

Abstract
Myofibroblasts are the major source of extracellular matrix components that accumulate during tissue fibrosis, and hepatic stellate cells (HSCs) are the major source of myofibroblasts in the liver. To date, robust systems to genetically manipulate these cells have not existed. We report that Pdgfrb-Cre inactivates genes in murine HSCs with high efficiency. We used this system to delete the αv integrin subunit because of the suggested role of multiple αv integrins as central mediators of fibrosis in multiple organs. Depletion of the αv integrin subunit in HSCs protected mice from CCl4-induced hepatic fibrosis, whereas global loss of αvβ3, αvβ5 or αvβ6 or conditional loss of αvβ8 on HSCs did not. Pdgfrb-Cre effectively targeted myofibroblasts in multiple organs, and depletion of αv integrins using this system was also protective in models of pulmonary and renal fibrosis. Critically, pharmacological blockade of αv integrins by a novel small molecule (CWHM 12) attenuated both liver and lung fibrosis, even when administered after fibrosis was established. These data identify a core pathway that regulates fibrosis, and suggest that pharmacological targeting of all αv integrins may have clinical utility in the treatment of patients with a broad range of fibrotic diseases.
Chronic tissue injury with fibrosis, loss of tissue architecture and organ dysfunction are characteristic features of many human diseases and major causes of morbidity and mortality worldwide. Currently, treatment of tissue fibrosis is severely limited and transplantation is often the only effective treatment for end-stage fibrotic diseases. However limited donor organ availability and the high cost and morbidity of transplantation underscore the urgent need for more effective therapies.
Secreted transforming growth factor beta (TGF-β) is arguably the major pro-fibrogenic cytokine and a central mediator of fibrosis in multiple organs. The molecular pathways that regulate TGF-β activity and signaling are therefore attractive targets for novel anti-fibrotic treatments. TGF-β is secreted as a latent complex that is present at high concentrations directly cross-linked to the extracellular matrix, and much of the regulation of TGF-β function in tissues is based on extracellular activation of this latent complex1,2. Two of the three mammalian TGF-β isoforms (TGF-β1 and 3) can be activated by members of the integrin family that interact with a linear arginine-glycine-aspartic acid (RGD) motif present in an amino terminal fragment of the TGF-β gene product called the latency associated peptide3,4,5. Inhibition and blockade of two of these integrins (αvβ6 and αvβ8) phenocopies all of the developmental effects of loss of TGF-β1 and 36, suggesting that these two integrins are required for most or all important roles of these TGF-β isoforms in development. However, the mechanisms of TGF-β activation that contribute to tissue pathology in adults are less well understood.
We and others have previously shown that TGF-β activation by the αvβ6 integrin plays an important role in models of fibrosis in the lungs, biliary tract and kidney3,7,8,9. However αvβ6 does not appear to be critical in all models of organ fibrogenesis, as ablation of αvβ6 was not protective in carbon tetrachloride (CCl4)-induced liver fibrosis7. Crucially, αvβ6 is largely restricted in its expression to a subset of epithelial cells7,10,11, and generates very localized activation of TGF-β that can only be detected by cells in direct contact with the integrin-expressing cell3. Moreover, tissue fibrosis in many organs is due to collagen production by myofibroblasts that are often at substantial distance from any αvβ6-expressing epithelial cells with implications for therapeutic design. Myofibroblasts express several αv integrins and are contractile cells, capable of exerting force on tethered ligands. The recently solved crystal structure of the small latent complex of TGF-β demonstrates that mechanical force generated by the contractile actomyosin cytoskeleton and transmitted by integrins is a common mechanism for activating latent TGF-β12. Furthermore, elegant in vitro studies of myofibroblasts have shown that they can utilize alternative αv integrins to activate TGF-β13, and we and others have shown that several integrins that share the αv subunit, including αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8 can recognize the same RGD peptide motif and, at least in some circumstances, can activate latent TGF-β3,4,14,15,16. Together these data suggest that myofibroblast αv expression may be critical to fibrosis and targetable. However, the precise importance in vivo of each of these integrins is undetermined.
The paucity of tools that allow reliable, specific inactivation of genes in myofibroblasts in vivo has greatly hindered progress in understanding the underlying biology of fibrotic diseases. In this study, we developed a strategy to inactivate genes in myofibroblasts in multiple organs. PDGFRβ (platelet derived growth factor receptor beta) induction is an early feature of myofibroblast activation in a broad range of tissues17,18,19,20,21,22,23,24. We therefore evaluated the efficiency of targeting these cells using Pdgfrb-Cre mice25, and used this system to investigate the role of αv integrins on myofibroblasts in regulating pathologic fibrosis in multiple solid organs.
Results
Pdgfrb-Cre effectively targets recombination in hepatic stellate cells
In the liver, hepatic stellate cells (liver specific pericytes) are the major source of extracellular matrix during hepatic fibrogenesis26,27. To date, no effective strategy has been developed to reliably delete genes specifically in HSCs. As PDGFRβ induction is an early feature of activation of HSCs to myofibroblasts17,18,19,20,21, we evaluated the efficiency of targeting HSCs using Pdgfrb-Cre mice. These mice, which express Cre recombinase under the control of a fragment of the gene encoding PDGFRβ, were previously developed to specifically target pericytes25. In the uninjured liver, HSCs are found in a peri-vascular location in close contact with underlying sinusoidal endothelial cells, and morphologically resemble pericytes in other organs.
Using mTmG reporter mice (a double-fluorescent reporter mouse that expresses membrane-targeted tandem dimer tomato (TdTomato) prior to Cre-mediated excision and membrane-targeted green fluorescent protein (mGFP) after excision)28 and Ai14 reporter mice (a single fluorescent reporter that expresses TdTomato after recombination)29 we found that Pdgfrb-Cre induced highly efficient recombination in a distribution appropriate for HSCs, both in uninjured control liver and after induction of hepatic fibrosis with repeated administration of CCl4 (Fig. 1a,b). To evaluate the specificity of recombination in HSCs we co-stained with desmin (a well characterized marker of HSCs) which was expressed in virtually all of the reporting cells (Fig. 1c, upper panels, Supplementary Fig. 1a). Co-staining with PDGFRβ confirmed both appropriate reporting by Pdgfrb Cre and also hitherto unknown expression of PDGFRβ by quiescent HSCs in uninjured liver (Fig. 1c, middle panels, Supplementary Fig. 1a). As expected α-SMA (a well characterized marker of activated HSCs) was seen only in larger blood vessel walls in uninjured liver (Fig. 1c, lower panels). In fibrotic liver, almost all of the reporting cells also expressed desmin and PDGFRβ, both in fibrous septae and the surrounding liver parenchyma (Fig. 1d, upper and middle panels, Supplementary Fig. 1a). Furthermore, virtually all of the α-SMA positive cells observed in fibrotic livers were also reporter positive (Fig. 1d, lower panels, Supplementary Fig. 1a) demonstrating a high degree of recombination in activated liver myofibroblasts. To further assess specificity of Pdgfrb-Cre recombination, we stained uninjured control and fibrotic livers from Ai14;Pdgfrb-Cre mice with antibodies to CD31 (endothelial cells), F4/80 (kupffer cells), CD3 (T cells) and pan-cytokeratin (biliary epithelium). No reporting cells expressed any of these markers (Supplementary Fig. 1b). As PDGFRβ expression by quiescent HSCs has not previously been reported we utilized an alternative strategy to further assess PDGFRβ expression in quiescent HSC. Analysis of Pdgfrb-BAC-eGFP knock-in reporter mice demonstrated that the eGFP positive cells in the livers from these mice report in an identical distribution to the TdTomato positive cells in the livers of Ai14;Pdgfrb-Cre mice (Supplementary Fig. 1c). Furthermore, to evaluate the specificity of reporting in quiescent HSCs we co-stained with desmin which was expressed in virtually all of the reporting cells (Supplementary Fig. 1c,d). Finally, co-staining with PDGFRβ demonstrated both appropriate reporting by the Pdgfrb-BAC-eGFP system and confirmed expression of PDGFRβ by quiescent HSCs (Supplementary Fig. 1c,d).
Figure 1. Pdgfrb-Cre effectively targets recombination in quiescent and activated hepatic stellate cells.

(a,b) Immunofluoresence micrographs of liver sections harvested from control (olive oil treated) or chronic CCl4 treated (x2 injections / week for six weeks) mTmG;Pdgfrb-Cre reporter mice or Ai14;Pdgfrb-Cre reporter mice (n = 4 male mice per group). Scale bars, 100 μm. (c,d) Immunofluoresence micrographs of liver sections from control (olive oil treated) (c) or CCl4 treated (d) Ai14;Pdgfrb-Cre mice (n = 4 male mice per group) stained for desmin, PDGFRβ or αSMA (green) with endogenous (not enhanced) Ai14-Td tomato report in red. (c) Scale bars, 25 μm. (d) Scale bar, 50 μm. Arrows indicate portal tracts in serial sections. (e) Gene expression profile of freshly sorted Td tomato positive cells from control (olive oil treated) or chronic CCl4 treated Ai14;Pdgfrb-Cre mice (n = 4 male mice per group). (f) Immunofluoresence staining of Td tomato positive cells sorted from the uninjured livers of Ai14;Pdgfrb-Cre mice and plated on tissue culture plastic for 7 days. Left panel shows Ai14 reporter (red), DAPI (blue), middle panel shows αSMA (green), DAPI (blue), right panel shows merged image. Scale bar, 100μm. Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
To further characterize reporting cells in Ai14;Pdgfrb-Cre mice we isolated non-parenchymal cells from uninjured control or fibrotic livers and purified Ai14 (Td tomato) positive cells by cell sorting. qPCR of mRNA obtained from live Td tomato positive cells showed marked induction of multiple genes associated with transition of quiescent HSCs to the activated, myofibroblast phenotype including desmin, PDGFRβ, αSMA, collagens I and III, MMPs and TIMP1 (Fig. 1e), and higher expression of a number of these genes was also confirmed at the protein level (Supplementary Fig. 1e–g). In addition, we found significantly higher expression of multiple αv integrin family genes following fibrosis induction (Fig. 1e). We also plated freshly isolated Td tomato positive cells from uninjured Ai14;Pdgfrb-Cre mouse livers on tissue culture plastic for five days to induce HSC activation. All of the cells in these sorted cultures expressed the myofibroblast marker αSMA (Fig. 1f). Taken together, these results suggest that Pdgfrb-Cre efficiently targets both quiescent and activated HSCs.
