

Abstract
DNA nanotechnology has been extensively explored to assemble various functional nanostructures for versatile applications. Mediated by Watson-Crick base-pairing, these DNA nanostructures have been conventionally assembled through hybridization of many short DNA building blocks. Here we report the noncanonical self-assembly of multifunctional DNA nanostructures, termed as nanoflowers (NFs), and the versatile biomedical applications. These NFs were assembled from long DNA building blocks generated via Rolling Circle Replication (RCR) of a designer template. NF assembly was driven by liquid crystallization and dense packaging of building blocks, without relying on Watson-Crick base-pairing between DNA strands, thereby avoiding the otherwise conventional complicated DNA sequence design. NF sizes were readily tunable in a wide range, by simply adjusting such parameters as assembly time and template sequences. NFs were exceptionally resistant to nuclease degradation, denaturation, or dissociation at extremely low concentration, presumably resulting from the dense DNA packaging in NFs. The exceptional biostability is critical for biomedical applications. By rational design, NFs can be readily incorporated with myriad functional moieties. All these properties make NFs promising for versatile applications. As a proof-of-principle demonstration, in this study, NFs were integrated with aptamers, bioimaging agents, and drug loading sites, and the resultant multifunctional NFs were demonstrated for selective cancer cell recognition, bioimaging, and targeted anticancer drug delivery.
INTRODUCTION
Owing to the unique feature of Watson-Crick base-pairing, DNA has emerged as building blocks for a wide variety of DNA nanostructures, in which the built-in functionalities enable the applications in biomedicine, biotechnology, and nanoelectronics. 1–5 The sequence programmability, automated controllable synthesis, high stability and intrinsic functionalities make DNA nanostructures advantageous over other counterparts in many biomedical applications. Conventional approaches to DNA nanostructure construction typically rely on Watson-Crick base-pairing between short DNA building blocks. However, these approaches have some intrinsic drawbacks, including 1) complicated design resulting from the myriad of different DNA strands needed to assemble relatively large and sophisticated nanostructures; 2) the large amount of DNA needed for bulky preparation; 3) the limited compaction resulted from steric hindrance of DNA strands, notwithstanding high DNA compaction is typically favored for nanotherapeutic and bioimaging nanoassemblies; 4) the extensive intrinsic nicks, i.e., broken phosphodiester bonds in the DNA backbone of each short building block, which serves as potential cleavage sites of many exonucleases,6,7 posing a threat to the biostability; and 5) dissociation that accompanies denaturation or extremely low concentrations, such as that in an in vivo circulation system, resulting in loss of nanostructure integrity. Therefore, it would be highly desirable to assemble densely compacted multifunctional DNA nanostructures using elongated non-nicked building blocks made from a low amount of only a few DNA strands, without relying on Watson-Crick base-pairing.
Towards this end, nature provides instructional examples. For instance, in the nuclei of a living organism, a tremendous amount of genomic dsDNA is densely compacted in a systematic manner that does not rely on Watson-Crick base-pairing. In a typical somatic human cell, 46 chromosomal dsDNAs with a total length of about one meter, carrying more than 30,000 functional genes, can be assembled into a single nucleus particle of tens of cubic micrometers.8 The dense DNA compaction is attributed to the highly ordered alignment of chromosomal DNA with the assistance of sophisticated cellular machinery, which allows long chromosomal DNA to be systematically assembled to nucleosomes, “beads-on-a-ring”, DNA fibers, and eventually chromosomes.9 Likewise, in a dinoflagellate, a type of flagellate protist, the DNA concentration in the nucleus was estimated to be up to 200 mg/mL, which is up to 80 times more than that in a human somatic cell.10 These densely packed genomic DNAs were documented to be liquid crystalline, a characteristic feature of highly ordered and densely packed molecular assemblies.10,11 Indeed, synthetic short dsDNAs with concentrations equivalent to genomic DNA in nuclei were reported to be liquid crystalline as well, and these highly concentrated and orderly aligned DNAs self-assembled into segregated structures in a manner of end-to-end stacking that does not rely on Watson-Crick base-pairing.12–15
Inspired by nature, we present herein the noncanonical self-assembly of hierarchical DNA nanoflowers (NFs) with densely packed DNA and built-in multifunctional moieties for biomedical applications. Using only a low amount of two DNA strands (one template and one primer), long building blocks were generated via Rolling Cycle Replication (RCR), an isothermal enzymatic reaction involving the replication of many circular genomic DNAs (e.g., plasmids or viral genomes16), the regulation of some eukaryotic tandem genes,17,18 and applications in biotechnology. 3,5,7,19–26 Without reliance on Watson-Crick base-paring, NFs were self-assembled through liquid crystallization of the resultant long building blocks, instead of conventionally used short DNA. The sparsity of nick sites in the elongated building blocks and the compactness of DNA in NFs are expected to increase the resistance of NFs to nuclease degradation, denaturation, or dissociation at low concentrations, and the exceptional biostability was demonstrated by the maintenance of NF structural integrity under the treatment with nucleases, human serum, high temperature, urea, or dilution, making NFs amenable for versatile biomedical situations. NFs are monodisperse and size-tunable. The ability to fine-tune NF sizes and construct NFs using essentially any designer templates suitable for RCR allows versatile future application of NFs. By rational design of RCR templates, the elongated DNA building blocks can carry a large amount of concatemer structural and functional moieties, which were further compacted into NFs. In our proof-of-principle study, we have successfully incorporated functionalities including aptamers, fluorophores, and drug loading sites into NFs, which were capable of selective cancer cell recognition, bioimaging, and targeted anticancer drug delivery. Overall, these noncanonically self-assembled DNA NFs are promising for versatile biomedical applications.
