.
| CM Subtype | N | % | BPM | MDP (mV) | Overshoot (mV) | APA (mV) | APD90 (ms) | Vmax (V / S) |
|---|---|---|---|---|---|---|---|---|
| Nodal-like | 2 | 5 | 72±10 | −32.6±5.4 | 33.4±3.2 | 67.4±6.3 | 244.5±30.6 | 2.9±1.4 |
| Atrial-like | 11 | 28 | 56±12 | −50.6±4.2 | 42.5±6.5 | 93.3±6.8 | 276.3±20.5 | 27.4±10.3 |
| Ventricular-like | 27 | 67 | 50±15 | −52.6±6.3 | 45.5±4.1 | 98.7±10.4 | 342.7±28.7 | 26.8±14.2 |
Mean ± SEM
CM = cardiomyocyte, BPM = beats per minute, MDP = maximal diastolic potential, APA = action potential amplitude, APD90 = action potential duration at 90%, Vmax = maximal upstroke velocity.
Baseline microelectrode array electrophysiology
Spontaneous beating and electrical activity were present in all plated EBs as shown in Supplemental Movies 2 - 6. By comparison, spontaneous beating activity was also observed in hiPSC-CMs derived from iPSC monolayer differentiation (Supplemental Movie 7). Baseline electrophysiological parameters recorded on day 75 are presented in Table 2. Representative traces for the subject's EKG (lead 4), hiPSC-CM's AP, and beating EB's FP are shown in Figure 3. The average beating frequency was 35 beats per minute (bpm) and the mean FPDc was 473 ms, suggesting ventricular-like CMs were predominant in our EBs (n=5), consistent with gene expression clustering and single-cell patch clamp data. The baseline is stable with little change in beating frequency and FPDc over a 10-min period (Supplemental Figure 5). Moreover, hiPSC-CMs derived using the iPSC monolayer differentiation method also exhibited electrophysiological parameters analogous to embryoid body-derived hiPSC-CMs (Supplemental Figure 6 & 7).
Table 2. Baseline electrophysiology in hiPSC-CM embryoid bodies obtained via MEAs.
| hiPSC-CM EBs | BPM | FPD (ms) | FPDc (ms) | Vmax (mV/s) |
|---|---|---|---|---|
| n = 5 | 34.95 ± 4.54 | 629.87 ±64.58 | 472.53 ± 58.99 | 185.97 ± 114.57 |
Mean ± SEM
hiPSC-CM EBs = Human induced pluripotent stem cell-derived cardiomyocyte embryoid bodies, BPM = beats per minute, FPD = field potential duration, FPDc = Corrected field potential duration, Vmax = maximal upstroke velocity.
Figure 3. QT interval, action potential duration, and field potential duration.

A, EKG (lead 4) obtained from the subject illustrating the QT interval. B, Action potential recorded via whole-cell patch-clamping of a representative ventricular hiPSC-CM from a dissociated EB showing the APD. C, Field potentials recorded from a representative EB via MEAs denoting the electrophysiological parameters of interest. Note the similarity between the EKG inset on the top left corresponding to the time course of a typical action potential recorded via conventional patch-clamping on the top right inset.
Neurohormonal responses
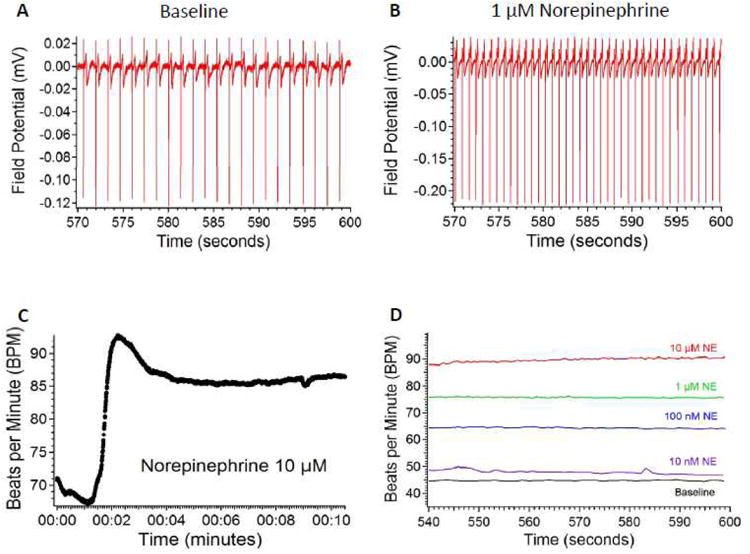
The autonomic nervous system plays an important role in the regulation of heart rate and contraction force. To assess the sensitivity of hiPSC-CMs to neurohormonal regulation, we administered the adrenergic catecholamine norepinephrine (NE), the mixed beta-agonist isoproterenol (ISO), and the muscarinic cholinomimetic carbamyl choline (CCH). NE had a clear dose-dependent effect on the beat frequency of all EBs tested (Figure 4A & 4B). The onset of responses with both adrenergic drugs was very quick, usually within 3 minutes (Figure 4C and Supplemental Figure 8A). Although there was occasional beat-to-beat variability and usually a rebound from the maximal effect, the increases in beat frequency stabilized and persisted until the end of the 10-minute recording (Figure 4D and Supplemental Figure 8B). Interestingly, the same EBs tested 9 days earlier at 67 days post-induction with ISO had clear responses in only 75% of the EBs tested (n=4) (Supplemental Figures 8C & 8D), and only half of them demonstrated clear dose-dependence with a half-maximal excitatory concentration (EC50) of 72.03 ± 16.80 nM (Supplemental Figure 9A), suggesting that the adrenoreceptor sensitivity of some of these EBs was immature. Notably, the EC50 for ISO's effect on beat frequency is very close to the EC50 for ISO's effect on adult human heart slice contractility of 118 ± 20nM34. The half-maximal excitatory concentration (EC50) for NE's effect on beat frequency was 41.66 ± 16.50 nM (n=4) (Figure 5A), though responses were often evident at 1 nM for both ISO and NE (data not shown). ISO and NE also had a clear dose-dependent effect on FPD, shortening it consistently (Figures 5B & 5C, Supplemental Figures 9B & C).