HSC αv integrin depletion protects mice from hepatic fibrosis
To determine whether Pdgfrb-Cre could be used to identify molecular mechanisms driving hepatic fibrosis, we focused on the integrin αv subunit because of the suggested role of multiple αv integrins in activating latent transforming growth factor β (TGF-β), a central mediator of fibrosis3,4,13,15,16.
Confirming our qPCR data (Fig. 1e) we found that mouse HSCs express multiple αv integrins (αvβ1, αvβ3, αvβ5 and αvβ8; Fig. 2a) and that αv integrin expression was significantly up-regulated during activation ex-vivo (Fig. 2b,c). We also found αv integrin expression on hepatic myofibroblasts in human fibrotic liver tissue (Supplementary Fig. 2a). αv integrin was efficiently deleted from itgavflox/flox;Pdgfrb-Cre (αv Cre) HSCs, as assessed by western blotting with an antibody raised against a portion of the αv extracellular domain (Fig. 2d). Furthermore itgavflox/flox;Pdgfrb-Cre mice were significantly protected from CCl4-induced fibrosis, as determined by collagen staining, hydroxyproline content and αSMA staining (Fig. 2e–h). This was not due to changes in the degree of initial injury caused by CCl4 (Supplementary Fig. 2b,c), the number of HSCs prior to injury (Supplementary Fig. 2d,e), differences in inflammatory infiltrates (Supplementary Fig. 2f–i) or changes in the vasculature (Supplementary Fig. 2j) of itgavflox/flox;Pdgfrb-Cre livers compared to control. In addition, αv integrin expression was not affected in purified hepatocytes, liver sinusoidal endothelial cells or kupffer cells from itgavflox/flox;Pdgfrb-Cre mice compared to control further confirming the specificity of Pdgfrb-Cre for HSCs (Supplementary Fig. 3a). Furthermore, HSCs cultured for 5 days from control and itgavflox/flox;PDGFRβ-Cre mice demonstrated robust expression of PDGFRβ, however as expected hepatocytes, liver sinusoidal endothelial cells and kupffer cells did not express PDGFRβ (Supplementary Fig. 3a). These data suggest that αv integrins on HSCs promote CCl4-induced liver fibrosis, and that Pdgfrb-Cre effectively targets gene deletion in these cells during fibrogenesis.
Figure 2. Depletion of the αv integrin on hepatic stellate cells protects mice from CCl4-induced hepatic fibrosis.

(a) Co-immunoprecipitation studies: Sorted Td tomato positive cells from uninjured livers of Ai14;Pdgfrb-Cre mice cultured for 7 days express αvβ1, αvβ3, αvβ5 and αvβ8. (b) Western blot and (c) qPCR analysis demonstrates induction of αv integrin expression during transition from the quiescent (day 0) to the culture activated phenotype (day 5,10) in wild type HSCs. (d) Western blotting of αv expression in control and itgavflox/flox;Pdgfrb-Cre (αv Cre) HSCs culture activated for 5 days. (e) Picrosirius red staining (collagen deposition) (upper panels) and αSMA immunohistochemistry (lower panels) of liver tissue after olive oil or chronic CCl4 treatment of control and itgavflox/flox;Pdgfrb-Cre mice (n = 8 male mice per group). Scale bar, 200μm. (f) Digital image analysis quantification of collagen staining. (g) Hydroxyproline analysis. (h) Digital image analysis quantification of αSMA staining. Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
HSC αv integrin depletion inhibits fibrosis via reduced TGF-β activation
To investigate whether loss of αv integrins affected activation-induced induction of extracellular matrix protein gene expression, control and αv null HSCs were culture activated for 5 days. αSMA, Col1A1 and Col3A1 expression was significantly reduced in itgavflox/flox;Pdgfrb-Cre HSCs (Fig. 3a–c). Furthermore, treatment with an anti-αv blocking antibody (RMV-7) also inhibited expression of these pro-fibrotic genes (Fig. 3d–f). Culture activated HSCs from control and itgavflox/flox;Pdgfrb-Cre mice had similar TGFβ1 mRNA levels (Fig. 3g). However, co-culture of HSCs from control and itgavflox/flox;Pdgfrb-Cre mice with mink lung epithelial reporter cells (TMLC) expressing firefly luciferase under the control of the TGF-β sensitive PAI-1 promoter30 demonstrated a significant reduction in the levels of active TGF-β by αv null HSCs compared to control. This difference was eliminated by TGF-β blocking antibody and rescued with addition of activated TGF-β1 (Fig. 3h), identifying a decrease in TGF-β activation by αv null HSCs. Furthermore, we found no significant difference in levels of total TGF-β1 protein in cell lysates and supernatants from control and itgavflox/flox;Pdgfrb-Cre HSCs cultured with and without TMLC, confirming that the reduction in TGF-β activation by itgavflox/flox;Pdgfrb-Cre HSCs is not secondary to a decrease in total TGF-β1 expression levels (Supplementary Fig. 3b,c). Moreover, addition of TGF-β1 to control and itgavflox/flox;Pdgfrb-Cre HSCs restored pro-fibrotic gene expression levels in αv null HSCs to TGF-β1-treated control levels (Supplementary Fig. 3d,e). As multiple cell types can secrete TGF-β, we investigated the cellular sources of TGF-β during liver fibrosis by purifying HSCs, hepatocytes, liver sinusoidal endothelial cells and kupffer cells from fibrotic livers, and found that the major cellular sources of TGF-β were HSCs and kupffer cells (Supplementary Fig. 4a). Previous studies have shown that TGF-β activation occurs in a highly spatially restricted manner3, but that integrins can activate TGF-β produced by any cells in the local vicinity.
Figure 3. αv integrin depletion on hepatic stellate cells inhibits pro-fibrotic gene expression via a reduction in transforming growth factor beta (TGF-β) activation.

(a) qPCR (left panel) and western blot analysis of αSMA expression (right panel) in control and itgavflox/flox;Pdgfrb-Cre (αv Cre) HSCs culture activated for 5 days. (b,c) qPCR analysis of Col1A1 and Col3A1 expression in control and itgavflox/flox;Pdgfrb-Cre (αv Cre) HSCs culture activated for 5 days. (d) qPCR (left panel) and western blot analysis of αSMA expression (right panel) in control and itgavflox/flox;Pdgfrb-Cre HSCs treated with 20 μg ml−1 isotype control or αv integrin specific (clone RMV-7) antibodies for 5 days post plating. (e,f) qPCR analysis of Col1A1 and Col3A1 expression in control and itgavflox/flox;Pdgfrb-Cre HSCs treated with 20 μg ml−1 isotype control or αv integrin specific (clone RMV-7) antibodies for 5 days post plating. (g) qPCR analysis of Tgfb1 expression in control and itgavflox/flox;Pdgfrb-Cre HSCs culture activated for 5 days. (h) TGF-β activation by control or itgavflox/flox;Pdgfrb-Cre HSCs alone, or in the presence of TGF-β blocking antibody (clone 1D11, 40 μg ml−1) or activated TGF-β (300 pg ml−1). (i) Immunofluoresence micrographs of liver sections from control and itgavflox/flox;Pdgfrb-Cre mice following chronic CCl4 treatment (n = 8 male mice per group). Scale bars, 25 μm. (j) Digital image analysis quantification of phospho-SMAD3 staining. Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
We also examined whether adhesion and migration defects in αv null HSCs might contribute to the protection from CCl4-induced fibrosis observed in itgavflox/flox;Pdgfrb-Cre mice. However there was no significant difference in adhesion to multiple matrix proteins present in normal or fibrotic liver and no difference in cell migration between control and itgavflox/flox;Pdgfrb-Cre HSCs (Supplementary Fig. 4b,c). Furthermore the rate of apoptosis (assessed by TUNEL staining) was similar in control and itgavflox/flox;Pdgfrb-Cre HSCs (Supplementary Fig. 4d,e). We also assessed whether depletion of the αv integrin on HSCs might alter the expression of other β1 subunit-containing integrins, however expression of α1, α5 and β1 was similar in control and itgavflox/flox;Pdgfrb-Cre HSCs (Supplementary Fig. 5a–c).
As we had shown that itgavflox/flox;Pdgfrb-Cre HSCs are defective in their ability to activate TGF-β in vitro, we extended our findings in vivo by examining canonical TGF-β signaling by immunostaining for phospho-SMAD3 (Fig. 3i). Digital image quantitation demonstrated significantly reduced phospho-SMAD3 signaling in the livers of itgavflox/flox;Pdgfrb-Cre mice compared to controls following chronic CCl4 administration (Fig. 3j). Taken together these results strongly suggest that the protection from CCl4-induced hepatic fibrosis observed in itgavflox/flox;Pdgfrb-Cre mice is at least in part a consequence of reduced TGF-β activation by αv deficient HSCs.