RESULTS AND DISCUSSION
Self-assembly of monodisperse, size-tunable, and densely packed multifunctional DNA NFs
Elongated DNA was generated through RCR using Φ29 DNA polymerase, which is capable of DNA replication with a circular template. The high efficiency and strand-displacement ability of Φ29 is critical to generate long non-nicked DNA. Since the templates for RCR can be tailor-designed, various structural and functional DNA moieties can be incorporated into templates and further built in RCR products. Indeed, DNA functional moieties have been intensively explored, including specific recognition elements (e.g., aptamers), therapeutics (e.g., antisense oligonucleotides,27 aptamers,28 and immunostimulatory CpG motifs29), and preferable drug association sequences (e.g., double-stranded CG or GC sequences for anthracycline drugs30). In this study, to construct NFs with built-in multifunctionalities, the RCR template (T-1, sequence in Table S1, predicted structure in Figure S1A) was designed such that the resultant RCR products would be decorated with a series of aptamers and drug loading sites for Doxorubicin (Dox), a widely used chemotherapeutic for many types of cancer. The concatemer aptamers in elongated ssDNA are expected to enhance the binding affinity of the resultant NFs to target cells through synergistic multivalent binding, and provide insight for future construction of NFs incorporated with therapeutic aptamers. The tremendous drug-association sequences in NFs are expected to endow NFs with high drug payload capacity. Aptamer sgc8, which can specifically recognize PTK7 over-expressed on many cancer cells,31,32 was used as a model. A flow cytometry study verified the selective recognition ability of the monomeric template complement to target CEM cells (T cell leukemia), but not to nontarget Ramos cells (B cell lymphoma) (Figure S2). T-1 was circularized using a ligation template and a T4 DNA ligase. The resultant circular template was then used in RCR, in which the previous ligation template also served as the primer (Figure 1) and elongated by Φ29 to generate periodic DNA with a myriad of aptamers and drug loading moieties (Figure S1B). RCR was verified using gel electrophoresis (Figure S3A). Despite the high stability of the 3-way junction structure of T-1 (Figure S1A), the robust strand displacement ability of Φ29 allowed it to overcome the topological constraints and efficiently generate DNA, as also reported before.33
Figure 1.
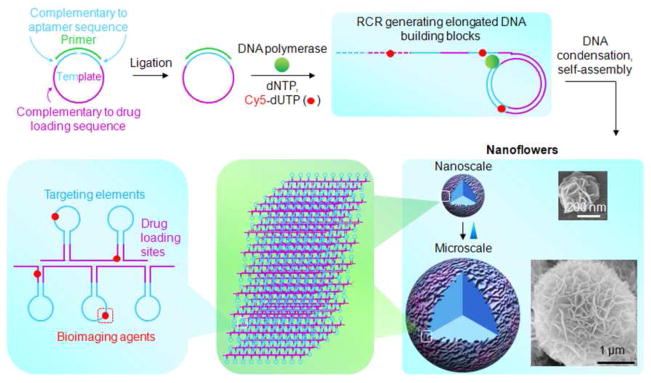
Schematic illustration of noncanonical self-assembly of multifunctional DNA nanoflowers (NFs). The linear DNA template was first ligated to form a circular template for RCR using Φ29 DNA polymerase and a primer. RCR generated a large amount of elongated non-nicked concatemer DNA with each unit complementary to the template. These DNAs then served as building blocks to self-assemble monodisperse, densely-packed, and hierarchical DNA NFs. NF sizes are tunable with diameters ranging from approximately 200 nm to several micrometers, as shown by the representative SEM images. NF assembly does not rely on Watson-Crick base-pairing between DNA building blocks, enabling tailored design of the template to carry multiple complements of functional nucleic acids, e.g., aptamers and drug loading sites. Functionalities could also be incorporated via primers or modified deoxynucleotides, such as Cy5-dUTP for bioimaging. The multifunctional NFs were applied for selective target cancer cell recognition, bioimaging, and targeted drug delivery.
The above-generated DNA then served as building blocks to assemble NFs. RCR products obtained after reaction for n hours were denoted as RCRn. Scanning electron microscopy (SEM) observation indicated that NFs were formed in RCR10 with diameters of about 200 nm and petal-like structures on the surfaces (Figure 2A). NFs from RCR10 are monodisperse, as shown by SEM images in a zoomed-out view and size determination with dynamic light scattering (DLS) in a bulky solution (Figure 2B). DLS results revealed an average radius of 112 ± 12 nm, which is consistent with the SEM data. Characterization of RCR10 using transmitted electron microscopy (TEM) (Figure 2C) and atomic force microscopy (AFM) (Figure S3B) further verified the size and morphology. TEM images displayed hierarchical internal structures in NFs. Even though large sticky RNA microsponges and bulky DNA metamaterial hydrogel have been constructed through similar strategies,19,34 our results represent the first to build monodisperse DNA nanomaterials with diameters as small as 200 nm through RCR. The ability to build monodisperse nanoassemblies with dimensions within a few hundred nanometers is highly significant, especially for biomedical applications. For instance, due to enhanced permeation and retention (EPR) effect, nanomaterials with diameters of as small as a few hundred nanometers are more likely to penetrate leaky blood vasculature and have enhanced retention at tumor sites.35,36 The one-step self-assembly of small monodisperse NFs avoids the otherwise physical cutting for aggregate partition or physical compaction for size reduction, which could be difficult to precisely manipulate and could damage DNA functionalities in NFs.19,34
Figure 2.

