

Abstract
ADAR2 is a member of a family of RNA editing enzymes found in metazoa that bind double helical RNAs and deaminate select adenosines. We find that when human ADAR2 is overexpressed in the budding yeast Saccharomyces cerevisiae it substantially reduces the rate of cell growth. This effect is dependent on the deaminase activity of the enzyme, suggesting yeast transcripts are edited by ADAR2. Characterization of this novel set of RNA substrates provided a unique opportunity to gain insight into ADAR2’s site selectivity. We used RNA-Seq. to identify transcripts present in S. cerevisiae subject to ADAR2-catalyzed editing. From this analysis, we identified 17 adenosines present in yeast RNAs that satisfied our criteria for candidate editing sites. Substrates identified include both coding and noncoding RNAs. Subsequent Sanger sequencing of RT-PCR products from yeast total RNA confirmed efficient editing at a subset of the candidate sites including BDF2 mRNA, RL28 intron RNA, HAC1 3′UTR RNA, 25S rRNA, U1 snRNA and U2 snRNA. Two adenosines within the U1 snRNA sequence not identified as substrates during the original RNA-Seq. screen were shown to be deaminated by ADAR2 during the follow-up analysis. In addition, examination of the RNA sequence surrounding each edited adenosine in this novel group of ADAR2 sites revealed a previously unrecognized sequence preference. Remarkably, rapid deamination at one of these sites (BDF2 mRNA) does not require ADAR2’s dsRNA-binding domains (dsRBDs). Human glioma-associated oncogene 1 (GLI1) mRNA is a known ADAR2 substrate with similar flanking sequence and secondary structure to the yeast BDF2 site discovered here. As observed with the BDF2 site, rapid deamination at the GLI1 site does not require ADAR2’s dsRBDs.
RNA editing reactions modify, insert or delete nucleotides and can change the coding properties of an RNA molecule.(1, 2) Hydrolytic deamination of adenosine (A) in RNA generates inosine (I) at the corresponding nucleotide position. Since inosine is decoded as guanosine during translation, this modification can lead to codon changes (recoding) and the introduction of amino acids into a gene product not encoded in the gene.(3, 4) Several recoding sites are found in mRNAs for proteins important in the central nervous system (CNS) such as glutamate receptors(3) and serotonin receptors.(4) Recoding within these mRNAs contributes to the protein structural diversity required for proper CNS function and altered editing of these RNAs has been linked to CNS disorders.(5–10) Recent high-throughput sequencing efforts have identified many other editing sites in the human transcriptome, including a recoding site in the pre-mRNA for a DNA repair enzyme.(11, 12) Furthermore, mutations in a gene encoding an adenosine-to-inosine RNA editing enzyme have been linked to the genetic autoimmune disorder Aicardi Goutieres Syndrome and the inherited skin disease dyschromatosis symmetrica hereditaria.(13, 14)
Two different enzymes carry out A to I editing in humans, ADAR1 and ADAR2. ADAR1 is expressed in a long form (p150) that is interferon-induced and present in the nucleus and cytoplasm while a constitutively expressed short form (p110) is found exclusively in the nucleus.(15) ADAR2 is a smaller protein with a different N-terminal domain structure.(16) ADARs 1 and 2 are expressed in most tissues, whereas a related protein referred to as ADAR3 is expressed exclusively in the brain.(17) To date, no editing substrate has been identified for ADAR3. Although our understanding of the ADAR mechanism and regulation has advanced in recent years,(18–20) important questions remain about the basis for substrate recognition and the role of the different protein domains in directing the editing reaction. ADARs recognize their RNA substrates, at least in part, via double stranded RNA-binding domains (dsRBDs) (Figure 1). The dsRBD typically spans two minor grooves at a binding site made up of ~16 base pairs.(21–23) Protein contacts are primarily at 2′-hydroxyls and phosphodiesters, making binding largely insensitive to duplex sequence. ADAR1 has three dsRBDs in its N-terminal RNA-binding domain whereas ADAR2 has two dsRBDs (Figure 1). The presence of dsRBDs in ADARs explains the requirement for double-stranded secondary structure of a defined length in known RNA editing substrates. However, the observed selectivity for certain adenosines within a duplex substrate remains difficult to fully explain and may be influenced by local sequence preferences of the zinc-containing C-terminal deaminase domain (Figure 1).(24) In addition, while a high resolution structure for the human ADAR2 deaminase domain has been reported, relatively little is known about how this domain interacts with RNA or how these interactions influence editing site selectivity.(25)
Figure 1.