Assessment of individual αv integrin heterodimers in CCl4-induced hepatic fibrosis
The αv integrin subunit has five possible β subunit binding partners (β1, β3, β5, β6 and β8)31, each of which has been reported to bind and / or activate latent TGF-β3,4,14,15,16, and four of which we found were expressed on HSCs (αvβ1, αvβ3, αvβ5 and αvβ8 – Fig. 2a).
To further assess the potential contribution of each αv integrin heterodimer during hepatic fibrogenesis we evaluated the response to CCl4 in mice globally lacking αvβ3, αvβ5, αvβ6 (previously been shown to be important in biliary tract fibrosis7) and in mice lacking αvβ8 on HSCs (itgb8flox/flox;Pdgfrb-Cre). We used conditional deletion of itgb8 from HSCs because global loss of itgb8 is embryonic lethal32. Individual depletion of any one of these integrin subunits failed to protect mice from CCl4-induced hepatic fibrosis (Fig. 4). These results suggest that either multiple αv integrins contribute to hepatic fibrogenesis and TGF-β activation by HSCs, or that the principal relevant integrin is αvβ1. It is not currently possible to specifically evaluate the role of αvβ1 in liver fibrosis in vivo, since global loss of itgb1 is lethal in mice after embryonic day 5.533,34 and itgb1flox/flox;Pdgfrb-Cre mice manifest early postnatal lethality35.
Figure 4. Global loss of αvβ3, αvβ5 or αvβ6 or conditional loss of αvβ8 on hepatic stellate cells does not protect mice from CCl4-induced hepatic fibrosis.

(a–d) Digital image analysis quantification of collagen staining in control and β3 KO, β5 KO, β6 KO and itgb8flox/flox;Pdgfrb-Cre (β8 Cre) mice (n = 6 male mice per group) after control (olive oil) or chronic CCl4 treatment (x2 injections / week for 6 weeks). (e–h) Hydroxyproline analysis of liver tissue from control and β3 KO, β5 KO, β6 KO and itgb8flox/flox;Pdgfrb-Cre mice. (i) Western blotting of integrin β8 subunit expression in control and itgb8flox/flox;Pdgfrb-Cre HSCs culture activated for 7 days.
Selective αv integrin depletion is protective in multiple models of organ fibrosis
Myofibroblasts in the lung and kidney are also the major mediators of extracellular matrix deposition and organ scarring during tissue injury, and have previously been shown to express high levels of PDGFRβ22,23,24,36. We therefore suspected that Pdgfrb-Cre might broadly mark myofibroblasts in multiple organs and allow manipulation of specific genes in these cells. To further evaluate this possibility we initially examined the lungs of mTmG;Pdgfrb-Cre reporter mice 28 days after intratracheal instillation of saline (control) or bleomycin. We found high levels of reporter expression in a distribution that closely resembled lung pericytes in control saline treated lungs (Fig. 5a, left panel). We also saw a marked expansion of reporter cells in fibrotic regions following induction of pulmonary fibrosis with bleomycin (Fig. 5a, right panel). To confirm that reporter expression was occurring specifically in lung pericytes in control saline-treated lung we co-stained with desmin and PDGFRβ (two well characterized markers of pericytes37) and found a high degree of co-localization with each (Fig. 5b, Supplementary Fig. 5d). Following bleomycin almost all of the reporting cells expressed desmin and PDGFRβ (Fig. 5c, upper and middle panels, Supplementary Fig. 5d), and most co-expressed α-SMA (Fig. 5c, lower panels, Supplementary Fig. 5d) demonstrating a high degree of recombination in lung myofibroblasts. qPCR from sorted Ai14-Td tomato positive cells from control or bleomycin-treated animals showed marked induction of multiple genes associated with lung fibrogenesis including αSMA, collagens I and III, MMP2 and TIMP1 at both day 14 and day 28 post-bleomycin instillation (Fig. 5d), and higher expression of αSMA protein confirmed myofibroblast induction in sorted Ai14-Td tomato positive cells following bleomycin treatment (Supplementary Fig. 6a). Furthermore, all sorted reporter cells (isolated from uninjured Ai14;Pdgfrb-Cre mouse lungs) that were activated in vitro by 7 day culture on tissue culture plastic expressed αSMA (Supplementary Fig. 6b). Control or itgavflox/flox;Pdgfrb-Cre (αv Cre) mice were thus treated with saline or bleomycin and lungs harvested 28 days after instillation. Bleomycin induced the expected fibrosis in control mice (as determined by picrosirius red staining and hydroxyproline content) but itgavflox/flox;Pdgfrb-Cre mice were completely protected in this model (Fig. 5e,f). These data demonstrate a central regulatory role for αv integrins on lung pericytes and/or myofibroblasts during lung fibrogenesis.
Figure 5. Pdgfrb-Cre-mediated depletion of the αv integrin is protective in multiple models of solid organ fibrogenesis.

(a) Immunofluoresence micrographs of lung sections from saline treated (left panel) or bleomycin treated (right panel) mTmG;Pdgfrb-Cre mice (n = 4 female mice per group) 28 days after instillation. Scale bar, 50 μm. (b,c) Immunofluoresence micrographs of lung sections from saline treated (b) or bleomycin treated (c) Ai14;Pdgfrb-Cre mice (n = 4 female mice per group). Scale bars, 25 μm. (d) Gene expression profile of freshly sorted Td tomato positive cells from saline treated (28 days post instillation) or bleomycin treated (14 or 28 days post instillation) Ai14;Pdgfrb-Cre mice (n = 4 female mice per group). (e) Picrosirius red staining of lung tissue 28 days after bleomycin instillation in control and itgavflox/flox;Pdgfrb-Cre (αv Cre) mice (n = 12 female mice per group). Scale bar, 100μm. (f) Hydroxyproline analysis of lung tissue. (g) Immunofluoresence micrographs of left kidney sections from mTmG;Pdgfrb-Cre mice (n = 4 male mice per group) following sham operation or unilateral ureteric obstruction (UUO) for 14 days. Scale bar, 100 μm. (h) Gene expression profile of freshly sorted Td tomato positive cells from sham operated or UUO (day 7) Ai14;Pdgfrb-Cre mice (n = 4 male mice per group). (i) Picrosirius red staining of kidney tissue 14 days after UUO in control and itgavflox/flox;Pdgfrb-Cre mice (n = 6 male mice per group). Scale bar, 100μm. (j) Digital image analysis of collagen staining. Data are mean ± s.e.m. *P < 0.05, **P < 0.01 (Student’s t test).
As stated above, renal myofibroblasts express PDGFRβ and are key effector cells in the deposition of extracellular matrix during renal fibrogenesis24. To investigate whether Pdgfrb-Cre efficiently targets renal myofibroblasts we used the unilateral ureteric obstruction (UUO) model of kidney fibrosis. mTmG;Pdgfrb-Cre reporter mice underwent sham operation or UUO and left kidneys were harvested 14 days after surgery. In sham operated mice mGFP positive reporter cells were noted to be perivascular, intra-glomerular and within the renal interstitium (Fig. 5g). UUO resulted in a massive expansion of reporter positive cells throughout the renal interstitium (Fig. 5g). As in the liver and lung, qPCR from sorted live Ai14-Td tomato positive cells from Ai14;Pdgfrb-Cre reporter mice showed induction of a panel of genes known to be induced when renal myofibroblasts activate (Fig. 5h) and higher expression of αSMA protein confirmed myofibroblast induction in sorted Ai14-Td tomato positive cells following UUO (Supplementary Fig. 6c). Furthermore, freshly isolated Ai14-Td tomato positive cells (isolated from uninjured Ai14;Pdgfrb-Cre mouse kidneys) cultured on tissue culture plastic for seven days all expressed the myofibroblast marker αSMA (Supplementary Fig. 6d). As was demonstrated in the liver and lung fibrosis models above, myofibroblast-specific deletion of itgav was protective against UUO-induced kidney fibrosis, as determined by picrosirius red staining and digital morphometric analysis (Fig. 5i,j). Taken together these data demonstrate that αv integrins on tissue myofibroblasts contribute to pathological tissue fibrosis in multiple solid organs.
We have previously shown an important role for the epithelial expressed αvβ6 integrin in bleomycin-induced pulmonary fibrosis3 and UUO-induced kidney fibrosis8,9, and in this study we demonstrate that myofibroblast-expressed αv integrins also contribute to lung and kidney fibrosis. Together, our findings show both cell lineages play an active role in pulmonary and renal fibrogenesis, further underlining the complex cellular interplay defining the program of tissue fibrosis. To exclude an indirect role of loss of αv on myofibroblasts on αvβ6 expression on adjacent epithelial cells, we assessed αvβ6 expression in control and fibrotic lung, liver and kidney in control and itgavflox/flox;Pdgfrb-Cre mice. We found no significant difference in αvβ6 integrin expression between control and itgavflox/flox;Pdgfrb-Cre mice in uninjured or fibrotic lung, liver and kidney tissue (Supplementary Fig. 6e–j). These data demonstrate that the protection from fibrosis seen in the itgavflox/flox;Pdgfrb-Cre mice is not secondary to altered αvβ6 integrin expression.