Characterization of small monodisperse hierarchical DNA NFs. (A) SEM images of NFs from RCR10 displaying monodisperse DNA NFs with petal-like structures on NF surfaces. (B) DLS data revealing the size distribution of NFs from RCR10. (C) A uranyl acetate-stained TEM image displaying the NF structure of RCR10, with hierarchical internal structures.
We next examined the NF assembly process, which could be influenced by such factors as tertiary structures of DNA templates, starting template concentration, substrate (dNTP) concentration, and reaction time. The influence of reaction time was first investigated. We expect that a reaction for a longer time until the exhaust of substrate would generate more DNAs for NF assembly. To study this, RCR for a series of different time lengths was performed, followed by SEM imaging to reveal the structures obtained at each time point. Results shown in Figure 3A and Figure S4 suggest a progressive process of NF assembly. In particular, amorphous bulky structures were observed in RCR0.5 (Figure S4). RCR1 displayed some crystal-like structures on the surfaces of bulky matrices (Figure 3A). These crystal-like structures proceeded to form petal-like structures on the surfaces of RCR2 (Figure S4), and flower-like spherical structures gradually formed on the matrices of RCR3 through RCR10 (Figure 3A). In the meantime, the NFs were gradually pinched off from the surfaces of bulky matrices and separated from each other, forming monodisperse NFs with diameters of around 200 nm in RCR10. We further examined RCR15 (d=ca. 700 nm), RCR20 (d=ca. 1.5 μm), and RCR30 (d=ca. 3–4 μm) and found that NFs kept growing with longer reaction time, presumably until the exhaust of substrate. NFs with diameter of ca. 4 μm were observed in RCR30. These data clearly indicated a time-dependent process of NF self-assembly, allowing us to fine-tune NF sizes for versatile applications simply by adjusting RCR time. To simplify the notation, NFs with diameters of approximate n μm are denoted as NFn. The monodisperse NF2s from RCR24 are shown in Figure 3B as an example of microscale NFs with pleated petals on the surfaces. To examine the internal structures of NFs, RCR24 was sonicated and chopped into smaller DNA particles. Under SEM observation, the hierarchical structures on the surfaces of the sonicated DNA particles provide clear evidence of the internal hierarchical structures of the original NFs and the dense DNA packaging in NFs (Figure S5). These internal hierarchical structures and dense DNA packaging could be used for high-payload encapsulation in porous structures or integration on DNA of nanoscale or molecular functional moieties, such as therapeutics, imaging agents, or catalytic agents. Overall, the progressive assembly of NFs allows for fine-tuning the sizes of monodisperse and densely packed DNA NFs for versatile applications.
Figure 3.

Noncanonical and progressive self-assembly of NFs through DNA liquid crystallization. (A) SEM images of RCR1 to RCR30, indicating the progressive NF self-assembly: 1) petal-shaped structures formed on the surfaces of bulky matrices (1–3 h); 2) spherical structures starting to form on matrix surfaces (3–10 h); 3) spherical structures separated from each other to form small monodisperse NFs (10 h); and 4) larger NFs formed with longer RCR reaction time (>10 h). This suggests the NF size-tunability. (B) SEM images of the hierarchical NFs from RCR24 (diameters: ca. 2 μm) with characteristic petal-shaped surface structures. (C) SEM images showing monodisperse NFs (diameters: ca. 500 nm) self-assembled in RCR10 using T-2. (D) Optical microscopy images of NFs observed under bright field (upper), and between crossed polarizers (lower), where spherulite NFs displayed disc-shaped optical textures resulted from birefringence of liquid crystalline NFs.
We next studied the effect of template sequences on NF assembly. Based on the above-designed RCR template (T-1), NF building blocks could be crosslinked via inter- and intrastrand hybridization (Figure S1B). On one hand, the crosslinking could facilitate DNA association during NF assembly; on the other hand, the crosslinked DNA networks could hinder the enzymatic reaction inside these networks and hence retard NF assembly. To examine the influence of template sequence, we designed another template, T-2, by substituting all sequences in T-1 with deoxyadenosine (A), except the primer complement used for template circularization and primer hybridization. Therefore, the resultant RCR products would carry a long polyT motif that is not favored in DNA hybridization. Again, T-2 was circularized and used for RCR. SEM imaging indicates that NFs were also formed in RCR10 using T-2, and the NF sizes (Figure 3C; diameter of ca. 500 nm) were much larger than those formed using T-1 for the same reaction time. The larger NF sizes could be attributed to the relatively loose structure of the circular T-2 and the resultant less hindrance to enzymatic activity, as well as less inter- and intrastrand DNA hybridization of RCR products leading to more loose DNA compaction. The ability to form NFs using a template that is not amenable to DNA hybridization provides strong evidence that the NF assembly does not rely on conventional Watson-Crick base-pairing. This essentially avoids the conventional complicated sequence design in nanostructure construction by DNA hybridization, and allows the incorporation of versatile DNA functional moieties into NFs, thereby making this strategy of building DNA nanostructures noncanonical and widely applicable. Furthermore, these data imply that NF sizes can also be tuned by adjusting template sequences.
