

Abstract
Primate immunodeficiency viruses, including HIV-1, are characterized by the presence of accessory genes such as vif, vpr, vpx, vpu, and nef. Current knowledge indicates that none of the primate lentiviral accessory proteins has enzymatic activity. Instead, these proteins interact with cellular ligands to either act as adapter molecules to redirect the normal function of host factors for virus-specific purposes or to inhibit a normal host function by mediating degradation or causing intracellular mislocalization/sequestration of the factors involved. This review aims at providing an update of our current understanding of how Vif, Vpu, and Vpx control the cellular restriction factors APOBEC3G, BST-2, and SAMHD1, respectively.
Overview
Since its identification as the causative agent of AIDS, HIV-1 has been made responsible for the deaths of millions of people worldwide. And there is no light at the end of the tunnel just yet. One might think that viruses should not have an inherent interest in killing their hosts; after all, why bite the hand that feeds you? In fact, many viruses have adapted to their hosts in a manner that allows a relatively peaceful coexistence. In addition, many viral infections are effectively controlled by preventive vaccination. So why is it that HIV-1 or other viruses such as Influenza still cause worldwide pandemics? The answer to that question is quite simple: (i) preventive vaccines are unavailable or ineffective and (ii) our immune system has not (yet) adapted to these viral challenges. In the case of HIV, the virus infects and kills CD4+ T cells thereby profoundly damaging the host’s ability to mount an effective adaptive T- or B-cell response [1,2]. On top of that, HIV can integrate into memory cells and long-lived macrophages, thereby establishing a latent or chronic infection. That is not to say that humans are not equipped with tools to fight HIV infections. Indeed, there is a growing list of host restriction factors that can, at least in principle, target human immunodeficiency viruses, including Apolipoprotein B mRNA-editing enzyme 3G (APOBEC3G), bone marrow stromal cell antigen 2 (BST-2), cyclophilin A, and tripartite motif protein 5alpha (Trim5α) (for review see [3]). The most recent addition to this growing list of antiviral restriction factors is SAMHD1, which was identified in a proteomics screen as a Vpx-interacting factor. SAMHD1 is now known to be responsible for the Vpx-sensitive restriction of HIV and SIV in myeloid cell types such as macrophages and dendritic cells [4,5]. APOBEC3G, SAMHD1, BST-2, Trim5α, or cyclophilin A are all part of the innate or intrinsic immune system and represent a rapid response system to fight infections.
Natural infection of African green monkeys by SIV typically does not induce AIDS-like symptoms despite persistent virus infection [6]. So why is it that SIV-infected monkeys live happily ever after while infection of humans by the closely related HIV-1 causes severe and generally fatal immune deficiency? Again, the answer - at least in part - is lack of adaptation. HIV-1 is believed to have evolved from chimpanzee immunodeficiency virus (SIVcpz) and was only recently introduced into humans [7]. While there is evidence for positive selection of host restriction factors [8], population-wide adaptation of the innate and intrinsic immune system to new viral challenges is slow and probably needs to be measured in centuries rather than weeks. This is probably the reason why in the case of HIV-1, viral countermeasures do currently have the upper hand.
When HIV-1 was first cloned and sequenced, researchers were surprised to find that in addition to the prototypical retroviral gag, pol, and env genes, this virus contained several additional open reading frames. Collectively, they were termed accessory genes due to a lack of understanding of their biological functions. HIV-1 has four such accessory genes, vif, vpr, vpu, and nef. HIV-2 and SIV also encode vif, vpr, and nef but they lack for the most part vpu. Instead, HIV-2 and most SIVs encode a vpx gene. However, it is important to point out that Vpu and Vpx are not functional homologues and they target different host factors as described in more detail below. While each accessory protein targets different cellular factors, the strategies employed are strikingly similar: none of the HIV accessory proteins has enzymatic activity; instead, they all seem to act as molecular adapters to manipulate the host cell, resulting in most instances in the proteolytic degradation of the cellular target (Fig. 1).
Figure 1.