Blockade of αv integrins attenuates liver and lung fibrosis
Taken together our data indicate that myofibroblast αv inhibition might represent a valuable therapeutic target. Therefore we utilized a novel small molecule inhibitor of αv integrins, CWHM 12, (Supplementary Fig. 7a) to determine whether pharmacologic blockade of αv integrins could attenuate fibrosis. CWHM 12 is a synthetic small molecule RGD peptidomimetic antagonist that consists of a cyclic guanidino-substituted phenyl group as the arginine mimetic, and a phenyl substituted beta amino acid as the aspartic acid mimetic, both linked via glycine (a detailed description of the design and synthesis of CWHM 12 is provided in the Methods section). CWHM 12 demonstrated high potency against all of the five possible β subunit binding partners (αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8) in in vitro ligand-binding assays, with somewhat less potency against αvβ5 than against the other αv integrins (Supplementary Fig. 7c–g,k). We also assessed a control small molecule, CWHM 96, which is the R-enantiomer of CWHM 12, and differs only in the orientation of it’s carboxyl (CO2H) group (Supplementary Fig. 7b, Methods section). In contrast to CWHM 12, CWHM 96 did not inhibit any of the five possible β subunit binding partners (αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8) in in vitro ligand-binding assays (Supplementary Fig. 7c-g,k). Furthermore, to assess the specificity of CWHM 12 we analysed the effect of CWHM 12 in in vitro ligand-binding assays for the following non-αv integrins: αIIbβ3, α2β1 and α10β1. We found that CWHM 12 had no effect on ligand binding for each of these integrins (Supplementary Fig. 7h–j).
We initially examined the potential of CWHM 12 to prevent liver fibrosis by inserting Alzet osmotic minipumps containing either CWHM 12 or vehicle control into mice, followed by CCl4 injections twice weekly for 6 weeks (Fig. 6a). Treatment with CWHM 12 significantly reduced liver fibrosis, as determined by collagen (picrosirius red) and αSMA staining and hydroxyproline content (Fig. 6a–d). We next asked whether CWHM 12 could prevent further progression of established fibrosis. We treated mice with CCl4 for 3 weeks to establish fibrotic disease and then treated with CWHM 12 or vehicle for the final 3 weeks of CCl4 (Fig. 6e). CWHM12 significantly reduced liver fibrosis even after fibrotic disease had been established (Fig. 6e–h). In addition, we examined canonical TGF-β signaling in the livers of control and CWHM 12 treated mice following chronic CCl4 administration by immunostaining for phospho-SMAD3 (Supplementary Fig. 8a,b). Digital image quantitation demonstrated significantly reduced phospho-SMAD3 signaling in the livers of CWHM12 treated mice compared to controls, demonstrating that the protection from CCl4-induced hepatic fibrosis observed in CWHM 12 treated mice is due at least in part to a reduction in TGF-β activation by αv integrins (Supplementary Fig. 8a,b). Furthermore, treatment with CWHM 96 (the control R-enantiomer of CWHM 12) had no effect on liver fibrosis, as determined by collagen (picrosirius red) staining and hydroxyproline content (Supplementary Fig. 8c–f). As αv integrin depletion on myofibroblasts also had anti-fibrotic effects in the lung (Fig. 5e,f), we assessed whether CWHM 12 administration beginning 14 days after intratracheal bleomycin could prevent further progression of lung fibrosis (Fig. 6i). Similar to our findings in the liver, administration of CWHM 12 significantly inhibited progression of pulmonary fibrosis (Fig. 6j). Of course, since we have shown that loss or functional blockade of the epithelial-restricted αvβ6 integrin also prevents pulmonary fibrosis in this model, we cannot be sure that CWHM 12 was not protective in the lung because of its inhibitory effects on αvβ6. Nonetheless, our findings suggest that inhibition of αv integrins by the novel small molecule CWHM 12 has potential clinical utility in the treatment of patients with a broad range of fibrotic diseases.
Figure 6. Blockade of αv integrins by a novel small molecule (CWHM 12) attenuates liver and lung fibrosis.

(a) Dosing regime in the prophylactic liver fibrosis model (left panel). Alzet minipumps containing CWHM 12 or vehicle were inserted, followed by CCl4 I.P. twice weekly for 6 weeks. Picrosirius red (upper) and αSMA immunohistochemistry (lower) of liver tissue from control and CWHM 12 treated mice (n = 6 female mice per group) after chronic CCl4 treatment. Scale bar, 200μm. (b) Digital image analysis of picrosirius red staining. (c) Hydroxyproline analysis. (d) Digital image analysis of αSMA staining. (e) Dosing regime in the therapeutic liver fibrosis model (left panel). Mice were given CCl4 I.P. twice weekly for 3 weeks, then Alzet minipumps containing either CWHM 12 or vehicle were inserted, followed by a further 3 weeks of CCl4 I.P. twice weekly. Picrosirius red (upper) and αSMA immunohistochemistry (lower) of liver tissue from control and CWHM 12 treated mice (n =14 female mice per group) after chronic CCl4 treatment. Scale bar, 200μm. (f) Digital image analysis of picrosirius red staining. (g) Hydroxyproline analysis. (h) Digital image analysis of αSMA staining. (i) Dosing regime in the therapeutic lung fibrosis model. Alzet minipumps containing either CWHM 12 or vehicle were inserted 14 days after treatment with bleomycin or saline, and lungs were harvested at 28 days. (j) Picrosirius red staining of lung tissue from control and CWHM 12 treated mice 28 days after bleomycin instillation (n = 15 female mice per group). Scale bar, 100μm (left panel). Hydroxyproline analysis (right panel). Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Student’s t test).
Discussion
Myofibroblasts are a major source of extracellular matrix that accumulates during fibrosis36. However, the paucity of tools for reliable inactivation of genes in myofibroblasts in vivo has greatly impeded progress in dissecting the molecular mechanisms driving organ fibrosis, thereby slowing the discovery of novel, mechanistically targeted anti-fibrotic treatments. In this study, we developed a strategy (Pdgfrb-Cre) to genetically manipulate myofibroblasts in multiple organs. Using this system we identified a key role for myofibroblast αv integrins in the regulation of fibrosis and then validated our approach by using a small molecule to target αv integrins. We have thus identified a novel and targeted approach to treat fibrosis.
As a first test of the effectiveness of this system, we deleted the αv integrin subunit from myofibroblasts and found that the loss of myofibroblast αv significantly inhibited fibrosis in the liver, lung and kidney, identifying myofibroblast αv integrins as components of a core pathway widely shared by pathological fibrosis in multiple solid organs. Of note, we were unable to effectively inhibit liver fibrosis by individual depletion of four of the β subunit partners of αv (β3, β5, β6 or β8) suggesting that this protection was either due to loss of αvβ1 (which cannot be studied with the tools currently available) or, more likely, that inhibition of multiple αv integrins is required to effectively treat liver fibrosis. Using a novel small molecule inhibitor, CWHM 12, we showed that therapeutically targeting all αv integrins effectively treated fibrosis in multiple organs. Notably, a recent study using cilengitide (an antagonist mainly selective for αvβ3 and αvβ5, with less potency towards αvβ6) demonstrated a 30% increase in hepatic collagen in two models of liver fibrosis38 supporting the idea that blockade of all αv integrins may be required to obtain significant anti-fibrotic effects. Furthermore, inhibition of TGF-β1 signaling via pharmacologic blockade of αv integrins with a small molecule such as CWHM 12 may yield the desired anti-fibrotic effects without the unwanted potential side-effects of pan-TGF-β blockade (autoimmunity and carcinogenesis). However, comprehensive toxicology testing with CWHM 12 will be required before consideration of clinical trials.
As discussed above one potential mechanism of protection for eliminating or blocking αv integrins is inhibition of activation of TGF-β. Our findings that loss of αv led to a reduction in in vivo phospho-SMAD3 immunostaining, expression of TGF-β inducible genes and TGF-β activation in culture by HSC supports a role for this mechanism. TGF-β is a major profibrogenic cytokine and is a central mediator of fibrosis in many tissues39,40,41. However, our results do not exclude other functions of αv integrins on myofibroblasts in contributing to tissue fibrosis.
It is of interest that activation of TGF-β appears to be a common function of multiple αv integrins. Many of the functional roles for distinct β subunit partners of αv are clearly unique, as demonstrated by the markedly different phenotypes described by us and others for mice globally lacking β3, β5, β6 and β8. Because of the absence of useful reagents to specifically examine the roles of αvβ1 in vivo it remains formally possible that αvβ1 could be the major integrin responsible for TGF-β activation by myofibroblasts, but previously published in vitro studies in different systems suggest that αvβ3, αvβ5 and αvβ8 all have the potential to partially mediate TGF-β activation by these cells.
In summary, we have developed a system which allows gene manipulation in myofibroblasts in multiple tissues and used this system to demonstrate that αv integrins on myofibroblasts are components of a core cellular and molecular pathway that contributes to pathologic fibrosis in multiple solid organs. Our results suggest that pharmacological targeting of all αv integrins may have clinical utility in the treatment of patients with a broad range of fibrotic diseases.