A Domain maps of human ADAR proteins, including hADAR2-D, a truncation mutant featuring only the deaminase domain. B Model of hADAR2 constructed from the crystal structure of hADAR2 deaminase domain(25) and NMR structures of its dsRBDs.(62)
Here we describe a novel set of ADAR substrates discovered in the budding yeast Saccharomyces cerevisiae. This discovery arose from our observation of an effect on the growth rate of yeast from overexpression of human ADAR2 (hADAR2). Our results suggested this effect was due to editing within yeast transcripts. Identifying edited regions in yeast transcripts could reveal novel editing sequence and/or structural preferences for ADAR2. Furthermore, once established, the approach could be applied to the study of other editing enzymes. In addition, we(26, 27) and others(28, 29) have used ADAR2-catalyzed editing of reporter constructs in yeast to carry out functional screens of ADAR mutants, so it was important to define the extent of editing within endogenous yeast transcripts resulting from expression of the human enzyme. Therefore, we used whole transcriptome sequencing (RNA-Seq.) to identify yeast transcripts exhibiting A to I changes catalyzed by hADAR2. Substrates identified include both coding and noncoding RNAs. Three S. cerevisiae RNAs in particular, the BDF2 mRNA as well as the U1 and U2 snRNAs, were efficiently modified by hADAR2. Analysis of the RNA sequence surrounding the yeast hADAR2 sites revealed a previously unrecognized sequence preference for this enzyme. Remarkably, rapid deamination (single turnover kobs > ~ 3 min −1) at the BDF2 mRNA site, whose flanking sequence perfectly matches the preferred consensus, does not require ADAR2’s dsRBDs, indicating the sequence preference is a property of the catalytic domain alone. Furthermore, we determined that the human ADAR substrate GLI1(30) mRNA is also rapidly edited by the hADAR2 catalytic domain in the absence of dsRBDs.
Materials and Methods
Yeast strains and plasmids
Saccharomyces cerevisiae strains INVSc1 (MATa/MATα, his3Δ1/his3Δ1, leu2/leu2, trp1–289/trp1–289, ura3–52/ura3–52), BCY123 (MATa, CAN1, ade2, trp1, ura3–52, his3, leu2–3, 112, pep4::HIS+, prb1::LEU+, bar1::HisG+, lys2::pGAL1/10-GAL4+), BY4743 (MATa/a, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0, bdf2::kanMX4/ bdf2::kanMX4), and BY4743 (MATa/MATα his3Δ0/his3Δ0, leu2Δ/leu2Δ0, met15Δ0/MET15, LYS2/lys2Δ 0, ura3Δ 0/ura3Δ 0) were used. The two BY4743 strains were acquired from EUROSCARF (acc. no. Y33767 and Y20000). pYES3/CT (Invitrogen) containing the hADAR2 substrate modified human glutamate receptor B editing substrate (GluR/B) upstream of the α-galactosidase coding sequencing and YEpTOP2PGAL1 plasmids encoding hADAR2 wild type, hADAR2 R455S T375S, hADAR2 R455S T375G, hADAR2 R455G T375S, hADAR2 E396A, and hADAR2 deaminase domain (aa306–701, hADAR2-D)(26) were prepared as described previously.(25) Full length BDF2 and HAC1 cDNA was generated using the Access RT-PCR Kit (Promega) using total RNA extracted from INVSc1 using RiboPure™-Yeast (Ambion). The following primers were used to amplify full length BDF2: 5′-AACCGGATCCGGCAATGTCTCGTACTAACATG-3′ and 5′-CCATTCTCGAGTGTTAATCACTGTCACTGTCG-3′. The following primers were used to amplify full length HAC1: 5′CAACAAGGATCCACATGGAAATGACTG-3′ and 5′-CTTGCGCTCGAGTTCATGAAGTGATGAAG-3′. BDF2 and HAC1 cDNA was inserted into linearized pYES3/CT and ligated using Quick Ligation™ Kit (New England Biolabs). Ligation product was used to transform XL 10 gold Escherichia coli (Agilent). Bacteria colonies were harvested and grown in Luria-Bertani (LB) ampicillin selective media overnight at 37 °C with shaking. Plasmid DNA was isolated using a QIAprep Spin Miniprep kit (Qiagen). cDNA insertion was confirmed by Sanger sequencing. The 192 bp truncation of BDF2 incorporating the BDF2 editing site was subsequently generated via PCR amplification with Phusion® High-Fidelity DNA Polymerase (New England Biolabs) using the full-length BDF2 plasmid as the template. The following primers were used to amplify the 192 bp truncation of BDF2: 5′-GCACATGGATCCGCAATGCCACCAAG-3′ and 5′-CACTGCCTCGAGTTTGGGTGGATG-3′. The 192 bp BDF2 DNA was subsequently inserted into pYES3/CT, amplified, and sequenced as described above. Yeast cells were transfected with plasmids using a lithium acetate protocol. pYES3/CT transformants were selected for on complete media (CM) –tryptophan +2% glucose plates, YEpTOP2PGAL1 transformants were selected for on CM –uracil +2% glucose plates, and double transformants were selected for on CM –uracil –tryptophan +2% glucose plates. A to G, A to C, and A to U changes at site 807 in BDF2 RNA were subsequently generated in pYES3/CT using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent) and confirmed via Sanger sequencing.