Figure 4. Norepinephrine causes sustained increases in hiPSC-CM beating frequency.
A, Rhythmic spontaneous field potentials at baseline during the last 30 seconds of a 10 minute recording. B, Effect of 1 μM norepinephrine (NE) on beat frequency. C, Graph plotting the timecourse of increase in beat frequency due to 10 μM NE in a representative EB. D, Steady-state concentration-dependent increases on absolute beating frequency. Note the stability of the response during the last 30 seconds of a 10-minute recording. n=8 EBs for the NE testing.
Figure 5. Norepinephrine induces a dose-dependent increase in BPM and consequently a decrease in FPD.

A, The dose dependent effect of NE on beat rate could be fitted well with the Hill equation (red line), yielding a half maximal excitatory concentration of 41.66 ± 16.50 nM (n=4). B, Representative traces illustrating the dose-dependent shortening of FPD secondary to NE. Arrow points to the left-shifting peak of the repolarizing wave to indicate shortening of the FPD. C, Graph plotting NE concentration - FPD response and fit to the Hill equation (red line connecting the data points, n=3). The IC50 was 19.38 nM. D, NE concentration vs. FPD corrected according to the Bazett formula and Hill fit (red line). n=8 EBs for the NE testing.
Heart rate is an important variable affecting the QT interval and correction formulae have been used since 1920, with the Bazett formula being the most popular35. Indeed, the Bazett correction produced a flat Hill fit to the NE frequency response (Figure 5D). Nevertheless, it is well known that the Bazett formula overcorrects at high beat frequencies, as our results with ISO illustrate (Supplemental Figure 9D) 36. CCh produced a dose-dependent decrease in beat frequency, regardless of prior adrenergic stimulation (Supplemental Figures 10A-D), and consequently resulted in FPD prolongation (Supplemental Figure 11A). The CCh-induced decreases in beat rate with or without ISO administration were not significantly different at any concentration tested. On average, maximal decreases in beat frequency exceeded 50% at 100 μM and one third of the EBs lost their spontaneous activity regardless of prior adrenergic stimulation. CCh also increased FP amplitude (FPA) in a dose-dependent manner (Supplemental Figure 11B).
Responses to hERG blockers
A variety of drugs can block the hERG channel due to its unique structural “promiscuity” 37, 38. We tested the clinically relevant drugs sotalol and quinidine, which are currently prescribed for atrial fibrillation and ventricular tachycardia. First we tested the mixed antiarrhythmic drug sotalol to gauge the time course of responses. The onset of the effect on the EB's FPD was comparable to that reported for patch-clamped hESC-CMs 13. As shown in Figure 6A, the onset of sotalol's effect at 30 μM was almost immediate and typically stabilized within approximately 7 minutes. As with ISO, the time course of responses to sotalol was concentration-dependent. For example, we observed the stabilization of the effect within 2 minutes with 100 μM (data not shown). As expected, the increase in FPD was concentration-dependent (Figure 6B). Sotalol also produced a dose-dependent decrease in beat frequency (Figure 6C), likely due to its activity as a beta-adrenoreceptor blocker, making a correction procedure necessary. The average increase in FPDc due to sotalol was statistically and physiologically significant at 100 μM and had a maximal prolongation of 36% at the highest sotalol concentration tested of 500 μM (n=4), with a half maximal effect at 214.1 ± 102.0 μM (Figure 6D). A very clear sotalol-induced, dose-dependent, FPDc prolongation was also obtained using age-matched monolayer-derived hiPSC-CMs (Supplementary Figure 12A). Consistent with APD studies on hESC-CMs, quinidine also induced FPDc prolongation that was statistically significant at 100 nM and physiologically significant at 300 nM, with a half maximal effect on FPDc prolongation at 333.1 nM (Supplemental Figures 13A & 13B) 13. A dose-dependent decrease in FPA and maximal upstroke velocity (Vmax) was also observed, likely due to quinidine's well-known capability to block the sodium current (Supplemental Figure 13C & 13D). Most importantly, hERG blockers induced the arrhythmic activity that results in TdP. We observed the characteristic notched or bifurcated repolarizing waves described in LQT2 patients and the LQT2 dog model with 100 μM sotalol (Figure 7A) 39, 40. At higher concentrations, we observed ectopic beats and the short-long-short rhythm known to precede TdP (Figure 7B) 10, 41, 42. One third of our sample tested with sotalol (n=6) displayed the aforementioned arrhythmias (Supplemental Figures 12B & 12C). Quinidine also induced arrhythmic activity in 40% of the EBs tested (n=5), eliciting early afterdepolarizations (EADs) and ectopic beats at concentrations as low as 1 uM (Figure 7C and Supplemental Figure 14B), well within the free effective therapeutic plasma concentration (ETPC) of 0.9 −3.2 μM 7. Interestingly, the notched waveform morphology seen with sotalol is very similar to that seen in the quinidine-induced EADs. Finally, we also observed sotalol-induced EADs at a higher dose (100 μM) when using monolayer-derived hiPSC-CMs (n=3) (Supplemental Figure 15). Untreated control EBs did not demonstrate EADs or ectopic beats (data not shown).