Methods
Mice
mTmG (Td tomato / EGFP)28 and Ai14 (Rosa-CAG-LSL-tdTomato-WPRE)29 mice were obtained from Jackson laboratories and crossed with Pdgfrb-Cre mice25 (obtained from Ralf Adams, University of Münster, Germany). Itgavflox/flox mice42 were obtained from Adam Lacy-Hulbert (Harvard Medical School, Boston, USA) and itgb8flox/flox mice43 were obtained from Dr Louis Reichardt (University of California, San Francisco) and all were maintained on C57BL/6 background. Itgb3−/− mice on 129/svJae background44 were obtained from Richard Hynes (Massachusetts Institute of Technology, Cambridge, USA), Itgb5−/− mice on 129/svJae background45 and Itgb6−/− mice on a C57BL/6 background46 were generated and maintained in our laboratory as previously described. Pdgfrb-BAC-eGFP reporter mice (on a C57BL/6 background) carry one copy of a BAC transgene expressing enhanced green fluorescent protein (GFP) under the control of Pdgfrb regulatory elements [obtained from GENSAT (Gene Expression Nervous System Atlas) and deposited in MMRRC [(Mutant Mouse Regional Resource Center - STOCK Tg(Pdgfrb-EGFP)JN169Gsat/Mmucd)]. Genotyping of all mice was performed by PCR. Wild type C57/BL6 mice were purchased from Jackson Laboratories. Mice used for all experiments were 8–12 weeks old and were housed under specific pathogen–free conditions in the Animal Barrier Facility of the University of California, San Francisco. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Fibrosis models
CCl4 liver injury was induced as described previously47 with eight to ten week old sex-matched mice. For acute liver injury – mice were injected i.p. with 1 μl/g body weight sterile CCl4 in a 1:3 ratio with olive oil or olive oil (control) after overnight fast (with free access to water). For chronic CCl4-induced liver fibrosis mice were injected i.p. with 1 μl/g body weight CCl4 or olive oil as above twice weekly for 6 weeks. Livers were harvested 24 hrs after the last injection. To induce pulmonary fibrosis, eight to ten week old sex-matched mice were anesthetized, and saline or bleomycin (1.5 U/kg Blenoxane) was instilled intratracheally. Lungs were harvested 14 or 28 days later. For renal fibrosis, unilateral ureteric obstruction (UUO) was induced by ligation of the left ureter as described previously48. Sham-operated mice underwent an identical surgical procedure except ligation of the ureter was not performed. Eight to ten week old male mice were used in these studies. Kidneys were harvested 7 and 14 days after surgery.
Primary cell isolation and fluoresence activated cell sorting
Mouse liver was perfused through the inferior vena cava sequentially with liver perfusion media (Invitrogen), 0.3% pronase (Roche) and 0.02% collagenase (Serva). The liver was excised, minced with scissors and further digested in 0.044% pronase and 0.008% DNAse (Roche). The cell suspension was shaken (200-250rpm) at 37 °C for 10 min and strained through sterile gauze. To remove hepatocytes the cell suspension was centrifuged at 90g for 2 min, supernatant collected, DNAse added and this procedure was repeated twice. Supernatant was centrifuged at 700g for 7 min to collect the non-parenchymal cell fraction. Single cell suspensions from mouse lung and kidney were prepared using a gentleMACS dissociator (Mitenyi Biotec) as per manufacturer’s instructions. Following live/dead staining with sytox blue (Invitrogen), live single Td tomato positive cells from Ai14;Pdgfrb-Cre mice were sorted using a FACSAria (BD Biosciences). Td tomato positive cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with glucose, L-glutamine, penicillin/streptomycin and 15% fetal calf serum. Primary mouse HSCs were isolated and passaged as described previously47. Primary mouse hepatocytes were isolated by retrograde perfusion of the liver with Liver Perfusion Medium (Invitrogen), followed by Liver Digest Medium (Invitrogen) at 37 °C. When hepatocytes were visually dispersed within the liver capsule, the liver was removed to a sterile dish and minced with scissors to release the crude cell isolate. The cells were then suspended in DME/F-12 medium and pelleted twice. Hepatocytes were purified from the washed pellets by resuspension in culture medium and centrifugation through 50% Percoll (GE Healthcare). To isolate liver sinusoidal endothelial cells (LSECs) and kupffer cells single cell suspensions from mouse liver were prepared using a gentleMACS dissociator (Mitenyi Biotec), followed by selection using CD146 (LSEC) or CD11b (kupffer cells) microbeads as per manufacturer’s instructions (Miltenyi Biotec).
Immunohistochemistry and Immunofluoresence
Paraffin-embedded sections were processed for immunohistochemistry as described previously47. The following primary antibodies were used for immunohistochemistry: αSMA A5228, 1:1000 (Sigma), GR1 MAB1037, 1:750 (R&D); F4/80 Ab6640, 1:100 (Abcam), CD31 sc1506, 1:80 (Santa Cruz Biotechnology), and αvβ6 mAb, 1:25 (human/mouse chimeric 2A1, a kind gift from Dr S. Violette, Biogen Idec, Cambridge, MA). 5 μM sections were stained with picrosirius red or antibody and results quantified using Nikon Elements software. Six random fields from each section were analyzed at a final magnification of 40×. For neutrophil counting, twenty random portal tracts per mouse liver were assessed. For immunofluoresence staining, liver tissue was fixed in 4% paraformaldehyde overnight at 4 °C, immersed in graded sucrose solutions, embedded in OCT (Tissue Tek) and stored at −80 °C. Frozen sections were incubated with the following antibodies: F4/80 MCA497R, 1:100; CD3 MCA500GT, 1:200 (Serotec), CD31 550274, 1:50 (BD Pharmingen), αSMA A5228, 1:200 (Sigma), PDGFRβ, 1:200 (a kind gift from Dr W. Stallcup, Sanford-Burnham Medical Research Institute, La Jolla), phospho-SMAD3 1880-1, 1:100 (Epitomics), cytokeratin WSS Z0622, 1:100 (Dako), αv integrin ab76609, 1:60; desmin ab8592, 1:250 (Abcam) and Alexa Fluor 488-conjugated and Alexa Fluor 555-conjugated secondary antibodies (Invitrogen). Confocal imaging was performed on a Zeiss LSM5 Pascal microscope. Digital morphometric measurements of P-SMAD3 were performed using Image J. Twelve random fields of areas of scar from each section were analyzed at a final magnification of 400×. Digital morphometric measurements of desmin and PDGFRβ immunostaining were performed using Image J. Eight random fields from each section were analyzed at a final magnification of 100×.
Human tissues
Human liver tissue was obtained from explant livers of patients with end-stage fibrotic liver disease. The use of human tissues for this study was approved by the Local Ethics Committee, University of Edinburgh. All samples collected were from subjects who gave informed consent for their tissues to be used for research purposes.
Hydroxyproline assays
Mouse liver tissue (200 mg) or the left lung was homogenized, precipitated with trichloroacetic acid and baked overnight at 110°C in HCl. Samples were reconstituted in water, and hydroxyproline content was measured using a colorimetric chloramine T assay.
Immunoprecipitation and western blotting
Cell sorted Td tomato positive cells from the uninjured livers of Ai14;Pdgfrb-Cre mice were plated on tissue culture plastic for 7 days and then lysed. Cell lysates were centrifuged at 14,000 rpm for 10 min at 4 °C and the supernatant collected. 10 μg of antibody to αv integrin (clone RMV-7, a kind gift from Dr H Yagita, Juntendo University, Japan) was added to the supernatant and this was rotated at 4 °C for 2 h, followed by the addition of 30 μl of prewashed protein G sepharose slurry (GE Healthcare) for a further 1 h at 4 °C. Following centrifugation, the beads were washed three times with PBS/protease inhibitor mixture, and once with PBS only. Laemmli sample buffer was added and the samples were boiled for 5 min followed by SDS-PAGE and western blotting using the following antibodies: αv integrin 611012, 1:500 (BD Biosciences), β1 integrin 1798-1, 1:1000 (Epitomics), β3 integrin ab75872, 1:1000; β5 integrin ab15459, 1:1000 (Abcam) and β8 integrin 1:3000 (a kind gift from Dr J McCarty, University of Texas)49. Western blotting was also undertaken using the following antibodies: αSMA A5228, 1:5000; β-actin A2228, 1:5000 (Sigma), desmin MAB3430, 1:1000 (Millipore), PDGFRβ 1469-1, 1:10,000 (Epitomics).
TGF-β activation assay
Control and itgavflox/flox;Pdgfrb-Cre HSCs were isolated and cultured for five days on tissue culture plastic, then plated at 50K cells/well in 96-well plates with mink lung epithelial cells expressing firefly luciferase downstream of a TGF-β sensitive portion of the plasminogen activator inhibitor 1 promoter30 (15K cells/well). Cells were co-cultured for 16 h and TGF-β activity was calculated by measurement of luminescence. Recombinant human TGF-β1 was reconstituted as per manufacturer’s instructions (R&D).