Deamination assays with hADAR2 mutants
For determining the deamination activity of various hADAR2 mutants compared in the growth assays, a 27 nt RNA duplex incorporating the GluR/G editing site radiolabeled with [γ−32P] at the edited adenosine was prepared as described previously.(31) The final product consisted of 5′-CAUUAAGGUGGGUGGAAUAGUAUAACA-3′ in duplex with 5′-UGUUAUAGUAUCCCACCUACCCUGAUG-3′ (Glu R/G site in bold). Single turnover deamination reactions were carried out with 266 nM hADAR2 and 27 nM RNA substrate in 15 mM Tris-HCl, pH 7.1, 3% glycerol, 0.5 mM DTT, 60 KCl, 1.5 mM EDTA, 0.003% NP-40, 160 units/mL RNasin, and 1.0 μg/mL yeast tRNAPhe. Reactions were incubated at 30 °C for varying times and were quenched by the addition of 0.5% SDS at 95 °C. The RNA substrates were subsequently digested with P1 nuclease. The resulting mononucleotide mixtures were resolved using thin-layer chromatography (TLC), visualized by exposure to storage phosphor imaging plates (Kodak), and analyzed by volume integration using the ImageQuant and KaleidaGraph software as described previously.(31)
RNA-Seq
INVSc1 hADAR2 wt + GluR/B and hADAR2 E396A + GluR/B were generated using a lithium acetate protocol. Yeast was initially grown from a single colony in CM –uracil –tryptophan + 2% glucose media at 30 °C with shaking for 24 hours. Following incubation, 1 mL of the initial cultures was pelleted, washed twice in ddH2O, and re-suspended in CM –uracil –tryptophan +3% galactose to induce expression. Galatose cultures were grown at 30 °C with shaking for 48 hours. In order to prevent a stress response in the yeast, cell count was periodically estimated by measuring the optical density at 600 nm (OD600) of the cultures in a Beckman DU7400 diode array spectrophotometer. If the OD600 rose above 1.5 the culture was diluted with additional media. Total RNA was extracted using RiboPure™ Yeast (Ambion).
Total RNA (5 μg) of was used to prepare the libraries. The Illumina mRNA sample preparation kit was used according to the manufacturer’s instructions. Following library quantification, the library was plated onto a single read flow cell for sequencing on an Illumina Genome Analyzer IIx. Sequencing was carried out for 40 cycles. Cluster formation was carried out with version 2 reagent kits and sequencing with version 3 sequencing kits from Illumina, all manipulations done according to manufacturer’s instructions. Image analysis and base calling was performed during the sequencing run using RTA 1.5. The subsequent alignments were carried out with version 1.5 of the Genome Analyzer Pipeline using the S. cerevisiae genome (version SGD/sacCer2, June 2008 Assembly) from the UCSC Genome Browser (http://genome.ucsc.edu) as a reference.