Figure 6. hERG blocker sotalol prolongs FPD despite reducing BPM.
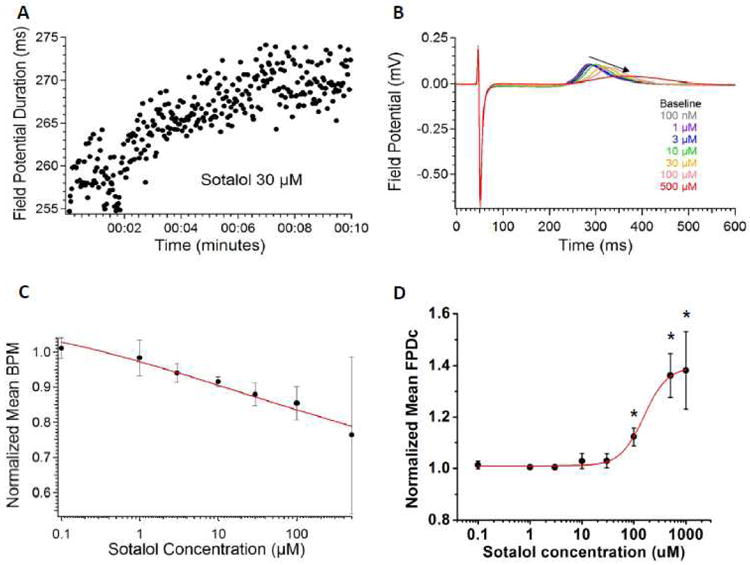
A, The effect of sotalol (30 μM) on the FPD of a representative EB begins almost immediately after drug administration and stabilizes within approximately 7 minutes. B, Dose-dependent prolongation of field potentials caused by sotalol in a representative EB. Arrow points to the right-shifting peak of the repolarizing wave to indicate lengthening of the FPD. C, Sotalol also affects BPM, necessitating a normalization procedure because FPD is related to beat frequency (n=4). D, The dose dependent effect of sotalol on FPD (corrected according to the Bazett formula) could be fitted nicely with the Hill equation (red line) (n=4). Asterisks note the FPDc prolongation was statistically and physiologically significant at concentrations of 100 uM and higher.
Figure 7. hERG block induces arrhythmic activity in hiPSC-CMs.

A, Typical notched or bifurcated repolarizing wave seen in LQTS2 and sotalol cardiotoxicity (n=6). B, Ectopic beats in a short-long-short rhythm were observed at a sotalol concentration of 500 μM and higher (n=6). C, Early afterdepolarizations (EADs) induced by quinidine (1 μM). Arrows point to the different arrhythmic events (n=5).
Responses to a “false positive” hERG blocker
Verapamil is a phenylalkylamine L-type calcium channel blocker that also has a potent effect on hERG. Blocking of L-type calcium channel leads to QT shortening while blocking of hERG results in QT prolongation. Given the dual roles to shape the QT, however, verapamil is a well-known “false-positive” hERG blocker with no reports of QT prolongation or TdP in humans 7. In our hiPSC-CM sample, verapamil induced progressive FPD shortening with increasing concentrations (Figure 8A). The half maximal effect on FPDc occurred at a concentration of 169 ± 24 nM (n=4) (Figure 8B), reasonably close to the free ETPC of 25-81 nM7. Interestingly, half of our sample's spontaneous electrical activity was abolished at 1 μM and all our EBs stopped beating at 10 μM (Supplemental Figure 15A). Spontaneous beating was 4 times more sensitive to verapamil at earlier developmental stages (half-maximal inhibitory concentration, or IC50, at 82 days post-induction 410.65 ± 40.80 nM vs. 103.20 ± 6.03 in 30 day-old EBs). Verapamil also decreased the initial fast depolarizing spike's amplitude in a dose-dependent fashion (Supplemental Figure 15B).