In vivo CWHM 12 and CWHM 96 studies
For all studies CWHM 12 and CWHM 96 were solubilized in 50% DMSO (in sterile water) and dosed to 100mg/kg/day. Drug or vehicle (50% DMSO) were delivered by implantable ALZET osmotic minipumps (Durect, Cupertino, CA). For CCl4-induced fibrosis, pumps were inserted subcutaneously either before the first dose of CCl4 (prophylactic) or after 3 weeks of treatment (therapeutic) and livers were harvested after 6 weeks. For bleomycin-induced fibrosis pumps were inserted 14 days after treatment with bleomycin or saline and lungs were harvested at 28 days (therapeutic only).
qRT-PCR
Total RNA was isolated using an RNeasy kit (Qiagen). cDNA was analyzed by SYBR-Green real-time PCR with an ABI 7900HT thermocycler and normalized to β-actin or 18S expression. Primers used were as follows: β-actin forward: TGTTACCAACTGGGACGACA, β-actin reverse: GGGGTGTTGAAGGTCTCAAA; 18S forward: TAGAGGGACAAGTGGCGTTC, 18S reverse: CGCTGAGCCAGTCAGTGT; Itgav forward: CCGTGGACTTCTTCGAGCC, Itgav reverse: CTGTTGAATCAAACTCAATGGGC; Desmin forward: GTGGATGCAGCCACTCTAGC, Desmin reverse: TTAGCCGCGATGGTCTCATAC; Pdgfrb forward: TCCAGGAGTGATACCAGCTTT, Pdgfrb reverse: CAGGAGCCATAACACGGACA; GFAP forward: CGGAGACGCATCACCTCTG, GFAP reverse: TCTCGGAGGCATAGGAGCG; α-SMA forward: GTCCCAGACATCAGGGAGTAA, α-SMA reverse: TCGGATACTTCAGCGTCAGGA; Col1A1 forward: GCTCCTCTTAGGGGCCACT, Col1A1 reverse: CCACGTCTCACCATTGGGG; Col 3A1 forward: AACCTGGTTTCTTCTCACCCTTC, Col 3A1 reverse: ACTCATAGGACTGACCAAGGTGG; TGFb1 forward: CTCCCGTGGCTTCTAGTGC, TGFb1 reverse: GCCTTAGTTTGGACAGGATCTG; Mmp-2 forward: CAAGTTCCCCGGCGATGTC, Mmp-2 reverse: TTCTGGTCAAGGTCACCTGTC; Mmp-3 forward: ACATGGAGACTTTGTCCCTTTTG, Mmp-3 reverse: TTGGCTGAGTGGTAGAGTCCC; Mmp-9 forward: CTGGACAGCCAGACACTAAAG, Mmp-9 reverse: CTCGCGGCAAGTCTTCAGAG; Mmp 13 forward: CTTCTTCTTGTTGAGCTGGACTC, Mmp 13 reverse: CTGTGGAGGTCACTGTAGACT; Timp-1 forward: TGCAACTCGGACCTGGTCATA, Timp-1 reverse: CGCTGGTATAAGGTGGTCTCG; Pparg forward: GGAAGACCACTCGCATTCCTT, Pparg reverse: GTAATCAGCAACCATTGGGTCA; Itgb1 forward: CTACTTCTGCACGATGTGATGAT, Itgb1 reverse: TTGGCTGGCAACCCTTCTTT; Itgb3 forward: CCACACGAGGCGTGAACTC, Itgb3 reverse: CTTCAGGTTACATCGGGGTGA; Itgb5 forward: GAAGTGCCACCTCGTGTGAA, Itgb5 reverse: GGACCGTGGATTGCCAAAGT; Itgb8 forward: CTGAAGAAATACCCCGTGGA, Itgb8 reverse: ATGGGGAGGCATACAGTCT.
Adhesion assay
Control and itgavflox/flox;Pdgfrb-Cre HSCs were cultured for 5 days and then seeded into 48 well tissue culture plates precoated with fibronectin, collagen I, collagen IV, laminin and fibrinogen or BSA treated controls (CytoSelect 48-well adhesion assay, ECM array, Cambridge Biosciences). Cells were allowed to adhere for 90 min, washed × 3 with PBS and stained and eluted as per manufacturer’s instructions. Adhesion is expressed as a % of unwashed cells adhered to 1% poly-L-lysine.
Migration assay
Control and itgavflox/flox;Pdgfrb-Cre HSCs were cultured for 5 days and then seeded into the upper chambers of 8 um pore size modified Boyden chambers as per manufacturer’s instructions (CytoSelect 24 well cell migration assay, Cambridge Biosciences). Fetal calf serum (10%) was added to the lower chamber and cells were allowed to migrate for 6h at 37 °C. Cells remaining in the upper chamber were wiped with a cotton tip and cells attached to the underside of the membrane were fixed, stained and eluted as per manufacturer’s instructions. Chemotaxis is expressed as % of an unwiped control.
ELISA
Total TGF-β1 and TIMP1 were measured using ELISA kits (R&D) as per manufacturer’s instructions.
TUNEL assay
Control and itgavflox/flox;Pdgfrb-Cre HSCs were plated overnight on glass chamber slides and apoptosis was detected using the In Situ death detection kit (Roche) as per manufacturer’s instructions. Cells were counterstained with DAPI and viewed under a Zeiss fluorescent microscope.
Assessment of recombination efficiency
Six confocal images (225 μm2, single optical sections) were randomly acquired throughout non-adjacent cryosections of liver or lung (n = 4 male mice per group). Antibody staining with desmin, PDGFRβ and αSMA and Ai14-TdTomato recombination reporter were separately subjected to threshold processing using NIH Image J software, then the percentage of Ai14-TdTomato positive, antibody stained cells was quantified.
Flow cytometry
Cells were harvested with TrypLE™ Select (Life Technologies, UK) and washed twice with PBS and re-suspended in FACS buffer (PBS supplemented with 2% FCS). 4×105 cells were then incubated with a conjugated antibody (or isotype control) at 4 °C for 30 mins in the dark. Cells were then washed and re-suspended in FACS buffer and analysed on a Becton Dickinson LSR Fortessa II. Antibodies used: PE-conjugated antibody to CD49a (integrin α1) and isotype control (BD Pharmingen). PE-conjugated antibody to CD49e (integrin α5), antibody to CD29 (integrin β1) and isotype control (eBioscience).
In vitro integrin functional assays
The effects of CWHM 12 and CWHM 96 on cell adhesion mediated by αvβ3, αvβ5, αvβ6, and α5β1 were measured as previously described with minor modifications50,51. Briefly, stably transfected human 293 cells over-expressing human αvβ3 or αvβ5 were pre-incubated in HBSS buffer (Sigma) containing 200 μM MnCl2 for 30 min at 37 °C with 3-fold dilutions of compound. Each sample was then added to triplicate wells of a 96-well plate which had been coated overnight at 4 °C with a predetermined optimal concentration of purified vitronectin (Calbiochem), washed, blocked by 1 hr incubation with BSA, and washed again. Cells were allowed to attach for 30 min at 37 °C, and non-adherent cells were removed by washing. Remaining attached cells were measured by endogenous alkaline phosphatase activity using para-nitrophenyl phosphate and reading absorbance signal at 405 nM. The same procedure was used to measure adhesion of αvβ6-expressing human HT-29 cells to purified human latency associated peptide (LAP; R&D Systems), and α5β1-expressing human K562 cells to human plasma fibronectin (Calbiochem). In all cell-based assays, binding by the expected integrin was verified by testing activity of corresponding isotype-matched positive (function-blocking) and negative control antibodies. Functions of integrins αvβ1, αvβ8, α2β1 and α10β1 were measured using cell-free receptor-ligand interaction assays using purified recombinant human integrins purchased from R&D Systems. Ligands used were human fibronectin (R&D Systems) for αvβ1, human LAP (R&D Systems) for αvβ8, bovine collagen II (Sigma) for α2β1, and murine laminin I (Cultrex, Trevigen) for α10β1. 96-well plates were coated with the predetermined optimal concentration of ligand overnight, washed 3X with TBS+++ (25 mM Tris pH7.4, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mM MnCl2, 1mM CaCl2), and blocked with TBS+++/1%BSA. Purified integrin was diluted in TBS+++/0.1%BSA with or without compounds, and the solution added to empty wells of the washed ligand-coated plate according to a standard template, with each sample repeated in triplicate. After incubation for 2 hr at room temperature, the plate was washed 3X with TBS+++. Biotin-labeled antibody against the αv subunit (αvβ1, αvβ8 assays) or β1 subunit (α2β1, α10β1 assays) (R&D Systems) was applied for 1 hr. The plate was washed 3X with TBS/0.1%BSA. Streptavidin-conjugated horseradish peroxidase (R&D Systems) was added to the wells, and the plate incubated for 20 min at room temperature. Following a 3X TBS+++ wash, bound integrin was detected using streptavidin-conjugated horseradish peroxidase and TMB substrate with absorbance measured at 650 nm. For assay of αIIbβ3 (IIbIIIa) function, plates were coated with the purified human integrin (Abcam) overnight, washed 3X with TBS+++, and blocked with TBS+++/1%BSA. Alexa Fluor647-labeled purified human fibrinogen (Life Technologies) was diluted in TBS+++/0.1%BSA with or without compounds, and the solutions were added to the integrin-coated plate. After 2 hr incubation, the plate was washed 3X with TBS+++, and bound ligand was detected by absorbance measured at 640/668nm. For all assays, concentration-response curves were constructed by non-linear regression analysis and IC50 values were calculated using GraphPad Prism software.
Synthesis of CWHM12
Instrumentation and General Methods
Chemical ionization mass spectra were recorded, at 70 eV ionizing voltage, on a Hewlett-Packard 5973 CI quadrupole mass spectrometer connected to a Hewlett-Packard 6890 gas chromatograph fitted with an Agilent Tech 12 m × 0.2 mm × 0.33 μm DB-1 (cross linked methyl silicone) column.