NF assembly driven by DNA liquid crystallization
As mentioned above, both genomic DNA and synthetic short DNA have been documented to be liquid crystalline at high local concentration.10–15,37 Given the capability of RCR to create highly concentrated DNA, we then studied whether NFs were also assembled through DNA liquid crystallization. Polymer (e.g., DNA) liquid crystals (LC) are formed above a critical concentration.12 Under the hypothesis that RCR with an increasing template concentration within a specified time period would produce an increasing local DNA concentration under optimal conditions and that LC would be formed when DNA reached above the critical concentration, the effect of template DNA concentration on NF formation was investigated. As shown in Figure S6, with increasing template (T-1) concentrations (10 nM, 30 nM, 100 nM, and 200 nM) in RCR, NF-shaped structures were not formed within 10 h until T-1 concentration increased up to 100 nM. Moreover, the NFs from RCR with 100 nM and 200 nM template were not as monodisperse as those observed with 300 nM template. Combined with the result that NFs were not formed until a sufficient time of RCR to accumulate local DNA, this result suggests that NFs were only assembled when the local DNA concentration achieved a critical concentration, providing the basis of NF assembly through DNA liquid crystallization. The liquid crystalline structures of NFs were directly validated using polarized light microscopy, which revealed that the spherulite NFs observed between crossed polarizers were birefringent, an optical property of anisotropic materials (Figure 3D). The optical texture of one NF, characterized by a disc-shaped phase, was consistent with that observed from the nematic liquid crystalline phase of segregated short DNA (6–20 bases) domains resulted from dense DNA packaging and ordering. 13,14,37 Additional evidence for LC formation in NFs include 1) TEM imaging of NFs revealing ultrathin sheet sections on DNA NFs (Figure S7), and 2) both SEM imaging and TEM imaging illustrated hierarchical structures on NF surfaces or inside NFs. The ultrathin sheet sections and hierarchical structures suggest an anisotropic process of NF assembly, consistent with characteristic anisotropic molecular alignment in polymer crystallization. Overall, these results demonstrate that NFs were self-assembled noncanonically through liquid crystallization of DNA, and in turn suggest NFs as excellent carriers with high-payload capacity for DNA functional moieties.
Exceptional stability of DNA NFs
The stability of nanoassemblies is critical to its practical application, especially under complicated environments of biomedical applications. For instance, when delivered systematically to a physiological environment, DNA nanoassembly degradation or dissociation could be facilitated by ubiquitous nucleases, dilution to very low concentrations by large volumes of circulating blood, and strong shear force. To assess the stability of NFs under nuclease treatment, NFs were incubated with DNase I for 24 h, followed by SEM imaging to examine the structural integrity of NFs. DNase I is a ubiquitous endonuclease that can cleave both ssDNA and dsDNA,6 and is responsible for at least 90% of the deoxynuclease activity in human plasma.38 Results in Figure 4A–B verified the structural integrity of NFs after treatment with up to 5 U/mL DNase I, which is considerably higher than the DNase I concentration in human blood (less than 1 U/mL).6,38 The stability of NFs was further evaluated using human serum, and SEM observation verified that the resultant NFs still had flower shape after treatment for 24 h (Figure 4C). Conventional DNA nanostructures assembled from short DNA building blocks tend to dissociate at a fairly low concentration after systematic injection into the human body.39 To evaluate the structural integrity in diluted solutions, NF solutions were diluted 100 times in water and stayed for 0.5 h. The SEM results shown in Figure 4D again verified the structural integrity of NFs. In addition, we evaluated the stability of NFs under denaturation conditions, which could disrupt hydrogen bonding and eventually cause nanoassembly dissociation,40 and SEM results confirmed the stability of NFs at high temperature (170 °C. Figure 4E) or treated with 5 M urea (Figure 4F). These data clearly demonstrate the remarkable stability of DNA NFs. The exceptional stability is presumably attributed to several features of NFs, including 1) long building blocks, which avoid the otherwise many nick sites sensitive to nuclease cleavage; 2) high density of DNA packed in each NF, reducing nuclease accessibility; and 3) extensive inter- and intrastrand weaving of long DNA building blocks, preventing denaturation or dissociation. Furthermore, in case the outer layer of NFs is dissociated or cleaved, it should be noted that the inner layers could maintain their functionalities. The high stability makes NFs promising for applications under complicated biomedical situations, such as clinical diagnosis, biomedicine, and biotechnology.
Figure 4.

Exceptional stability of NFs. (A–C) SEM images of NFs treated with DNase I (A,B. 5 U/mL) and human serum (C, 10% diluted) for 24 h at 37 °C. (D–F) SEM images displaying NFs heated at 170 °C for 0.5 h (D), diluted 100 times (E), and treated with urea (5 M) for 0.5 h (F). The maintenance of NF structural integrity indicates the high stability of NFs.