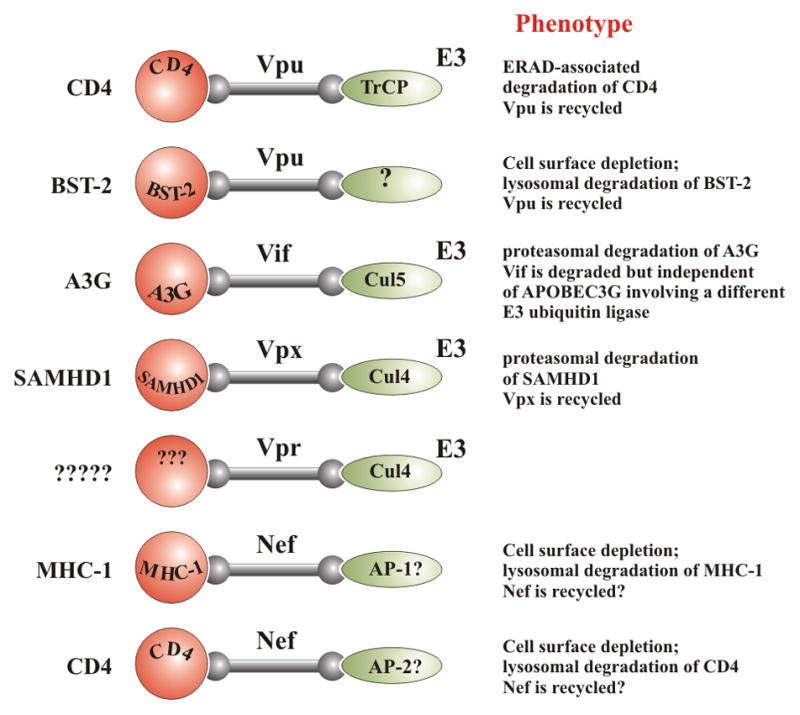
HIV accessory proteins function as adapter molecules. HIV accessory proteins have no enzymatic activity. Instead, they act as adaptor molecules to connect cellular substrates to other cellular pathways such as E3 ubiquitin ligases that then trigger ubiquitination and degradation of the substrates. In some cases, they connect their substrate to adapter protein (AP) complexes that trigger internalization and subsequent lysosomal degradation of the substrate. In most cases, the viral adapter molecule is recycled. Substrates are shown on the left and effectors on the right. For recent reviews on the function of Nef and Vpr see [78–81].
Vif
Vif (Viral infectivity factor) is critical for the production of infectious virus in vivo. While vif-deficient viruses can replicate unimpaired in many tissue culture systems, there are no known replication competent vif defective primary HIV or SIV isolates [9]. Vif targets APOBEC3G, a cellular cytidine deaminase that in the absence of Vif is packaged into virions and causes severe damage to the viral genome by deaminating cytidine residues during reverse transcription of the viral genome [10]. Deamination of cytidine produces deoxyuridine that is misread by the reverse transcriptase as thymidine during second strand cDNA synthesis and results in the insertion of alanine instead of guanine (reviewed in [11]). The presence of deoxyuridine in single-stranded viral cDNA can also lead to activation of the cellular DNA repair machinery and result in lethal fragmentation of the viral cDNA. In the presence of Vif, APOBEC3G is excluded from virions, thus allowing the virus to replicate unharmed in APOBEC3G-expressing cells. This makes Vif an interesting target for the development of novel antivirals. Indeed, several small molecule inhibitors targeting Vif/APOBEC3G have been identified [12,13]. However, none of them has proven very potent when tested in tissue culture. Dominant-negative mutants of Vif that interfere with the activity of virus-encoded Vif may offer an alternative approach but their potential for development into clinically useful antivirals remains to be explored [14].
One of the important points to consider when designing Vif-based antivirals is that incomplete inhibition of Vif could be counterproductive and provide the virus with a selective advantage. Indeed, naturally occurring HIV-1 variants with partially defective vif genes rapidly developed drug resistance when put under selection pressure [15]. The reason is that sublethal levels of APOBEC3G will not completely block virus replication but will promote deamination-induced mutagenesis of the viral genome, which in turn accelerates viral evolution in response to environmental challenges such as antiviral drug therapy. So how does Vif neutralize APOBEC3G? The commonly accepted and most widely studied mechanism is proteasomal degradation. Vif interacts with APOBEC3G and at the same time assembles a Cul5-based E3 ubiquitin ligase complex [16]. This molecular adapter function of Vif results in ubiquitination of APOBEC3G and subsequent degradation by the cellular proteasomal machinery (Fig. 1). It was also reported that Vif can inhibit the packaging of APOBEC3G through degradation-independent pathway(s) [17]. Although not well understood at the molecular level, a degradation-independent mechanism may be especially critical early in infection when a virus enters a cell that is loaded with pre-existing APOBEC3G. Controlling pre-existing APOBEC3G is not a trivial job. Experimental evidence indicates that Vif preferentially degrades newly synthesized APOBEC3G while pre-existing APOBEC3G, which is presumably engaged in high-molecular mass complexes with other host proteins and RNA, is relatively insensitive to Vif-induced degradation [18]. Yet, such pre-existing APOBEC3G is efficiently packaged into HIV virions and potently blocks viral infectivity unless Vif is present to prevent APOBEC3G encapsidation. Long-term exposure of cells to Vif will eventually result in depletion of APOBEC3G [19]. However, early on when Vif levels are still low, APOBEC3G clearly outnumbers Vif. Thus, if APOBEC3G degradation where the only mechanism available to Vif, one would expect viruses produced early during infection, when there are still significant amounts of APOBEC3G in a cell, to be less infectious than later on when Vif has reached steady-state levels and cells are depleted of APOBEC3G. Such a phenomenon was, however, never observed experimentally. In fact, the relative infectivity of viruses produced from HIV-infected macrophages decreased rather than increased with time even though levels of APOBEC3G in the cultures gradually decreased [19].