Figure 8. MEA/hiPSC-CM platform identifies false-positive and false-negative hERG blockers.

A, Dose-dependent shortening of field potentials caused by verapamil in a representative EB. Arrow points to the left-shifting peak of the repolarizing wave to indicate shortening of the FPD. B, The dose-dependent decreases in the normalized mean FPDc could be fitted nicely with the Hill equation (red line), evidencing a half maximal effect at 169.28 ± 24.00 nM (n=4). C, Alfuzosin induced dose-dependent FPD prolongation in our hiPSC-CMs as shown in this graph of a representative EB's responses. Arrow points to the right-shifting peak of the repolarizing wave to indicate lengthening of the FPD. D, FPDc prolongation was statistically and physiologically significant at 300 nM and higher concentrations as noted by the asterisks (n=4).
Responses to a “false negative” hERG blocker
Alfuzosin is a selective alpha-adrenoreceptor blocker approved by the Food and Drug Administration (FDA) for the treatment of benign prostatic hyperplasia. Alfuzosin is a rare drug that delays cardiac repolarization by increasing sodium current43. This mechanism of QT prolongation is similar to the hereditary arrhythmia, LQT3. Alfuzosin very weakly inhibits hERG current tested in CHO or HEK293 cells stably overexpressing hERG, hence the readout is “safe” using the hERG blockade assay. However, it can delay the cardiac repolarization (QT prolongation) by increasing the sodium current, thus leading to “false negative” readout. Using our MEA/hiPSC-CM platform, we were able to record dose-dependent increases in FPD (Figure 8C). Increases in FPDc were statistically and physiologically significant at the clinically relevant concentration of 300 nM (Figure 8D) (n=4), consistent with previous studies43. The average FPDc had a maximal prolongation of 65% at 1 uM and spontaneous electrical activity was abolished in half of our sample at this concentration. The largest increase in FPDc was 220% at 10 μM. Interestingly, we did not see any EADs, but this is consistent with the rabbit PF and Langendorff-perfused heart43.
Responses to other ion channel blockers
To further characterize the pharmacology of our hiPSC-CMs, we investigated the sensitivity to other ion channel blockers. First we tested nifedipine, a potent dihydropyridine L-type calcium channel blocker. A concentration-dependent decrease in FPD was observed consistent with the effects seen with verapamil, and was both physiologically and statistically significant at concentrations of 10 nM and higher (Supplementary Figure 16A and 16B). This is also consistent with the effect on hESC-CM APD 13. Nifedipine produced an important reduction in FPA with increasing dosage. Beat frequency was also sensitive to nifedipine, consistent with verapamil's effects,. The loss of spontaneous activity was observed in 75% of our sample at 100 nM, and at 300 nM all our EBs stopped beating, similar to previous reports1. The IC50 for FPDc shortening was 41.96 ± 4.62 nM, consistent with the IC50 for Ca current block in patch-clamped hiPSC-CMs and reasonably close to the reported free EPTC of 3.1-7.7 nM 7, 21. For nifedipine treatment, similar dose-dependent FPDc shortening was obtained using age-matched hiPSC-CMs derived from the monolayer method (Supplementary Figure 17). We then tested lidocaine, an anesthetic that blocks fast voltage-gated sodium channels. Like quinidine, a dramatic dose-dependent decrease in the amplitude of the field potential's fast depolarizing spike and Vmax was observed (Supplementary Figure 16C and 16D). The reduction in Vmax was statistically significant at 10 μM, consistent with the effect on hESC-CM APDs13. In addition, half of the sample stopped beating at 300 μM, the maximal concentration tested, in line with prior studies 1, 44.
Discussion
The MEA/hiPSC-CM assay addresses the unmet need in the pharmaceutical industry for a humanized cardiac safety platform with better predictive power. Species differences and the lack of the complex channel interactions in hERG-transfected cell lines limit the sensitivity and specificity of the standard methodologies for cardiac toxicity-drug safety screening 13, 45. hiPSC-CMs express virtually all known ion channels and subunits consistent with adult human ventricular tissues, as well as APs consistent with ventricular myocyte electrophysiology, despite hiPSC-CMs' immature morphology and spontaneous beating 21. Moreever, this spontaneous activity can be exploited to investigate chronotropic drug effects, an advantage of using a non-invasive system such as the MEA platform. In addition, our multicellular preparation reduces the vulnerability of the reductionist single-cell preparation and the intrinsic differences between cells. In fact, close examination of our dose-response studies reveals very small variability in the responses at baseline and initial doses. Furthermore, we did not observe EADs, ectopic beats, or any other arrhythmic activity in baseline studies.