Biomedical applications of multifunctional DNA NFs
NFs can be assembled using essentially any DNA templates suitable for RCR. Therefore, various DNA functional moieties can be incorporated during NF assembly for versatile applications. For example, during the de novo generation of NF building blocks, functionalities can also be incorporated via 1) chemically functionalized deoxynucleotides that can be utilized as enzymatic substrates in RCR and 2) customized design of templates and primers. In this study, we used both approaches to incorporating different fluorescent bioimaging agents, aptamers, and drug loading sites.
The incorporation of fluorophores into NFs was first studied. Fluorescence imaging is usually less invasive and has been extensively used in bioimaging. In the first approach (Figure 5A) to incorporating fluorophores, FITC, as a model, was chemically modified on one end of RCR primer, such that multiple copies of fluorophores can be integrated into one NF that is composed of many copies of DNA building blocks. In the second approach, bioimaging agents were incorporated enzymatically via chemically modified deoxynucleotides (Figure 5D). To demonstrate this principle, a Cyanine 5 (Cy5)-modified dUTP, which can work as a substrate for many DNA polymerases, including Φ29,41 was used in RCR to incorporate Cy5 into NFs. The morphology of these two types of NFs was verified by SEM (Figure 5B,E), and their fluorescence properties were validated through fluorescence microscopy (Figure 5C,F). The successful incorporation of fluorophores into NFs through different approaches provides the basis for the applications of NFs in bioimaging.
Figure 5.
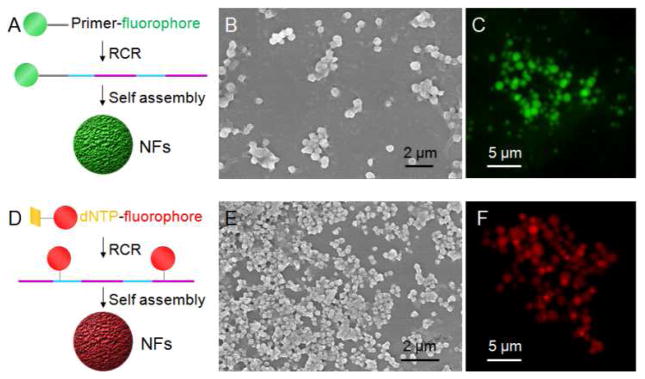
Versatile incorporation of fluorescent bioimaging agents into NFs. (A, D) Schematic representation of the incorporation of fluorophores into NFs via chemical attachment of FITC on primers (A), and via chemically modified deoxynucleotides (Cy5-dUTP) (D). The resultant NFs were characterized by SEM imaging (B: FITC-NF0.5s; E: Cy5-NF0.3s) and fluorescence microscopy imaging (C: FITC-NF2s; F: Cy5-NF2s).
Given that aptamer (sgc8) and drug loading sequences for Dox were incorporated in NFs and their small size promising for future in vivo application, NF0.2s were evaluated for selective cancer cell recognition and targeted drug delivery. Using flow cytometry, NF0.2s incorporated with FITC, drug loading sites, and sgc8, were demonstrated to selectively recognize target HeLa cells (Figure 6A) and CEM cells (Figure 6B), but not nontarget Ramos cells (Figure 6C). This provides the basis for potential applications in cancer theranostics. These NFs were then studied for bioimaging and targeted anticancer drug delivery. To achieve efficient intracellular drug delivery, nanocarriers have been developed that can be internalized into diseased cells. Nanomaterials within an appropriate size range can be internalized by many cancer cells,35,36 and our NFs can be tuned to fall into this range. In addition, molecular elements, e.g., aptamers,42,43 antibodies,44 and folic acid,45 have all been explored for receptor-mediated internalization. Specifically, sgc8 was shown to be internalized into target cancer cells via endocytosis.43 Using confocal microscopy imaging, the intracellular fluorescence (FITC) signals of NFs were identified in target HeLa cells as an example (Figure 6D), indicating the internalization of these NFs. This was further demonstrated using two-photon microscopy (TPM) (Figure S8), an imaging technique featured with less excitation light absorbed by tissues out of focal volume, deep tissue penetration, lower tissue autofluorescence, and less tissue photo-damage, compared to conventional fluorescence microscopy. These results demonstrate the application of fluorophore-incorporated NFs for bioimaging, and the revealed internalization ability of NFs into target cancer cells allows for efficient drug delivery mediated by NFs.
Figure 6.

Biomedical applications of multifunctional NFs for selective cancer cell recognition, intracellular bioimaging, and targeted anticancer drug delivery. (A–C) Flow cytometry results demonstrating the selective recognition of sgc8-incorporated NF0.2s (FITC-incorporated on primers) to target HeLa cells (A) and CEM cells (B), but not to nontarget Ramos cells (C). (D) Confocal microscopy images displaying that these NF0.2s were internalized into target HeLa cells after incubation at 37 °C for 2 h. (E–G) MTS assay results showing selective cytotoxicity of Dox delivered by NFs in target HeLa cells (E) and CEM cells (F), but much less in nontarget Ramos cells (G), in contrast to nonselective cytotoxicity of free Dox in both target cells and nontarget cells. The selective cytotoxicity of Dox delivered by NFs indicates its capability of targeted drug delivery.