More recently, Vif-induced degradation of APOBEC3G was found to involve CBFβ [20,21] a cellular transcription factor known to form hetero-dimeric complexes with RUNX proteins to enhance their DNA binding activity [22]. The interaction of CBFβ with Vif was identified in a proteomics screen [20,21]. How a cellular transcription factor enhances Vif-induced degradation of APOBEC3G is not entirely clear. However, CBFβ does not appear to directly interact with APOBEC3G or components of the E3 ubiquitin ligase complex (i.e., elongin B/C, or Cul5) but instead seems to ensure proper folding of Vif. CBFβ also seems to increase the stability of Vif [21,23]. Again, the molecular mechanism is unclear. It is worth noting though that even in the presence of CBFβ, Vif is unstable and subject to proteasomal degradation. The impact of CBFβ on the production of infectious virus has yet to be determined. Given that production of infectious viruses is not absolutely dependent on APOBEC3G degradation, the reported impact of CBFβ on APOBEC3G degradation may not be directly reflected in its effect on viral infectivity.
Vpu
Vpu is unique to HIV-1 and its predecessor SIV strains. The initial functional property attributed to Vpu was to facilitate virus release from infected cells [24]. It became apparent early on that this function of Vpu was host cell-dependent, suggesting the involvement of host cell factor(s). However, not until 20 years after the initial discovery of Vpu and its effect on virus release was the involvement of BST-2 unraveled [25,26]. Prior to the identification of BST-2, different functional models of how Vpu regulates virus release had been proposed. Among them were regulation of virus release by a Vpu ion channel activity or inactivation of a cellular background channel by Vpu [27–29]. However, the identification of BST-2 as a target for Vpu induced a major shift in research focus and investigating the role of Vpu in the antagonism of BST-2 is unquestionably the most active aspect of current Vpu-related research. Nevertheless, one should not ignore the fact that Vpu controls additional functions. These include degradation of the HIV receptor CD4 or the inhibition of NFκB activation. These functions of Vpu are relatively well understood on a mechanistic level but their significance for HIV replication is still not fully clear [24,30].
BST-2 inhibit virus release by tethering Vpu-defective virions to the plasma membrane [25,26]. BST-2 is a transmembrane protein with two membrane anchors. Based on its homology to rat BST-2 [31], human BST-2 is generally thought to contain a C-terminal glycosylphosphatidylinositol (GPI) anchor even though experimental evidence points to a second transmembrane domain [32]. The two membrane anchors are connected by an ectodomain containing three cysteine residues that are involved in the formation of functionally critical cysteine-linked dimers [33,34]. Interestingly, there is significant flexibility regarding size and structure of the BST-2 ectodomain [35]; in fact, an artificial BST-2 construct assembled from various heterologous proteins had tethering activity but was insensitive to Vpu [34]. BST-2 can be neutralized by three different lentiviral proteins: HIV-1 employs Vpu, HIV-2 uses its Env glycoprotein, and SIV uses Nef. All three proteins are thought to interfere with BST-2 function via direct physical interaction ultimately leading to depletion of BST-2 from the cell surface. However, the observation that Vpu can enhance virus release in acutely infected T cells in the absence of appreciable surface downmodulation [36], suggests that surface depletion of BST-2 is a downstream consequence of Vpu activity, caused at least in part by interference of Vpu with the resupply of surface depleted BST-2 from the pool of newly synthesized protein [24,37]. The fact that lentiviruses have evolved three proteins to independently target BST-2 highlights the importance of controlling BST-2 function. It remains a matter of debate, however, exactly why control of BST-2 is of such importance to HIV. One argument is that the expression of BST-2 on human target cells may limit transmission of HIV-1 from its predecessor SIV [38]. This is because Vpu from SIVcpz, while capable of degrading CD4, is unable to target chimpanzee or human BST-2. In these viruses, Nef has assumed the role of Vpu to enhance virus release. However, Nef cannot target human BST-2 because its target sequence (G/D)DIWK) located in the cytoplasmic domain of BST-2 has been lost in human BST-2. On the other hand, Vpu from certain HIV-1 isolates is capable of targeting macaque BST-2 [39] suggesting that species-specificity of Vpu is not as absolute as initially thought. It is not quite clear how or even why Vpu acquired anti-BST-2 activity. For instance HIV-2, which lacks a vpu gene, has learned to use its Env glycoprotein to antagonize human BST-2 [40,41]. Of note, even some HIV-1 isolates, such as the R5-tropic AD8 isolate, can make use of their Env protein to regulate virus release from human cells [42] raising the interesting question of whether following the initial zoonosis, the Env protein temporarily assumed Vpu-like function in HIV-1. At any rate, the ability to target BST-2 is not a feature of Vpu that arose only as a result of the transmission of SIV into humans. Indeed, Vpus encoded by SIVgsn, SIVmon, SIVmus, and SIVden are active against the BST-2 of their native hosts but not against human BST-2 [43,44]. Nevertheless,
Inhibition of virus release should not be mistaken for inhibition of virus replication. Even though tetherin potently inhibited replication of Mo-MLV in interferon-stimulated NIH 3T3 cells, no difference in plasma viremia was observed under physiological conditions between untreated normal or tetherin knockout MoMLV-infected mice [45]. Furthermore, while tetherin seemed to delay the cell-to-cell transmission of vpu-defective HIV-1 in short-term single cell analyses [46], the phenotype of a vpu defect in HIV-1 in spreading infections is characterized by reduced levels of cell-free virus but not by reduced viral replication kinetics [47,48]. This indicates that vpu-defective virus can employ a cell-to-cell mode of transmission that, at least in tissue culture, is as effective as cell-free transmission by wild type virus. Thus, it may be more appropriate to regard BST-2 as modulator of the mode of virus transmission (i.e. cell-free versus cell-to-cell) rather than a restriction factor inhibiting virus replication. If virus transmission from person to person occurs via cell-free virus, viruses unable to target BST-2 will certainly have a selective disadvantage. On the other hand, the fact that a number of patient-derived primary HIV-1 isolates, including HXB2, MAL, and AD8, carried mutations in the vpu translation initiation codon [49,50], suggests that Vpu expression may provide selective advantages or disadvantages at certain times during virus infection and may be turned on or off depending on the environmental pressures provided by the host. Such an “on-demand” regulation of virus release activity is also seen in HIV-2 where a single amino acid change in the Env TM subunit can switch its Vpu-like activity on or off [51].