Our findings have several important implications for the use of MEA/hiPSC-CM assay to detect drug-induced arrhythmias. This is the first quantitative characterization of the hiPSC-CMs' pharmacology and ability to detect drug-induced arrhythmia using MEAs and a comprehensive panel of drugs from various classes. We have shown that responses of hiPSC-CMs to neurohormonal drugs and ion channel blockers are qualitatively and quantitatively consistent with previous reports 1, 7, 13, 21, 34, 44, including APD and FPD measures on hESC-CMs. These findings correlate with the free EPTC in humans, confirming their suitability for testing drug effectiveness. We also show that the MEA/hiPSC-CM assay is sensitive to ion channel blockers affecting specific phases of the cardiac AP: (i) sodium channel blockers lidocaine and quinidine, which slow Vmax and reduce the FP amplitude in phase 0; (ii) L-type calcium channel blockers nifedipine and verapamil, which shorten the AP plateau (phase 2); and (iii) class III antiarrhythmic hERG blockers, which delay phase 3 repolarization. Furthermore, we have shown that the MEA/hiPSC-CM platform can clearly identify false negative and false positive torsadogenic drugs. Nonetheless, characterization of Kv4.3, KCNQ1, and Kir2.1, including their conductances and modulators, is necessary. For example, some studies suggest that underexpression of Kir2.1 (and thus the IK1 current) causes spontaneous beating in hESC-CMs and hiPSC-CMs 44, 46. As mentioned previously, this spontaneous activity can be exploited to investigate chronotropic drug effects; for example, we found that calcium currents have a pivotal role in the generation of spontaneous electrical activity in hiPSC-CMs. Notably, we recorded significant developmental changes in the sensitivity to verapamil, consistent with the developmental upregulation of ion channel expression that others have described 47, 48. The determination of the appropriate time points to test drug efficacy and safety research on hiPSC-CMs is imperative. In this study, hiPSC-CMs were employed to show the potential of these cells for patient-specific, drug screening purposes. We believe that hiPSC-CMs will eventually allow us to conduct “personalized” drug screening assays because these stem cell-derived cardiomyocytes have the same genomic background and express the same genetic mutations as the patient.
Arrhythmia detection using impedance measures and hiPSC-CMs is already being used by pharmaceutical companies and contract research organizations49-51. Here we present a refinement upon these methodologies and show that the low-impedance MEA/hiPSC-CM platform can be used to identify and quantify arrhythmic events such as EADs and ectopic beats, the actual underlying mechanisms of TdP, confirming the presence of the phase 2-3 window necessary for inward current to produce these arrhythmic events 6, 8, 52, 53. Importantly, the incidence of drug-induced arrhythmias was consistent with animal models, especially the dog. The incidence of sotalol-induced arrhythmias at a concentration of 100 μM was close to that of the dog LV wedge and dog mid-myocardial cardiomyocyte 40, 54 (Supplemental Figure 14A). The incidence of quinidine-induced arrhythmias at a concentration of 1 μM was very similar to that of the dog Purkinje fiber 55, 56 (Supplemental Figure 14B). Only 2 previous studies evaluated arrhythmia quantification, but these were on single hESC-CMs using patch-clamp or sharp electrodes 57, 58. Unfortunately, arrhythmic behavior is often encountered in both “normal” hESC-CMs and hiPSC-CMs 29, 57. The use of EADs or ectopic beats as an endpoint for drug safety has a greater prognostic value than hERG IC50 assays and could potentially reduce the attrition of drugs with therapeutic potential. In fact, in a thorough review of the literature, Redfern and colleagues found the hERG IC50 assay could only predict safety if the block occurred at a 30-fold concentration above the effective therapeutic plasma concentrations 7. At present, patch-clamping is the gold standard for assessing whether a drug affects a specific ion channel, such as hERG. However, because many drugs have pleiotropic effects, the MEA/hiPSC-CM assay is more likely to identify arrhythmogenic effects in human cardiac myocytes that go beyond simple hERG effects alone, as our results with alfuzosin demonstrate (Figure 5A). The MEA/hiPSC-CM assay thus may be a better first-line assay for determining cardiotoxicity that could complement patch-clamp studies to pinpoint the specific channel(s) underlying the QT prolongation or arrhythmic behavior.
Our findings further indicate that the MEA/hiPSC-CM assay may also be more cost-effective and efficient than either PF assays or patch-clamping single hiPSC-CMs. As shown in Figure 5A, the latency for sotalol prolongation is much shorter than the PF assay and comparable to patch-clamp assays13. As continuous perfusion is avoided to prevent mechanical stimulation that may affect beating frequency, there is approximately a 35-fold reduction in test article consumption compared to the PF assay and a 5-fold reduction compared to the patch-clamp assay. In addition, perforated patch-clamping techniques require 15 to 20 minutes to achieve a gigaseal and then a stable membrane resistance in addition to the drug exposure time. MEAs only require culturing the sample on the probe. This increases the productivity significantly because data are always obtained with MEAs.