For biomedical applications, DNA nanomaterials have attracted tremendous interest, owing to the typical biocompatibility and biodegradability. In particular, the biocompatibility of NFs was verified by an MTS assay that showed negligible inhibition of proliferation in cancer cells (Figure S9A). The high density of DNA drug loading sequences and internal hierarchical structures make NFs intriguing for high-capacity loading of therapeutics via either association with specific drug-loading sequences or by physical encapsulation in the internal porous structures. To load Dox into NFs, Dox and NF0.2s were mixed and stayed at room temperature to allow saturation of drug loading, followed by centrifugation and removal of free Dox in the supernatant. The amount of removed Dox was determined, and the drug loading capacity of the NFs was calculated accordingly. The resultant NF-Dox complexes, whose structural integrity was confirmed by SEM (Figure S9B), were then evaluated for targeted drug delivery. Specifically, target CEM cells and HeLa cells, and nontarget Ramos cells, were all treated with free Dox and NF-Dox, respectively, at the equivalent drug concentrations. After incubation of cells with NF-Dox for 2 h, the distribution of Dox delivered via NFs in HeLa cells, as an example, was examined using confocal microscopy, and results verified the intracellular uptake and accumulation of Dox (Figure S9C). The limited Dox distribution in nucleus is speculated to be resulted from the slow Dox release kinetics, due to dense and viscous macromolecular barriers along the release routes, as reported in a similar drug release study.19 Dox was also located around cell membrane, which can be explained by the limited internalization efficiency within this time period as well as the slow release kinetics. The cell viabilities were evaluated using an MTS assay. In target cells, Dox delivered by NFs induced only slightly less inhibition of cell proliferation than free Dox (Figure 6E,F), while in nontarget Ramos cells, NF-Dox complexes induced considerably less inhibition of cell proliferation than free Dox (Figure 6G). In contrast to nonselective cytotoxicity of free Dox in both target and nontarget cells, selective cytotoxicity induced by NF-Dox complexes in target cells demonstrated the applicability of NFs for targeted drug delivery. Overall, these results clearly demonstrated the capability of intracellular imaging and targeted drug delivery mediated by multifunctional NFs.
CONCLUSIONS
Inspired by the natural high packaging efficiency of genomic DNA or synthetic DNA in a manner that does not rely on Watson-Crick base-pairing, we herein presented a noncanonical strategy to build monodisperse multifunctional DNA nanostructures, termed as nanoflowers (NFs), and demonstrated their versatile biomedical applications. These hierarchical NFs were self-assembled from elongated DNA building blocks generated through Rolling Cycle Replication (RCR). Unlike a large amount of myriad DNA strands typically used in conventional DNA nanostructure construction, only a low amount of two strands (one template and one primer) are needed in NF assembly. The unnecessity of Watson-Crick base-pairing for NF assembly avoids the otherwise complicated design of DNA sequences for inter- or intrastrand hybridization in conventional nanostructure assembly. Further investigation demonstrated that NFs are assembled through DNA liquid crystallization, an anisotropic process for orderly alignment of highly concentrated polymers. The DNA in NFs is thus densely packed, a desirable feature for high-capacity encapsulation of DNA moieties, such as therapeutics and bioimaging agents. The NFs are size-tunable, with diameters ranging from approximately 200 nm to a few micrometers, by manipulating such factors as the RCR reaction time or template sequences. The unnecessity of Watson-Crick base-pairing and the size tunability enable NFs promising for versatile applications. DNA NFs are exceptionally resistant to nuclease treatment, dilution to low concentration, or denaturation by heating or urea treatment. The high stability of NFs is attributed to their characteristic features, including long and non-nicked DNA building blocks that provide relative less cleavage sites for nucleases, and dense DNA packaging. The high stability is critical for the biomedical and biotechnological applications of NFs. Importantly, various functionalities can be simply incorporated into NFs during self-assembly. As a proof-of-principle study, through template design, NFs were incorporated with 1) fluorescent bioimaging agents by attaching fluorophores either on primers or on deoxynucleotide triphosphates (dUTP), 2) cancer cell-targeting aptamers for selective cancer cell recognition, and 3) drug-loading sequences for targeted anticancer drug delivery. The resultant multifunctional NFs were further implemented for bioimaging, selective cancer cell recognition, and targeted anticancer drug delivery. Collectively, a noncanonical strategy was presented for the self-assembly of multifunctional DNA NFs in a way that does not rely on Watson-Crick base-pairing, and the resultant NFs were featured by size-tunability, dense DNA packaging, exceptional stability, and ease of functionalization. As such, these NFs are promising for versatile biomedical applications.