Vpx
Vpx is an accessory protein unique to HIV-2 and SIV. It is believed to have originated by gene duplication from Vpr [52]. Indeed, Vpr and Vpx proteins share several common features: (i) they are both specifically packaged into viral particles through an interaction with the p6 domain of the viral Gag precursor; and (ii) they both assemble an E3 ubiquitin ligase complex consisting of DCAF1 (previously known as Vpr-binding protein, VprBP), DDB1 (damage-specific DNA binding protein1), Cul4 (cullin4), and Rbx1 (Ring box protein1) [53,54]. The association of Vpr and Vpx with components of an E3 ubiquitin ligase complex indicted that these proteins, like Vif and Vpu, target cellular protein(s) for proteasomal degradation (Fig. 1). The fact that both proteins are specifically packaged into virions at significant levels further suggests they may play a role early during infection. Indeed, in macrophages and dendritic cells, infection by Vpx-defective virus is blocked at a pre-reverse transcription step. Interestingly, the inhibition of infection by Vpx-deficient virus was prevented when cells were preloaded with Vpx prior to infection using Vpx-containing VLPs [55,56]. While the cellular target for Vpr remains elusive, the substrate of Vpx-dependent proteolysis was recently identified as Sterile alpha motif and HD domain protein 1 (SAMHD1) [4,5,57]. Silencing of SAMHD1 in monocyte-derived macrophages or dendritic cells rendered them susceptible to infection by Vpx-defective virus [4,5,57]. Interestingly, while HIV-1 does not possess a Vpx protein, Vpx also enhances HIV-1 infection of myeloid and dendritic cells, as well as resting CD4+ T-cells [58–63]. In susceptible cell types, SAMHD1 has been shown to restrict infection of lentiviruses at the reverse transcription step. Vpx counteracts this restriction by binding to and causing the proteasomal degradation of SAMHD1 via interaction with a Cul4/DDB1/DCAF1 ubliquitin ligase complex [4,5,64]. Consistent with this, knockdown of SAMHD1 enhanced HIV-1 infection and rendered it Vpx independent [4,5,57,62].
Several splice variants of SAMHD1 were identified in PMA-treated THP-1 cells although their functional significance remains unclear [65]. Furthermore, SAMHD1 has recently been shown to possess nucleic acid binding properties [66–68] and in one study was also reported to have exonuclease activity [68]. However, its main catalytic activity described to date is its dGTP-dependent dNTPase activity that allows it to degrade cellular dNTPs [69,70]. In this way, SAMHD1 is thought to restrict HIV-1 replication by decreasing the levels of cellular dNTP pools to below that required for reverse transcription [71,72]. Interestingly, while SAMHD1 has been shown to restrict HIV-1 replication in non-dividing cell types such as MDMs, dendritic cells, resting CD4 T cells as well as PMA-differentiated THP-1 and U937 cells (the latter requiring exogenous expression of SAMHD1) [4,5,57,62,73], SAMHD1 restriction does not strictly correlate with its expression. Indeed, fully HIV-1 permissive cells, such as activated CD4+ T cells or undifferentiated THP-1 cells also express high amounts of the SAMHD1 protein [5,62]. SAMHD1 restriction could be alleviated by silencing its expression in restrictive cell types. Intriguingly, however, transfer of SAMHD1 into permissive cell types failed to induce a restrictive phenotype even when the protein was over-expressed [73]. This distinguishes SAMHD1 from APOBEC3G and BST-2 whose restrictive phenotypes were readily transferred by expression in permissive target cells. The inability of SAMHD1 to restrict viral replication when over-expressed in HeLa or 293T cells suggested that SAMHD1 either exerts its effect in conjunction with other thus far unidentified host factor(s) or, alternatively, that the antiviral activity of SAMHD1 is regulated at a post-translational level. Such a regulation of SAMHD1 activity could explain why levels of SAMHD1 observed in undifferentiated non-restrictive THP-1 cells and in differentiated restrictive THP-1 cells are similar. Indeed, SAMHD1 was very recently found to be regulated by phosphorylation (Welbourn & Strebel [submitted] and [74,75]). Specifically, phosphorylation at a threonine residue near the C-terminus of SAMHD1 (T592) appears to inhibit SAMHD1’s ability to restrict HIV infection. The sequence surrounding T592 (LIT*PQKK) conforms well to a cyclin-dependent kinase (CDK) consensus phosphorylation target site, S/T*PXR/K [76], suggesting T592 could be phosphorylated in vivo by a cyclin-dependent kinase. CDKs together with their partner cyclins promote cell proliferation by driving progression though the cell cycle [77]. The suggestion that regulation of SAMHD1 function is tied into the cell proliferation status is consistent with the observation that SAMHD1 restricts HIV-1 only in differentiated or non-dividing cells. During cell cycle progression, CDKs would be active to phosphorylate SAMHD1 at T592 and therefore render it inactive for HIV-1 restriction. On the other hand, SAMHD1 T592 would be in a largely unphosphorylated state in non-dividing/differentiated cells since CDKs would be inactive then. This model is supported by our finding that SAMHD1 phosphorylation is lower in differentiated THP-1 cells than in their cycling counterparts. It is tempting to speculate that phosphorylation of SAMHD1 at T592 impairs its catalytic ability to degrade dNTPs. Surprisingly, however, T592 phosphorylation did not significantly affect the catalytic dNTPase activity of SAMHD1 in vitro (Welbourn and Strebel, submitted). Further experimentation will be required to fully unravel the mechanism by which SAMHD1 blocks viral infection of non-dividing cells.