Edited sites were confirmed by amplifying ~300 base pair cDNA products from either total RNA of INVSc1 expressing hADAR2 wt and hADAR2 E396A (growth and induction as described above) using the Access RT-PCR Kit (Promega) or total RNA of INVSc1 incubated in the presence of 1.5 μM of hADAR2 and hADAR2 E396A for 4 hours under the conditions described in the deamination assay above (each reaction containing 1 μg RNA) and subjecting them to Sanger sequencing. Sequencing data was quantified using ImageJ. The following primers were used to amplify cDNA incorporating BDF2 position 807: 5′-GCAATGCCACCAAGAGTTTTACCCGC-3′ and 5′-GCAATCGGATCAACAGGTTGTAAAAAGGG-3′. The following primers were used to amplify cDNA incorporating U1 snRNA position 235: 5′-TCAGAGGAGATCAAGAAGTCC-3′ and 5′-GAAAGGCCCCAGCTCCCC-3′. The following primers were used to amplify cDNA incorporating U2 snRNA position 512: 5′-CCGAGCCGTTTATGTCCAACGCGGG-3′ and 5′-CCCCACCCTACACCCCCTCAAACC-3′. The following primers were used to amplify cDNA incorporating the RL28 predicted editing site: 5′-GGAAGAGGGAATGAAAGAACC-3′ and 5′-GACAAAGGCTATGCACACTTTCT-3′. The following primers were used to amplify cDNA incorporating 25S rRNA position A2104: 5′-GAGGGCCTTGGTCAGACGCAGC-3′ and 5′-GTCATAGTTACTCCCGCCGTTTACC-3′. The following primers were used to amplify cDNA incorporating HAC1 editing sites: 5′-CGTGGTTCTCTGATGGGGGAGGAGCC-3′, 5- CCTCCTCCCCCACCTACGACAACAACCG-3′ and 5′-GTTGAAAAGCTGCCCAACCTAAGG-3′.
Growth assays
Liquid media assays
The following INVSc1 strains were grown in permissive media at 30 °C with shaking: hADAR2 wt + BDF2, hADAR2 wt + GluR/B, hADAR2 T375S R455S + GluR/B, hADAR2 T375S R455G + GluR/B, hADAR2 T375G R455S + GluR/B, hADAR2 E396A + BDF2, hADAR2 E396A + GluR/B, hADAR2 wt + HAC1, hADAR2 E396A + HAC1, hADAR2 wt + pYES3/CT (empty), and hADAR2 E396A + pYES3/CT (empty). The following BDF2 knockout strains were grown in permissive media at 30 °C with shaking: BY4743 bdf2::MX4 + hADAR2 wt and BY4743 bdf2::MX4 + hADAR2 E396A. The following strains were grown in permissive media at 30 °C with shaking: BY4743 + hADAR2 wt and BY4743 + hADAR2 E396A. Cell density was estimated by measuring the OD600. All cultures were washed twice in ddH2O prior to taking their OD600.
Solid media assays
Cultures were pelleted, washed twice in ddH2O, and re-suspended in ddH2O to OD600 1.0 (~3 × 107 cells per mL). Serial dilutions of the yeast cultures were spotted (3 μL per spot) on CM –uracil –tryptophan + 3% galactose or CM –uracil + 3% galactose plates; duplicate CM –uracil –tryptophan + 2% glucose or CM –uracil + 2% glucose plates were prepared in each assay. After the spots had been absorbed into the agar media the plates were incubated at 30 °C. Images of the plates were taken with a Fujifilm FinePix Z20fd digital camera.
Immunoblotting and analysis
INVSc1 transfected with GluR/B and hADAR2 mutants or empty vector (YEpTOP2PGAL1) were incubated overnight in CM –uracil –tryptophan +2% glucose at 30 °C with shaking. Following incubation, 1 mL volumes of the cultures were washed twice in ddH2O, re-suspended in 27 mL of CM –uracil –tryptophan +3% glycerol/lactate, and grown for 16 hours at 30 °C with shaking. Expression was induced by adding 3% galactose to cultures. Six hours following the introduction of galactose, cell count was estimated by taking the OD600 of the cultures. The yeast cells were then pelleted and stored at −80 °C. Cell lysates were prepared as described previously.(32)
Crude extracts and purified hADAR2 wt protein were subjected to SDS-PAGE and immunoblot. Blots were initially probed using polyclonal rabbit antibodies specific to hADAR2 and goat anti-rabbit antibodies conjugated with alkaline phosphatase (Santa Cruz Biotechnology), visualized, and then re-probed using monoclonal mouse antibodies specific to yeast phosphoglycerate kinase (3-GPK) (Molecular Probes) and goat anti-mouse antibodies conjugated with alkaline phosphatase (GE Healthcare). Blots were incubated in ECL reagent and visualized on a Molecular Dynamics Typhoon 9400 or Typhoon FLA 9000. Bands were quantified and analyzed using ImageQuant software. Results were normalized to 3-GPK levels.