EXPERIMENTAL SECTION
Self-assembly of DNA NFs using RCR
A phosphorylated linear template (0.6 LM) and a primer (1.2 LM, serving as a ligation template) were mixed and annealed in DNA ligation buffer (5 mM Tris-HCl, 1 mM MgCl2, 0.1 mM ATP, 1 mM Dithiothreitol) by heating at 95 °C for 2 min, followed by gradual cooling to room temperature over 3 h. The annealed product was incubated with T4 DNA ligase (10 U/LL; New England Biolabs, Ipswich, MA) at room temperature for 3 h. For RCR, the resultant circularized template (0.3 LM, unless otherwise denoted) was incubated with Φ29 DNA polymerase (2 U/LL), dNTP (2 mM/LL), and BSA (1X) in buffer solution (50 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM MgCl2, 4 mM Dithiothreitol) (New England Biolabs, Ipswich, MA) at 30 °C. Cyanine 5-dUTP (200 LM; Enzo Lifescicence, Farmingdale, NY) was added to the reaction mixture, if applicable. Reactions were terminated by heating at 75 °C for 10 min. The NFs were then washed with double-distilled H2O, precipitated by centrifugation, and stored at 4 °C for future use.
Evaluation of the stability of NFs
The stability of NFs under nuclease cleavage, dilution to low concentration, heating, and urea denaturation was evaluated, respectively. Specifically, for nuclease cleavage, NFs were treated with DNase I (New England Biolabs, Ipswich, MA) of specified concentrations, or 10% human serum (Asterand, Detroit, MI) diluted in Dulbecco’s PBS (Sigma Aldrich, St. Louis, MO) at 37 °C for 24 h, prior to nuclease deactivation by heating at 75 °C for 10 min. For dilution, NFs were diluted 100 times in Dulbecco’s PBS from the original NF solution; for heating, NFs were kept at 170 °C for 0.5 h; and for denaturation, NFs were treated with urea (5 M) for 0.5 h, prior to washing with double-distilled water. The morphologies of the resultant NFs were then examined using SEM.
Specific recognition ability to target cancer cells
The binding abilities of aptamers or aptamer-incorporated NFs were determined using flow cytometry on a FACScan cytometer (BD Immunocytometry Systems). Aptamers (200 nM) or aptamer-incorporated NFs (10 LL equivalent NF reaction solution) were incubated with cells (2×105) in binding buffer (200 LL; 4.5 g/L glucose, 5 mM MgCl2, 0.1 mg/mL yeast tRNA (Sigma Aldrich, St. Louis, MO) and 1 mg/mL BSA (Fisher Scientific, Pittsburg, PA) in Dulbecco’s PBS) on ice for 30 min, followed by washing twice with washing buffer (1 mL, 4.5 g/L glucose and 5 mM MgCl2 in Dulbecco’s PBS). Cells were suspended in binding buffer (200 LL) prior to flow cytometric analysis. Data were analyzed with the FlowJo software (Tree Star, Inc., Ashland, OR). Random DNA sequences (lib) were used as a negative control.
Supplementary Material
Acknowledgments
The authors sincerely Ms. Lei Mei and Ms. Hao Liang for help with manuscript preparation, and thank Dr. Kathryn R. Williams for manuscript review and insightful discussions. This work is supported by grants awarded by the National Key Scientific Program of China (2011CB911000), the Foundation for Innovative Research Groups of NSFC (Grant 21221003), China National Instrumentation Program 2011YQ03012412 and by the National Institutes of Health (GM079359 and CA133086).
Footnotes
Notes
The authors declare no competing financial interest.
Additional materials and methods; DNA sequences; figures about characterization and application of NFs. This material is available free of charge via the Internet at http://pubs.acs.org.”
References
- 1.Seeman NC. Ann Rev Biochem. 2010;79:65. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bath J, Turberfield AJ. Nat Nanotechnol. 2007;2:275. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 3.Drmanac R, Sparks AB, et al. Science. 2010;327:78. [Google Scholar]
- 4.Genereux JC, Barton JK. Chem Rev. 2009;110:1642. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McManus SA, Li Y. J Am Chem Soc. 2013;135:7181. doi: 10.1021/ja311850u. [DOI] [PubMed] [Google Scholar]
- 6.Tamkovich SN, Cherepanova AV, Kolesnikova EV, Rykova EY, Pyshnyi DV, Vlassov VV, Laktionov PP. Ann New York Acad Sci. 2006;1075:191. doi: 10.1196/annals.1368.026. [DOI] [PubMed] [Google Scholar]
- 7.Hamblin GD, Carneiro KMM, Fakhoury JF, Bujold KE, Sleiman HF. J Am Chem Soc. 2012;134:2888. doi: 10.1021/ja2107492. [DOI] [PubMed] [Google Scholar]
- 8.Venter JC, Adams MD, et al. Science. 2001;291:1304. [Google Scholar]
- 9.Olins DE, Olins AL. Nat Rev Mol Cell Biol. 2003;4:809. doi: 10.1038/nrm1225. [DOI] [PubMed] [Google Scholar]
- 10.Chow MH, Yan KTH, Bennett MJ, Wong JTY. Eukaryotic Cell. 2010;9:1577. doi: 10.1128/EC.00026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livolant F. Tissue & Cell. 1984;16:535. doi: 10.1016/0040-8166(84)90029-6. [DOI] [PubMed] [Google Scholar]