Conclusion
HIV accessory genes, which were once thought to play minor roles in the virus replication cycle have turned out to be key players in the control of the host’s innate and intrinsic immune system. Among those, Vif is probably the most critical accessory protein since there are no primary HIV or SIV isolates that are defective in Vif. Vpu and Vpx, on the other hand, appear to have more specialized functions that control the mode of virus transmission or facilitate the infection of non-dividing cells, respectively. Non-dividing cells represent only a subset of natural target cells for HIV-1. However, their infection may be critical for the establishment of chronic infection. As such, treatment strategies targeting HIV accessory proteins may have great potential for inhibiting virus replication and preventing or delaying HIV-induced pathogenesis.
Host restriction factors are part of the innate immune system
HIV encodes accessory proteins to control host restriction factors
Vif degrades APOBEC3G and inhibits its encapsidation into virions
Vpu degrades CD4 and downmodulates BST-2
Vpx degrades SAMHD1
Acknowledgments
K. Strebel is supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (1 Z01 AI000669).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walker B, McMichael A. The T-cell response to HIV. Cold Spring Harb Perspect Med. 2012:2. doi: 10.1101/cshperspect.a007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perreau M, Levy Y, Pantaleo G. Immune response to HIV. Curr Opin HIV AIDS. 2013;8:333–340. doi: 10.1097/COH.0b013e328361faf4. [DOI] [PubMed] [Google Scholar]
- 3.Strebel K, Luban J, Jeang KT. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009;7:48. doi: 10.1186/1741-7015-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **4.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. This publication was published side-by-side with Ref 5 and describes the identification of SAMHD1 as a Vpx-interacting host factor. The authors report that Vpx of SIVmac251 induces the proteasomal degradation of SAMHD1. Silencing of SAMHD1 alleviates HIV-1 restriction and increases the susceptibility of monocyte-derived dendritic cells to infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **5.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. This publication was published side-by-side with Ref 4 and describes the identification of SAMHD1 as cellular target for degradation by Vpx. The authors show that SAMHD1 depletion from monocyte-derived macrophages facilitates replication of HIV-1 even though this virus does not encode Vpx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beer B, Denner J, Brown CR, Norley S, zur Megede J, Coulibaly C, Plesker R, Holzammer S, Baier M, Hirsch VM, et al. Simian immunodeficiency virus of African green monkeys is apathogenic in the newborn natural host. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:210–220. doi: 10.1097/00042560-199807010-00003. [DOI] [PubMed] [Google Scholar]
- 7.Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 8.Bozek K, Lengauer T. Positive selection of HIV host factors and the evolution of lentivirus genes. BMC Evol Biol. 2010;10:186. doi: 10.1186/1471-2148-10-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **9.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. This publication is the original report identifying CEM15 (now generally referred to as APOBEC3G) as the cellular target of Vif. The authors used a cDNA subtraction strategy comparing two closely related cell types (CEM & CEM-SS), one of which was sensitive and the other permissive for vif-defective HIV-1. Expression of CEM15 in normally permissive 293T cells transferred the restrictive phenotype. This is a landmark publication that has induced an avalanche of follow-up studies by multiple research groups. [DOI] [PubMed] [Google Scholar]
- 10.Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 12.Mohammed I, Parai MK, Jiang X, Sharova N, Singh G, Stevenson M, Rana TM. SAR and Lead Optimization of an HIV-1 Vif-APOBEC3G Axis Inhibitor. ACS Medicinal Chemistry Letters. 2012;3:465–469. doi: 10.1021/ml300037k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo T, Liu D, Lv W, Wang X, Wang J, Lv M, Huang W, Wu J, Zhang H, Jin H, et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol. 2012;86:5497–5507. doi: 10.1128/JVI.06957-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker RC, Jr, Khan MA, Kao S, Goila-Gaur R, Miyagi E, Strebel K. Identification of dominant negative human immunodeficiency virus type 1 Vif mutants that interfere with the functional inactivation of APOBEC3G by virus-encoded Vif. J Virol. 2010;84:5201–5211. doi: 10.1128/JVI.02318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulder LC, Harari A, Simon V. Cytidine deamination induced HIV-1 drug resistance. Proc Natl Acad Sci U S A. 2008;105:5501–5506. doi: 10.1073/pnas.0710190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wissing S, Galloway NL, Greene WC. HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors. Mol Aspects Med. 2010;31:383–397. doi: 10.1016/j.mam.