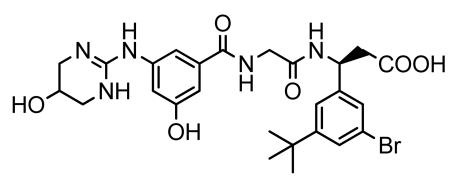
Preparation of (3S)-N-[3-hydroxy-5-[(1,4,5,6-tetrahydro-5-hydroxy-2-pyrimidinyl)amino] benzoyl]glycyl-3-(3-bromo-5-tert-butylphenyl)-β-alanine (CWHM12)
Step 1
Preparation of 3-Hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)aminobenzoic acid
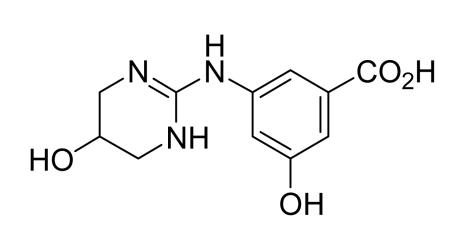
3-Hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)aminobenzoic acid was synthesized according to literature procedures52.
Step 2
Preparation of 2-(3-Hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)amino)benzamido) acetic acid
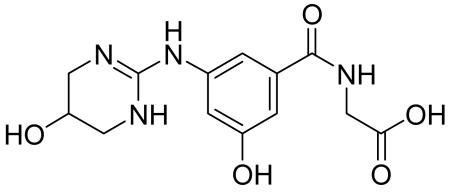
2-(3-Hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)amino)benzamido)acetic acid was prepared by the following procedure:
Coupling of 3-hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)aminobenzoic acid with glycine ethyl ester
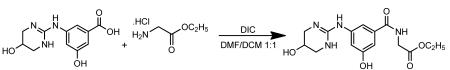
Glycine ethyl ester hydrochloride (5.02 g, 35.95 mmol) was added to a suspension of 3-hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)aminobenzoic acid (9.013 g, 35.87 mmol) in a 1:1 mixture of DMF (50 ml) and DCM (50 ml) and the mixture was stirred at room temperature under nitrogen atmosphere. Neat N,N’-diisopropylcarbodiimide (6.75 ml, 43.60 mmol) was added to above reaction mixture and the mixture was stirred at room temperature overnight to give a colorless suspension. The crude reaction mixture was used as such for the hydrolysis of the above ester.
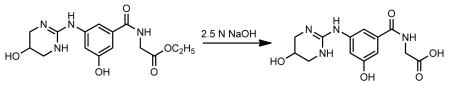
The above crude reaction mixture was cooled to 10°C (ice bath) and a 2.5 N NaOH solution (90 ml) was added slowly with stirring, the solution temperature was kept below 20°C, to give a pale yellow solution/suspension. The reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was acidified with 5N HCl with stirring to pH 5 to give a colorless precipitate and the mixture was stirred at room temperature for another 15 min and filtered to give a colorless solid. The solid was washed with water (1×25 ml) and then with acetonitrile (1×25 ml). The solid was dried in-vacuo to give a colorless powder (9.686 g, yield 88%).
1H NMR (400 MHz, D2O): δ 3.37 (dd, J = 12.7 and 3.1 Hz, 2H), 3.50 (dd, J = 12.7 and 2.8 Hz, 2H), 4.17 (s, 2H), 4.37 (m, 1H), 6.97 (t, J = 2.01 Hz, 1H), 7.17-7.26 (m, 2H). 1H NMR spectrum of the sample was consistent with the suggested structure of the product.
Step 3
Preparation of (S)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl)propionate hydrochloride
Step A
Preparation of 3-bromo-5-tert-butylbenzaldehyde
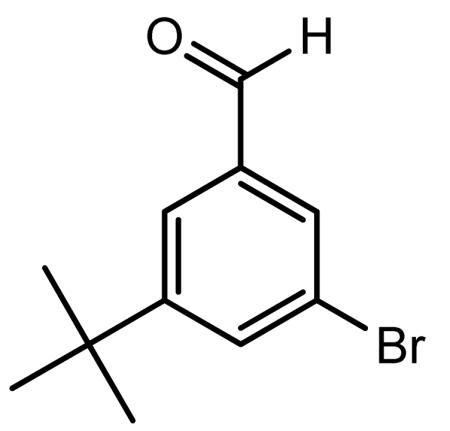
1,3-Dibromo-5-tert-butylbenzene (4.95 g, 16.95 mmol) was dissolved in anhydrous ether (25.0 ml) in a dried flask under nitrogen. The reaction mixture was cooled to −78°C and stirred under nitrogen atmosphere. A 1.6 M solution of n-BuLi in hexanes (10.60 ml, 16.95 mmol) was added dropwise to the above solution and the reaction mixture was stirred at −78°C for 30 min after complete addition of n-BuLi. After 30 min of stirring at −78°C, the reaction mixture was warmed to −30°C. DMF (1.60 ml, 20.66 mmol) was added to above reaction mixture dropwise, keeping the reaction mixture below −20°C. After addition of DMF was complete, the reaction mixture was warmed slowly to 0°C (30 min) and then stirring at room temperature overnight under nitrogen to give a yellow-orange solution. The reaction mixture was poured into 40 ml of chilled 10% aqueous HCl and the reaction mixture was stirred for 15 min. The ether layer was separated, washed with water (2 × 25 ml), dried over anhydrous MgSO4, filtered and evaporated in vacuo to give the product as a pale yellow viscous liquid. The crude product was dissolved in dichloromethane (25 ml) and passed through a small pad of silica gel (~100 mg). Evaporation of the solvent in vacuo gave the product as a very pale yellow viscous liquid (4.096 g). GC-MS analysis (CI mode/methane) shows the desired product’s mass: mz 240 (79BrM+) and m/z 242 (81BrM+);Calcd for C11H13BrO: 241.12.
Step B
Preparation of 3-amino-3-(3-bromo-5-tert-butylphenyl)propionic acid
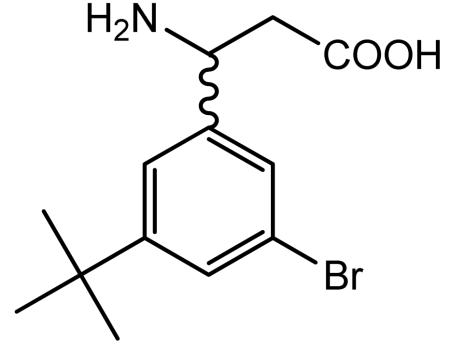
A suspension of 3-bromo-5-tert-butylbenzaldehyde (4.17 g, 17.30 mmol), malonic acid (2.15 g, 20.72 mmol) and ammonium acetate (2.66 g, 34.59 mmol) in isopropanol (35 ml) was heated at reflux under nitrogen for 3 h to afford a thick colorless solid. The solid was filtered hot, washed with hot isopropanol (2 × 25 ml) and dried in vacuo to give the desired racemic product as a colorless solid (2.68 g).
Step C
Preparation of ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl) propionate hydrochloride
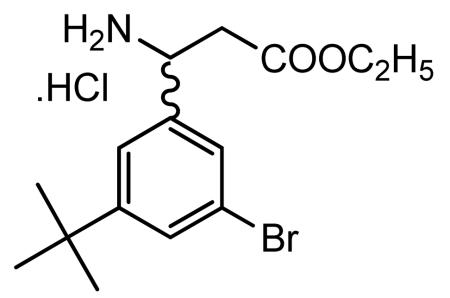
Absolute ethanol saturated with anhydrous HCl gas (75 ml) was added to 3-amino-3-(3-bromo-5-tert-butylphenyl)propionic acid (4.78 g, 15.92 mmol) and the reaction mixture was heated at reflux for 1.5 h to give a pale yellow solution. The solvent was removed in vacuo to give a colorless solid. The solid was slurried with diethyl ether and heptane (2×25 ml). After the solvent layer was decanted off, the residue was dried in vacuo to give the racemic β-amino ester hydrochloride salt as a cream solid (5.00 g).
Step D
Preparation of (S)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl)propionate hydrochloride
Enzymatic resolution of the racemic mixture
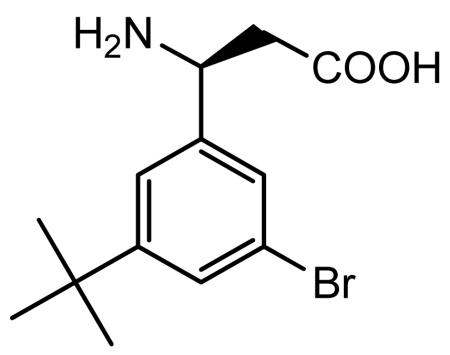
A suspension of ethyl 3-amino-3-(3-bromo-5-tert-butylpheny)propionate hydrochloride (4.54 g, 12.45 mmol) in water (10 ml) was basified with 2.5N NaOH (pH ~12) by dropwise addition to give a creamy oily residue. The pH of the aqueous layer was adjusted to pH 8.20 by the addition of 50 mM KH2PO4 solution. Amano lipase PS (5.24 g) was added to above reaction mixture and the reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered after 22 h and the solid was washed with acetone to give a colorless solid of the resolved (S)- acid (1.72 g).
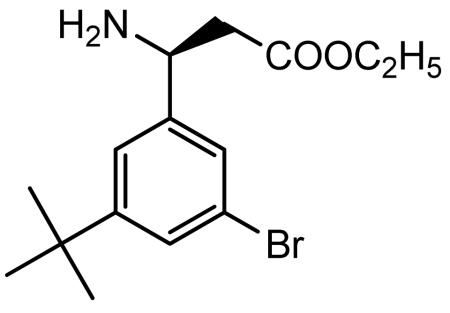
Absolute ethanol saturated with anhydrous HCl gas (35 ml) was added to (S)-3-amino-3-(3-bromo-5-tert-butylphenyl)propionic acid (1.83 g, 6.10 mmol) and the reaction mixture was heated at reflux for 2 h to give a colorless solution. The solvent was removed in vacuo to give a cream-yellow foamy solid. The solid was slurried with heptane. After the solvent was decanted off, the residue was dried in vacuo to give the desired (S)-β-amino ester hydrochloride salt as a pale yellow foamy solid (2.26 g).