Although patient-specific hiPSC-CMs hold tremendous potential in terms of their applications for personalized drug screening, there are several limitations at present. A major issue associated with iPSC-derived cells is their immaturity when compared to primary cells obtained from human adult tissues. A number of landmark in vitro disease modeling studies have shown that neurons, hepatocytes, endothelial cells, and cardiomyocytes derived from iPSCs are developmentally immature and do not perfectly replicate the physiological properties of their natural adult counterparts 59-62. Previous work has shown that hiPSC-CMs exhibit functional but incomplete calcium handling components in the sarcoplasmic reticulum, leading to an electrophysiological output that imperfectly replicates that of adult cardiomyocytes 62. Both hESC-CMs and hiPSC-CMs could be better characterized as resembling human fetal cardiomyocytes as opposed to adult cardiomyocytes in terms of their electrophysiology and gene expression 63-65. It is also important to note that whereas immature hiPSC-CMs can develop specific disease phenotypes after two months of in vitro culture, a patient's clinical phenotype can take years or decades to manifest. Finally, it is critical to investigate means of improving hiPSC-CM yield, cell homogeneity, and maturity during the in vitro differentiation process if these cells are to be used for large-scale, drug testing studies. In addition to the aforementioned method of differentiating embryoid bodies to iPSC-CMs, our group and others have started using a monolayer-based differentiation approach to obtain >80% yields of functioning hiPSC-CMs 66. Differentiation protocols will improve as we devise more effective methods of fine-tuning cardiac fate specification in vitro. In spite of the current limitations associated with hiPSC-CM technology, we believe that even fetal-like hiPSC-CMs hold a distinct edge over cells from transgenic animals when it comes to pharmacological drug screening assays for the simple fact that hiPSC-CMs are derived from human tissues and are being used to study human diseases.
In summary, we have recapitulated drug-induced arrhythmias with MEA on hiPSC-CMs. Our results support the use of the MEA/hiPSC-CM platform for drug development. These hiPSC-CMs represent a useful tool for safety pharmacology that can complement the current industry standards and have demonstrated potential for personalized therapeutics. We foresee the implementation of these methodologies in future drug safety practices and guidelines.
Supplementary Material
Acknowledgments
Funding Sources: We gratefully acknowledge funding support from Burroughs Wellcome Foundation, Leducq Foundation, NIH New Innovator Award DP2OD004437, NIH R01 HL113006, NIH U01 HL099776 (JCW), and California Institute for Regenerative Medicine (CIRM) RB3-05129 (SMW). The authors thank AutoMate Scientific and AlphaMED Scientific for their technical support.
Footnotes
Conflict of Interest Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:107–116. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Mandenius CF, Steel D, Noor F, Meyer T, Heinzle E, Asp J, Arain S, Kraushaar U, Bremer S, Class R, Sartipy P. Cardiotoxicity testing using pluripotent stem cell-derived human cardiomyocytes and state-of-the-art bioanalytics: A review. J Appl Toxicol. 2011;31:191–205. doi: 10.1002/jat.1663. [DOI] [PubMed] [Google Scholar]
- 3.Kannankeril PJ, Roden DM. Drug-induced long qt and torsade de pointes: Recent advances. Curr Opin Cardiol. 2007;22:39–43. doi: 10.1097/HCO.0b013e32801129eb. [DOI] [PubMed] [Google Scholar]
- 4.Denning C, Anderson D. Cardiomyocytes from human embryonic stem cells as predictors of cardiotoxicity. Drug Discovery Today: Ther Strategies. 2008;5(4):223–232. [Google Scholar]
- 5.Fermini B, Fossa AA. The impact of drug-induced qt interval prolongation on drug discovery and development. Nat Rev Drug Discov. 2003;2:439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- 6.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical qt interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long qt syndrome. Current opinion in cardiology. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Roden DM, Viswanathan PC. Genetics of acquired long qt syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kannankeril P, Roden DM, Darbar D. Drug-induced long qt syndrome. Pharmacol Rev. 2010;62:760–781. doi: 10.1124/pr.110.003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin RL, McDermott JS, Salmen HJ, Palmatier J, Cox BF, Gintant GA. The utility of herg and repolarization assays in evaluating delayed cardiac repolarization: Influence of multi-channel block. Journal of cardiovascular pharmacology. 2004;43:369–379. doi: 10.1097/00005344-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Masumiya H, Saito M, Ito M, Matsuda T, Noguchi K, Iida-Tanaka N, Tanaka H, Shigenobu K. Lack of action potential-prolonging effect of terfenadine on rabbit myocardial tissue preparations. Biol Pharm Bull. 2004;27:131–135. doi: 10.1248/bpb.27.131. [DOI] [PubMed] [Google Scholar]