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol. 2007;81:8236–8246. doi: 10.1128/JVI.02694-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goila-Gaur R, Khan MA, Miyagi E, Strebel K. Differential sensitivity of “old” versus “new” APOBEC3G to human immunodeficiency virus type 1 vif. J Virol. 2009;83:1156–1160. doi: 10.1128/JVI.01734-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyagi E, Schwartzkopff F, Plishka R, Buckler-White A, Clouse KA, Strebel K. APOBEC3G-independent reduction in virion infectivity during long-term HIV-1 replication in terminally differentiated macrophages. Virology. 2008;379:266–274. doi: 10.1016/j.virol.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **20.Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012;481:376–379. doi: 10.1038/nature10718. This study was published back-to-back with Ref 21 and identifies CBFβ as a Vif-interacting protein. The authors report that CBFβ is important for Vif-induced degradation of APOBEC3G. Silencing of CBFβ inhibits APOBEC3G degradation and reduces the infectivity of virions by raising the level of virus-associated APOBEC3G. CBFβ does not directly bind to APOBEC3G or Cullin5 but increases the affinity of Vif to Cullin5, a component of the E3 ubiquitin ligase responsible for ubiquitination of APOBEC3G. [DOI] [PubMed] [Google Scholar]
- **21.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012;481:371–375. doi: 10.1038/nature10693. This study was published back-to-back with Ref 20 and used a mass-spectrometry approach for the identification of novel Vif-interacting proteins. Among the identified proteins was CBFβ. Silencing of CBFβ inhibits the ability of Vif to induce degradation of APOBEC3G and to produce infectious virus. In contrast to Zhang et al (Ref 20), this study also reports a significant effect of CBFβ on the steady-state expression of Vif. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- 23.Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD. CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol Cell. 2013;49:632–644. doi: 10.1016/j.molcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrew A, Strebel K. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol Aspects Med. 2010;31:407–417. doi: 10.1016/j.mam.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 26.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 1996;398:12–18. doi: 10.1016/s0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- 28.Ewart GD, Sutherland T, Gage PW, Cox GB. The Vpu protein of human immunodeficiency virus type 1 forms cation- selective ion channels. J Virol. 1996;70:7108–7115. doi: 10.1128/jvi.70.10.7108-7115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. Mutual Functional Destruction of HIV-1 Vpu and Host TASK-1 Channel. Mol Cell. 2004;14:259–267. doi: 10.1016/s1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]
- 30.Akari H, Bour S, Kao S, Adachi A, Strebel K. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor kappaB-dependent expression of antiapoptotic factors. J Exp Med. 2001;194:1299–1311. doi: 10.1084/jem.194.9.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1. 24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- *32.Andrew AJ, Kao S, Strebel K. C-terminal Hydrophobic Region in Human Bone Marrow Stromal Cell Antigen 2 (BST-2)/Tetherin Protein Functions as Second Transmembrane Motif. J Biol Chem. 2011;286:39967–39981. doi: 10.1074/jbc.M111.287011. This study investigates the question whether human BST-2 carries a C-terminal GPI-anchor modification or a second transmembrane (TM) domain. GPI anchor modification was shown for rat BST-2; however, human and rat BST-2 share only 33 amino acid identity. This study fails to develop experimental evidence for GPI anchor modification of human BST-2. Instead, by replacing the presumed GPI anchor motif of human BST-2 with TM domains of unrelated proteins, the authors demonstrate that a second TM domain instead of a C-terminal GPI anchor is adequate to produce functional BST-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrew AJ, Miyagi E, Kao S, Strebel K. The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology. 2009;6:80. doi: 10.1186/1742-4690-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrew AJ, Berndsen CE, Kao S, Strebel K. The Size and Conservation of a Coiled-coil Structure in the Ectodomain of Human BST-2/Tetherin Is Dispensable for Inhibition of HIV-1 Virion Release. J Biol Chem. 2012;287:44278–44288. doi: 10.1074/jbc.M112.418822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A. 2009;106:2868–2873. doi: 10.1073/pnas.0813223106. The study confirms that long-term or high-level exposure of cells to Vpu, e.g. in infected macrophages or in transfected HeLa cells, can lead to cell surface depletion of BST-2. However, this study also demonstrates for the first time that Vpu can enhance the release of viruses from BST-2 expressing cells during acute spreading infection without loss of cell surface BST-2 expression. Thus, cell surface depletion of BST-2 by Vpu is not a prerequisite but a downstream consequence of Vpu function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dube M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA. HIV-1 Vpu Antagonizes BST-2 by Interfering Mainly with the Trafficking of Newly Synthesized BST-2 to the Cell Surface. Traffic. 