- 12.Strzelecka TE, Davidson MW, Rill RL. Nature. 1988;331:457. doi: 10.1038/331457a0. [DOI] [PubMed] [Google Scholar]
- 13.Nakata M, Zanchetta G, Chapman BD, Jones CD, Cross JO, Pindak R, Bellini T, Clark NA. Science. 2007;318:1276. doi: 10.1126/science.1143826. [DOI] [PubMed] [Google Scholar]
- 14.Zanchetta G, Nakata M, Buscaglia M, Bellini T, Clark NA. Proc Natl Acad Sci USA. 2008;105:1111. doi: 10.1073/pnas.0711319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livolant F, Levelut AM, Doucet J, Benoit JP. Nature. 1989;339:724. doi: 10.1038/339724a0. [DOI] [PubMed] [Google Scholar]
- 16.Johne R, Muller H, Rector A, van Ranst M, Stevens H. Trends Microbiol. 2009;17:205. doi: 10.1016/j.tim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Cohen S, Agmon N, Yacobi K, Mislovati M, Segal D. Nucleic Acids Res. 2005;33:4519. doi: 10.1093/nar/gki764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fire A, Xu SQ. Proc Natl Acad Sci USA. 1995;92:4641. doi: 10.1073/pnas.92.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JB, Peng S, Yang D, Roh YH, Funabashi H, Park N, Rice EJ, Chen L, Long R, Wu M, Luo D. Nat Nanotechnol. 2012;7:816. doi: 10.1038/nnano.2012.211. [DOI] [PubMed] [Google Scholar]
- 20.Zhao W, Cui CH, Bose S, Guo D, Shen C, Wong WP, Halvorsen K, Farokhzad OC, Teo GSL, Phillips JA, Dorfman DM, Karnik R, Karp JM. Proc Natl Acad Sci USA. 2012;109:19626. doi: 10.1073/pnas.1211234109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson JP, Reynolds BL, Baum K, Williams JG. Nano Lett. 2010;10:788. doi: 10.1021/nl9039718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin C, Xie M, Chen JJL, Liu Y, Yan H. Angew Chem, Int Ed. 2006;45:7537. doi: 10.1002/anie.200602113. [DOI] [PubMed] [Google Scholar]
- 23.Larsson C, Koch J, Nygren A, Janssen G, Raap AK, Landegren U, Nilsson M. Nat Meth. 2004;1:227. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Daubendiek SL, Zillman MA, Ryan K, Kool ET. J Am Chem Soc. 1996;118:1587. doi: 10.1021/ja952786k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan R, Zuo X, Wang S, Quan X, Chen D, Chen Z, Jiang L, Fan C, Xia F. J Am Chem Soc. 2013;135:4604. doi: 10.1021/ja311313b. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, Li B, Milligan JN, Bhadra S, Ellington AD. J Am Chem Soc. 2013;135:7430. doi: 10.1021/ja4023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alama A, Barbieri F, Cagnoli M, Schettini G. Pharmacol Res. 1997;36:171. doi: 10.1006/phrs.1997.0227. [DOI] [PubMed] [Google Scholar]
- 28.Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner GJ, Liu HM, Wooldridge JE, Dahle CE, Krieg AM. Proc Natl Acad Sci USA. 1997;94:10833. doi: 10.1073/pnas.94.20.10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu G, Meng L, Ye M, Yang L, Sefah K, O’Donoghue MB, Chen Y, Xiong X, Huang J, Song E, Tan W. Chem Asian J. 2012;7:1630. doi: 10.1002/asia.201101060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shangguan D, Li Y, Tang Z, Cao Z, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Proc Natl Acad Sci USA. 2006;103:11838. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H, Li Y, Tan W. J Prot Res. 2008;7:2133. doi: 10.1021/pr700894d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin C, Wang X, Liu Y, Seeman NC, Yan H. J Am Chem Soc. 2007;129:14475. doi: 10.1021/ja0760980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JB, Hong J, Bonner DK, Poon Z, Hammond PT. Nat Mater. 2012;11:316. doi: 10.1038/nmat3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 36.Petros RA, DeSimone JM. Nat Rev Drug Discov. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 37.Bellini T, Zanchetta G, Fraccia TP, Cerbino R, Tsai E, Smith GP, Moran MJ, Walba DM, Clark NA. Proc Natl Acad Sci USA. 2012;109:1110. doi: 10.1073/pnas.1117463109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nadano D, Yasuda T, Kishi K. Clin Chem. 1993;39:448. [PubMed] [Google Scholar]
- 39.Shu D, Shu Y, Haque F, Abdelmawla S, Guo P. Nat Nanotechnol. 2011;6:658. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herskovits TT. Biochemistry. 1963;2:335. doi: 10.1021/bi00902a027. [DOI] [PubMed] [Google Scholar]
- 41.Shalon D, Smith SJ, Brown PO. Genome Res. 1996;6:639. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- 42.Zhang K, Sefah K, Tang L, Zhao Z, Zhu G, Ye M, Sun W, Goodison S, Tan W. ChemMedChem. 2012;7:79. doi: 10.1002/cmdc.201100457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao Z, Shangguan D, Cao Z, Fang X, Tan W. Chem Eur J. 2008;14:1769. doi: 10.1002/chem.200701330. [DOI] [PubMed] [Google Scholar]
- 44.Chari RVJ. Acc Chem Res. 2007;41:98. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 45.Santra S, Kaittanis C, Santiesteban OJ, Perez JM. J Am Chem Soc. 2011;133:16680. doi: 10.1021/ja207463b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.