Step 4
Preparation of (3S)-N-[3-hydroxy-5-[(1,4,5,6-tetrahydro-5-hydroxy-2-pyrimidinyl)amino] benzoyl]glycyl-3-(3-bromo-5-tert-butylphenyl)-β-alanine (CWHM 12)
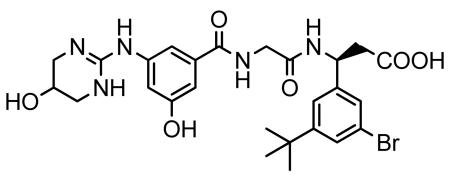
A mixture of 2-(3-hydroxy-5-((5-hydroxy-1,4,5,6-tetrahydropyrimidin-2-yl)amino)benzamido)acetic acid (from step 2) (1.92 g, 6.23 mmol), and (S)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl) propionate hydrochloride (from step 3) (2.25 g, 6.17 mmol) was dissolved in DMF (20 ml) and dichloromethane (10 ml) to give a cream suspension. 1-hydroxybenzatriazole hydrate was added to above reaction mixture and the reaction mixture was stirred in an ice-bath under nitrogen atmosphere for 10 min. N,N’-diisopropylcarbodiimide (DIC) was added and the reaction mixture was stirred at room temperature overnight under nitrogen atmosphere. The solvent was evaporated in-vacuo to give a pale yellow viscous gummy residue. The residue was dissolved in acetonitrile (50 ml) and filtered to remove precipitated urea. Evaporation of the filtrate in-vacuo afforded a pale yellow to cream foamy residue of the crude (3S)-N-[3-hydroxy-5-[(1,4,5,6-tetrahydro-5-hydroxy-2-pyrimidinyl)imino]benzoyl]glycyl-3-(3-bromo-5-tert-butylphenyl)-β-alanine ethyl ester. To a suspension of the crude ester in a 1:1 mixture of water/acetonitrile (14 ml) was added lithium hydroxide monohydrate (2.07 g, 49.38 mmol) at room temperature and the reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was acidified with TFA and evaporated in-vacuo to give a cream foamy solid. The solid was purified by reverse-phase preparative HPLC (10-90% water/acetonitrile containing 0.05% TFA) to give the desired product, after lyophilization, as a colorless powder (2.244 g). LC/MS shows the desired product’s mass: m/z 590 (79BrM+H) and 592 (81BrM+H); Calcd for C26H32BrN5O6: 590.47. 1H NMR (400 MHz, DMSO-d6):δ 1.27 (s, 9H, (CH3)3C-), 2.70 (d, J = 7.31 Hz, 2H, −CH2-COOH), 3.16 (dd, J = 12.22, 3.10 Hz, 2H), 3.33 (brd, J = 12.44 Hz, 2H), 3.87 (d, J = 5.81 Hz, 2H), 4.08 (appt/m,J = 2.92 Hz, 1H), 5.18 (q, J = 7.62 Hz, 1H, −NH-CH-CH2-COOH), 6.75 (t, J = 2.03 Hz, 1H), 7.13 (dt, J = 10.70 and 1.50 Hz, 2H), 7.34 (dt, J = 9.54 and 1.50 Hz, 2H), 7.39 (appt, J = 1.72 Hz, 1H), 8.09 (brs, 2H), 8.51 (d, J = 8.30 Hz, 1H), 8.61 (brt, J = 5.88 Hz,1H), 9.57 (s,1H), 10.00 (brs, 1H), 12.35 (brs, 1H, -COOH). 1H NMR spectrum of the sample was consistent with the suggested structure of the product.
Synthesis of CWHM 96
CWHM 96 was synthesized using the same method as described for CWHM 12, except that (R)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl) propionate hydrochloride is substituted for (S)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl) propionate hydrochloride in step 4. (R)-ethyl 3-amino-3-(3-bromo-5-tert-butylphenyl) propionate hydrochloride is isolated from the mother liquor in the enzymatic lipase reaction that produces (S)-3-amino-3-(3-bromo-5-tert-butylphenyl) propionate in the CWHM 12 method.
Statistics
All data are presented as mean ± S.E.M. Statistical significance was calculated using a 2-tailed Student’s t test. Differences with a P value of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by a Wellcome Trust Intermediate Clinical Fellowship (ref. 085187) to N.C.H., NIH grants HL102292, HL53949 and AI077439 (to D.S.), a UCSF Liver Center Tool and Technology grant (to N.C.H) and P30 DK026743 (UCSF Liver Center). We thank K. Thorn at the UCSF Nikon Imaging Center for assistance with image analysis. We also thank C. Her, N. Wu, and S. Huling (UCSF) and D. Rodrigues and R. Aucott (University of Edinburgh) for expert technical assistance. We also gratefully acknowledge the contribution of M. Singh (chemical synthesis of compounds CWHM 12 and CWHM 96), D. Tajfirouz, S. Freeman and M. Yates at Saint Louis University for technical assistance in conducting integrin functional assays to characterize compound activities.
Footnotes
Conflict of interest: P.G.R. and D.W.G. hold equity in Antegrin Therapeutics, LLC.
References
- 1.Gleizes PE, et al. TGF-beta latency: biological significance and mechanisms of activation. Stem Cells. 1997;15:190–197. doi: 10.1002/stem.150190. [DOI] [PubMed] [Google Scholar]
- 2.Munger JS, et al. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51:1376–1382. doi: 10.1038/ki.1997.188. [DOI] [PubMed] [Google Scholar]
- 3.Munger JS, et al. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 4.Mu D, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell. Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annes JP, Rifkin DB, Munger JS. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65–68. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 6.Aluwihare P, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell. Sci. 2009;122:227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang B, et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hahm K, et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma LJ, et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am. J. Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J. Histochem. Cytochem. 1993;41:1521–1527. doi: 10.1177/41.10.8245410. [DOI] [PubMed] [Google Scholar]
- 11.Breuss JM, et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J. Cell. Sci. 1995;108:2241–2251. doi: 10.1242/jcs.108.6.2241. [DOI] [PubMed] [Google Scholar]
- 12.Shi M, et al. Latent TGF-β structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell. Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol. Biol. Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asano Y, et al. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J. Immunol. 2005;175:7708–7718. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 16.Asano Y, Ihn H, Yamane K, Jinnin M, Tamaki K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006;168:499–510. doi: 10.2353/ajpath.2006.041306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J. Clin. Invest. 1989;84:1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J. Clin. Invest. 1989;84:1786–1793. doi: 10.1172/JCI114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–1569. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinzani M, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 21.Ikura Y, et al. Expression of platelet-derived growth factor and its receptor in livers of patients with chronic liver disease. J. Gastroenterol. 1997;32:496–501. doi: 10.1007/BF02934089. [DOI] [PubMed] [Google Scholar]
- 22.Coin PG, et al. Lipopolysaccharide up-regulates platelet-derived growth factor (PDGF) alpha-receptor expression in rat lung myofibroblasts and enhances response to all PDGF isoforms. J. Immunol. 1996;156:4797–4806. [PubMed] [Google Scholar]
- 23.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Chen YT, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 25.Foo SS, et al. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–173. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 26.de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified rat liver fat-storing cells in culture divide and contain collagen. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- 27.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. U S A. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 29.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abe M, et al. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 31.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhu J, et al. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fässler R, Meyer M. Consequences of lack of β1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 34.Stephens LE, et al. Deletion of β1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- 35.Abraham S, Kogata N, Fässler R, Adams RH. Integrin beta1 subunit controls mural cell adhesion, spreading, and blood vessel wall stability. Circ. Res. 2008;102:562–570. doi: 10.1161/CIRCRESAHA.107.167908. [DOI] [PubMed] [Google Scholar]
- 36.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Patsenker E, et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50:1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 40.Roberts AB, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 42.Lacy-Hulbert A, et al. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc. Natl. Acad. Sci. U S A. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. Vascular development of the brain requires β8 integrin expression in the neuroepithelium. J. Neurosci. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodivala-Dilke KM, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J. Clin. Invest. 1999;103:229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang X, Griffiths M, Wu J, Farese RV, Jr., Sheppard D. Normal development, wound healing, and adenovirus susceptibility in beta5-deficient mice. Mol. Cell. Biol. 2000;20:755–759. doi: 10.1128/mcb.20.3.755-759.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang XZ, et al. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J. Cell. Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson NC, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. U S A. 2006;103:5060–5065. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson NC, et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008;172:288–298. doi: 10.2353/ajpath.2008.070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarty JH, et al. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 50.Nagarajan SR, et al. R-isomers of Arg-Gly-Asp (RGD) mimics as potent alphavbeta3 inhibitors. Bioorg. Med. Chem. 2007;15:3783–3800. doi: 10.1016/j.bmc.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 51.Shannon KE, et al. Anti-metastatic properties of RGD-peptidomimetic agents S137 and S247. Clin. Exp. Metastasis. 2004;21:129–138. doi: 10.1023/b:clin.0000024764.93092.5f. [DOI] [PubMed] [Google Scholar]
- 52.Organic Process Research & Development. 2004;8:571–575. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.