2011;12:1714–1729. doi: 10.1111/j.1600-0854.2011.01277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauter D, Unterweger D, Vogl M, Usmani SM, Heigele A, Kluge SF, Hermkes E, Moll M, Barker E, Peeters M, et al. Human Tetherin Exerts Strong Selection Pressure on the HIV-1 Group N Vpu Protein. PLoS Pathog. 2012:8. doi: 10.1371/journal.ppat.1003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shingai M, Yoshida T, Martin MA, Strebel K. Some Human Immunodeficiency Virus Type 1 Vpu Proteins Are Able To Antagonize Macaque BST-2 In Vitro and In Vivo: Vpu-Negative Simian-Human Immunodeficiency Viruses Are Attenuated In Vivo. J Virol. 2011;85:9708–9715. doi: 10.1128/JVI.00626-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bour S, Strebel K. The human immunodeficiency virus (HIV) type 2 envelope protein is a functional complement to HIV type 1 Vpu that enhances particle release of heterologous retroviruses. J Virol. 1996;70:8285–8300. doi: 10.1128/jvi.70.12.8285-8300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology. 2010;7:51. doi: 10.1186/1742-4690-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schubert U, Bour S, Willey RL, Strebel K. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J Virol. 1999;73:887–896. doi: 10.1128/jvi.73.2.887-896.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 2009;6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikovics K, Dazza M-C, Ekwalanga M, Mammano F, Clavel F, Saragosti S. Counteraction of tetherin antiviral activity by two closely related SIVs differing by the presence of a Vpu gene. PLoS One. 2012:7. doi: 10.1371/journal.pone.0035411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liberatore RA, Bieniasz PD. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:18097–18101. doi: 10.1073/pnas.1113694108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casartelli N, Sourisseau M, Feldmann J, Guivel-Benhassine F, Mallet A, Marcelin AG, Guatelli J, Schwartz O. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 2010;6:e1000955. doi: 10.1371/journal.ppat.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strebel K, Klimkait T, Martin MA. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science. 1988;241:1221–1223. doi: 10.1126/science.3261888. [DOI] [PubMed] [Google Scholar]
- 48.Terwilliger EF, Cohen EA, Lu YC, Sodroski JG, Haseltine WA. Functional role of human immunodeficiency virus type 1 vpu. Proc Natl Acad Sci U S A. 1989;86:5163–5167. doi: 10.1073/pnas.86.13.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen EA, Terwilliger EF, Sodroski JG, Haseltine WA. Identification of a protein encoded by the vpu gene of HIV-1. Nature. 1988;334:532–534. doi: 10.1038/334532a0. [DOI] [PubMed] [Google Scholar]
- 50.Theodore TS, Englund G, Buckler-White A, Buckler CE, Martin MA, Peden KW. Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res Hum Retroviruses. 1996;12:191–194. doi: 10.1089/aid.1996.12.191. [DOI] [PubMed] [Google Scholar]
- 51.Bour S, Akari H, Miyagi E, Strebel K. Naturally occurring amino acid substitutions in the HIV-2 ROD envelope glycoprotein regulate its ability to augment viral particle release. Virology. 2003;309:85–98. doi: 10.1016/s0042-6822(02)00128-9. [DOI] [PubMed] [Google Scholar]
- 52.Tristem M, Marshall C, Karpas A, Petrik J, Hill F. Origin of vpx in lentiviruses. Nature. 1990;347:341–342. doi: 10.1038/347341b0. [DOI] [PubMed] [Google Scholar]
- 53.Planelles V, Barker E. Roles of Vpr and Vpx in modulating the virus-host cell relationship. Mol Aspects Med. 2010;31:398–406. doi: 10.1016/j.mam.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujita M, Otsuka M, Nomaguchi M, Adachi A. Multifaceted activity of HIV Vpr/Vpx proteins: the current view of their virological functions. Rev Med Virol. 2010;20:68–76. doi: 10.1002/rmv.636. [DOI] [PubMed] [Google Scholar]
- 55.Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008;4:e1000059. doi: 10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goujon C, Darlix JL, Cimarelli A. The Vpx Protein of Hiv-2. Virologie. 2009;13:259–269. doi: 10.1684/13-5.2011.14690. [DOI] [PubMed] [Google Scholar]
- 57.Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, et al. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 2011;7:e1002425. doi: 10.1371/journal.ppat.1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goujon C, Arfi V, Pertel T, Luban J, Lienard J, Rigal D, Darlix JL, Cimarelli A. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol. 2008;82:12335–12345. doi: 10.1128/JVI.01181-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goujon C, Riviere L, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix J-L, Cimarelli A. SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology. 2007;4:2–2. doi: 10.1186/1742-4690-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharova N, Wu Y, Zhu X, Stranska R, Kaushik R, Sharkey M, Stevenson M. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008;4:e1000057. doi: 10.1371/journal.ppat.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gramberg T, Sunseri N, Landau NR. Evidence for an activation domain at the amino terminus of simian immunodeficiency virus Vpx. J Virol. 