- 13.Peng S, Lacerda AE, Kirsch GE, Brown AM, Bruening-Wright A. The action potential and comparative pharmacology of stem cell-derived human cardiomyocytes. J Pharmacol Toxicol Methods. 2010;61:277–286. doi: 10.1016/j.vascn.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Cheng HC, Incardona J. Models of torsades de pointes: Effects of fpl64176, dpi201106, dofetilide, and chromanol 293b in isolated rabbit and guinea pig hearts. J Pharmacol Toxicol Methods. 2009;60:174–184. doi: 10.1016/j.vascn.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- 16.Belles B, Malecot CO, Hescheler J, Trautwein W. “Run-down” of the ca current during long whole-cell recordings in guinea pig heart cells: Role of phosphorylation and intracellular calcium. Pflugers Arch. 1988;411:353–360. doi: 10.1007/BF00587713. [DOI] [PubMed] [Google Scholar]
- 17.Hao LY, Kameyama A, Kameyama M. A cytoplasmic factor, calpastatin and atp together reverse run-down of ca2+ channel activity in guinea-pig heart. J Physiol. 1999;514(Pt 3):687–699. doi: 10.1111/j.1469-7793.1999.687ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao LY, Wang WY, Minobe E, Han DY, Xu JJ, Kameyama A, Kameyama M. The distinct roles of calmodulin and calmodulin kinase ii in the reversal of run-down of l-type ca(2+) channels in guinea-pig ventricular myocytes. J Pharmacol Sci. 2009;111:416–425. doi: 10.1254/jphs.09094fp. [DOI] [PubMed] [Google Scholar]
- 19.Ebert AD, Liang P, Wu JC. Induced pluripotent stem cells as a disease modeling and drug screening platform. Journal of Cardiovascular Pharmacology. 2012;60:408–416. doi: 10.1097/FJC.0b013e318247f642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mordwinkin NM, Burridge PW, Wu JC. A review of human pluripotent stem cell-derived cardiomyocytes for high-throughput drug discovery, cardiotoxicity screening, and publication standards. J Cardiovasc Transl Res. 2013;6:22–30. doi: 10.1007/s12265-012-9423-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301:H2006–2017. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: Human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR. Patient-specific induced pluripotent stem-cell-derived models of leopard syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long qt syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 26.Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long qt syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-qt syndrome. N Engl J Med. 2010 doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 28.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Nguyen PK, Wang PJ, Bers DM, Robbins RC, Wu JC. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease specific patterns of cardiotoxicity. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.001883. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caspi O, Itzhaki I, Kehat I, Gepstein A, Arbel G, Huber I, Satin J, Gepstein L. In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem cells and development. 2009;18:161–172. doi: 10.1089/scd.2007.0280. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a kdr+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 34.Brandenburger M, Wenzel J, Bogdan R, Richardt D, Nguemo F, Reppel M, Hescheler J, Terlau H, Dendorfer A. Organotypic slice culture from human adult ventricular myocardium. Cardiovasc Res. 2012;93:50–59. doi: 10.1093/cvr/cvr259. [DOI] [PubMed] [Google Scholar]
- 35.Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353–370. [Google Scholar]
- 36.Patterson S, Agin M, Anziono R, Burgess T, Chuang-Stein C, Dmitrienko A, Ferber G, Geraldes M, Ghosh K, Menton R, Natarajan J, Offen W, Sound J, Smith B, Suresh R, Zariffa N, A PRM. Investigating drug-induced qt and qtc prolongation in the clinic: A review of statistical design and analysis considerations: Report from the pharmaceutical research and manufacturers of america qt statistics expert team. Drug Inf J. 2005;39:243–265. [Google Scholar]
- 37.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long qt syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the herg channel drug binding site. J Biol Chem. 2004;279:10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- 39.Dausse E, Berthet M, Denjoy I, Andre-Fouet X, Cruaud C, Bennaceur M, Faure S, Coumel P, Schwartz K, Guicheney P. A mutation in herg associated with notched t waves in long qt syndrome. J Mol Cell Cardiol. 1996;28:1609–1615. doi: 10.1006/jmcc.1996.0151. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in lqt2 and lqt3 models of the long-qt syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 41.Roden DM. Drug-induced prolongation of the qt interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 42.El-Sherif N, Caref EB, Chinushi M, Restivo M. Mechanism of arrhythmogenicity of the short-long cardiac sequence that precedes ventricular tachyarrhythmias in the long qt syndrome. J Am Coll Cardiol. 1999;33:1415–1423. doi: 10.1016/s0735-1097(98)00700-1. [DOI] [PubMed] [Google Scholar]
- 43.Lacerda AE, Kuryshev YA, Chen Y, Renganathan M, Eng H, Danthi SJ, Kramer JW, Yang T, Brown AM. Alfuzosin delays cardiac repolarization by a novel mechanism. J Pharmacol Exp Ther. 2008;324:427–433. doi: 10.1124/jpet.107.128405. [DOI] [PubMed] [Google Scholar]