2010;84:1387–1396. doi: 10.1128/JVI.01437-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baldauf H-M, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med. 2012;18:1682–1687. doi: 10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology. 2012;9:87–87. doi: 10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahn J, Hao C, Yan J, Delucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J. HIV/Simian Immunodeficiency Virus (SIV) Accessory Virulence Factor Vpx Loads the Host Cell Restriction Factor SAMHD1 onto the E3 Ubiquitin Ligase Complex CRL4DCAF1. J Biol Chem. 2012;287:12550–12558. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Welbourn S, Miyagi E, White TE, Diaz-Griffero F, Strebel K. Identification and characterization of naturally occurring splice variants of SAMHD1. Retrovirology. 2012;9:86–86. doi: 10.1186/1742-4690-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White TE, Brandariz-Nunez A, Carlos Valle-Casuso J, Amie S, Nguyen L, Kim B, Brojatsch J, Diaz-Griffero F. Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology. 2013;436:81–90. doi: 10.1016/j.virol.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, Burckstummer T. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutieres syndrome-associated mutations. Hum Mutat. 2012;33:1116–1122. doi: 10.1002/humu.22087. [DOI] [PubMed] [Google Scholar]
- 68.Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, Yakunin AF. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J Biol Chem. 2013;288:8101–8110. doi: 10.1074/jbc.M112.431148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *69.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. This study shows that human SAMHD1 is a dGTP-stimulated triphosphohydrolase that converts deoxynucleoside triphosphates to the constituent deoxynucleoside and inorganic triphosphate. The crystal structure of the catalytic core of SAMHD1 reveals that the protein is dimeric and indicates a molecular basis for dGTP stimulation of catalytic activity against dNTPs. The authors propose that SAMHD1 may restrict HIV-1 replication by hydrolysing the majority of cellular dNTPs thus inhibiting reverse transcription and viral cDNA synthesis. [DOI] [PubMed] [Google Scholar]
- 70.Powell RD, Holland PJ, Hollis T, Perrino FW. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286:43596–43600. doi: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *71.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13:223–228. doi: 10.1038/ni.2236. Similar to Goldstone et al (Ref 69) this study reports that SAMHD1 restricts HIV infection by hydrolyzing intracellular dNTPs, lowering their concentrations to below those required for reverse transcription. SAMHD1-mediated restriction was alleviated by the addition of exogenous dNTPs. An HIV-1 RT mutant with low affinity for dNTPs was particularly sensitive to SAMHD1-mediated restriction. Vpx prevented the SAMHD1-mediated decrease in dNTP concentration by inducing the degradation of SAMHD1. The authors propose that nucleotide-pool depletion could be a general mechanism for protecting cells from infectious agents that replicate through a DNA intermediate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. Tight Interplay among SAMHD1 Protein Level, Cellular dNTP Levels, and HIV-1 Proviral DNA Synthesis Kinetics in Human Primary Monocyte-derived Macrophages. J Biol Chem. 2012;287:21570–21574. doi: 10.1074/jbc.C112.374843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.St Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, Wu L. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology. 2012;9:105–105. doi: 10.1186/1742-4690-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. The Retroviral Restriction Ability of SAMHD1, but Not Its Deoxynucleotide Triphosphohydrolase Activity, Is Regulated by Phosphorylation. Cell Host Microbe. 2013;13:441–451. doi: 10.1016/j.chom.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cribier A, Descours B, Valadao ALC, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by Cyclin A2/CDK1 Regulates Its Restriction Activity toward HIV-1. Cell Rep. 2013;3:1036–1043. doi: 10.1016/j.celrep.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 76.Endicott JA, Noble ME, Tucker JA. Cyclin-dependent kinases: inhibition and substrate recognition. Curr Opin Struct Biol. 1999;9:738–744. doi: 10.1016/s0959-440x(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 77.Nigg EA. Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol. 1993;3:296–301. doi: 10.1016/0962-8924(93)90011-o. [DOI] [PubMed] [Google Scholar]
- 78.Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res. 2011;9:496–504. doi: 10.2174/157016211798842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doria M. Role of the CD4 down-modulation activity of Nef in HIV-1 infectivity. Curr HIV Res. 2011;9:490–495. doi: 10.2174/157016211798842125. [DOI] [PubMed] [Google Scholar]
- 80.Abraham L, Fackler OT. HIV-1 Nef: a multifaceted modulator of T cell receptor signaling. Cell Commun Signal. 2012;10:39–39. doi: 10.1186/1478-811X-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Romani B, Cohen EA. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol. 2012;2:755–763. doi: 10.1016/j.coviro.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]