- 44.Satin J, Kehat I, Caspi O, Huber I, Arbel G, Itzhaki I, Magyar J, Schroder EA, Perlman I, Gepstein L. Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J Physiol. 2004;559:479–496. doi: 10.1113/jphysiol.2004.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin RL, McDermott JS, Salmen HJ, Palmatier J, Cox BF, Gintant GA. The utility of herg and repolarization assays in evaluating delayed cardiac repolarization: Influence of multi-channel block. Journal of cardiovascular pharmacology. 2004;43:369–379. doi: 10.1097/00005344-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 46.Doss MX, Di Diego JM, Goodrow RJ, Wu Y, Cordeiro JM, Nesterenko VV, Barajas-Martinez H, Hu D, Urrutia J, Desai M, Treat JA, Sachinidis A, Antzelevitch C. Maximum diastolic potential of human induced pluripotent stem cell-derived cardiomyocytes depends critically on i(kr) PLoS One. 2012;7:e40288. doi: 10.1371/journal.pone.0040288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otsuji TG, Minami I, Kurose Y, Yamauchi K, Tada M, Nakatsuji N. Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem Cell Res. 2010;4:201–213. doi: 10.1016/j.scr.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Sartiani L, Bettiol E, Stillitano F, Mugelli A, Cerbai E, Jaconi ME. Developmental changes in cardiomyocytes differentiated from human embryonic stem cells: A molecular and electrophysiological approach. Stem Cells. 2007;25:1136–1144. doi: 10.1634/stemcells.2006-0466. [DOI] [PubMed] [Google Scholar]
- 49.Jonsson MK, Wang QD, Becker B. Impedance-based detection of beating rhythm and proarrhythmic effects of compounds on stem cell-derived cardiomyocytes. Assay Drug Dev Technol. 2011;9:589–599. doi: 10.1089/adt.2011.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, Chiao E, Kolaja KL. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. 2011;123:281–289. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]
- 51.Abassi YA, Xi B, Li N, Ouyang W, Seiler A, Watzele M, Kettenhofen R, Bohlen H, Ehlich A, Kolossov E, Wang X, Xu X. Dynamic monitoring of beating periodicity of stem cell-derived cardiomyocytes as a predictive tool for preclinical safety assessment. Br J Pharmacol. 2012;165:1424–1441. doi: 10.1111/j.1476-5381.2011.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.January CT, Riddle JM. Early afterdepolarizations: Mechanism of induction and block. A role for l-type ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 53.January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: Induction with the ca2+ channel agonist bay k 8644. Circ Res. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- 54.Abi-Gerges N, Ji GJ, Lu ZJ, Fischmeister R, Hescheler J, Fleischmann BK. Functional expression and regulation of the hyperpolarization activated non-selective cation current in embryonic stem cell-derived cardiomyocytes. J Physiol. 2000;523(Pt 2):377–389. doi: 10.1111/j.1469-7793.2000.t01-2-00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roden DM, Hoffman BF. Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine purkinje fibers. Relationship to potassium and cycle length. Circ Res. 1985;56:857–867. doi: 10.1161/01.res.56.6.857. [DOI] [PubMed] [Google Scholar]
- 56.Davidenko JM, Cohen L, Goodrow R, Antzelevitch C. Quinidine-induced action potential prolongation, early afterdepolarizations, and triggered activity in canine purkinje fibers. Effects of stimulation rate, potassium, and magnesium. Circulation. 1989;79:674–686. doi: 10.1161/01.cir.79.3.674. [DOI] [PubMed] [Google Scholar]
- 57.He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: Action potential characterization. Circulation research. 2003;93:32–39. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- 58.Jonsson MK, Duker G, Tropp C, Andersson B, Sartipy P, Vos MA, van Veen TA. Quantified proarrhythmic potential of selected human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:189–200. doi: 10.1016/j.scr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 59.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810–1819. doi: 10.1002/hep.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taura D, Sone M, Homma K, Oyamada N, Takahashi K, Tamura N, Yamanaka S, Nakao K. Induction and isolation of vascular cells from human induced pluripotent stem cells--brief report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:1100–1103. doi: 10.1161/ATVBAHA.108.182162. [DOI] [PubMed] [Google Scholar]
- 62.Lee YK, Ng KM, Lai WH, Chan YC, Lau YM, Lian Q, Tse HF, Siu CW. Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem cell reviews. 2011;7:976–986. doi: 10.1007/s12015-011-9273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beqqali A, Kloots J, Ward-van Oostwaard D, Mummery C, Passier R. Genome-wide transcriptional profiling of human embryonic stem cells differentiating to cardiomyocytes. Stem Cells. 2006;24:1956–1967. doi: 10.1634/stemcells.2006-0054. [DOI] [PubMed] [Google Scholar]
- 64.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends in molecular medicine. 2011;17:475–484. doi: 10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 65.Cao F, Wagner RA, Wilson KD, Xie X, Fu JD, Drukker M, Lee A, Li RA, Gambhir SS, Weissman IL, Robbins RC, Wu JC. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS One. 2008;3:e3474. doi: 10.1371/journal.pone.0003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee P, Klos M, Bollensdorff C, Hou L, Ewart P, Kamp TJ, Zhang J, Bizy A, Guerrero-Serna G, Kohl P, Jalife J, Herron TJ. Simultaneous voltage and calcium mapping of genetically purified human induced pluripotent stem cell-derived cardiac myocyte monolayers. Circ Res. 2012;110:1556–1563. doi: 10.1161/CIRCRESAHA.111.262535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.