Overexpression and purification of hADAR2 and hADAR2-derived mutants
N-terminal histidine-tagged hADAR2 wt, hADAR2-D, and all mutant hADAR2 proteins were expressed in Saccharomyces cerevisiae strain BCY123 and purified as described previously(33) with the following modifications. Proteins were not eluted through a heparin ion exchange column following Ni-NTA agarose column purification. Protein concentrations were estimated using BSA standards visualized by SYPRO Orange staining of SDS-polyacrylamide gels. Purified proteins were stored in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 20% glycerol, 1 mM 2-mercaptoethanol at −80 °C.
Sequence alignment
Candidate transcript sequences were analyzed using the WebLogo program(34) (http://weblogo.berkeley.edu/). The sequence logo was generated from 7 nt upstream and downstream the 19 edited sites identified in S. cerevisiae.
Evaluation of second nearest neighbor preference in vivo
INVSc1 strains hADAR2 wt + BDF2, hADAR2 wt + bdf2 A807G, and hADAR2 E396A + BDF2 were initially grown from a single colonies in CM –uracil –tryptophan + 2% glucose media at 30 °C with shaking for 24 hours. Following incubation, 1 mL of the initial cultures were pelleted, washed twice in ddH2O, and re-suspended in CM –uracil –tryptophan +3% galactose to induce expression. Galactose cultures were grown at 30 °C with shaking for 48 hours. Total RNA was then extracted using RiboPure™-Yeast (Ambion). This was repeated three times (n = 3).
Edited sites were confirmed by amplifying ~300 base pair cDNA products from total RNA using the Access RT-PCR Kit (Promega). cDNA products were sequenced using Sanger sequencing and quantified using ImageJ.
In vitro transcription of RNA
A truncation of BDF2 incorporating 93 bps upstream and 98 bps downstream of the edited site was amplified from full length BDF2 plasmid and inserted into pYES3/CT (Bdf200). Bdf200 was subsequently subjected to site-directed mutagenesis to generate A to C, A to G, and A to U 3′ next-nearest neighbor mutants (bdf200 nnnAC, bdf200 nnnAG, and bdf200 nnnAU). Bdf200 plasmids were linearized using XhoI (New England Biolabs) and used as templates to generate 232 nt bdf2 RNAs via in vitro transcription. A ~350 RNA substrate derived from 5-HTSer2cR was prepared as described previously.(35) A truncation of GluR/B incorporating 70 nt upstream and 58 nt downstream of the edited adenosine was amplified from the Glu R B reporter plasmid described previously(26) and inserted into pYES/CT. The plasmid containing the truncated GluR/B sequence was subsequently subjected to site-directed mutagenesis to change the editing site’s 3′ next nearest neighbor from G to A and the neighbor’s base-pair partner from C to T (GluR/B G2292A, C2335T). Both GluR/B plasmids were linearized using XhoI (New England Biolabs) and used as templates to generate 192 nt GluR/B RNAs via in vitro transcription. A truncation of hGLI incorporating 81 bp upstream and 65 bp downstream of the editing site was amplified from genomic DNA derived from HeLa cells (ATCC) using the following primers: 5′-GTCGATTAATACGACTCACTATAGGGACAGAACTTTGATCCTTACCTCC -3′ and 5′-CATATAGGGGTTCAGACCACTG -3′. This truncation was subsequently used as a template to generate 150 nt hGLI1 RNA via in vitro transcription. The transcription reactions were run for 3–5 hours at 37 °C and contained 30 ng/μL template, 40 mM Tris-HCl pH 7.9 at room temperature, 10 mM NaCl, 6 mM MgCl2, 2 mM spermidine, 10 mM DTT, 1 units/μL RNasin® Plus RNase Inhibitor (Promega), 0.5 mM NTPs, and 0.4 units/μL T7 RNA polymerase (Promega). Transcribed RNAs were purified by denaturing polyacrylamide gel electrophoresis and visualized using UV shadowing. RNA bands were excised from the gel, crushed, and soaked overnight at 4 °C in 0.5 M NH4OAc, 0.1% SDS, and 0.1 mM EDTA. Polyacrylamide fragments were removed using a 0.2 μm Centrex filter (Whatman) followed by phenol-chloroform extraction and ethanol precipitation. The RNA solutions were lyophilized to dryness, re-suspended in nuclease-free water, quantified by absorbance at 260 nm and stored at −80 °C. The RNAs were later heated at 95 °C for 5 minutes and then slowly cooled to room temperature in 10 mM Tris-HCl, 0.1 mM EDTA pH 7.5, 100 mM NaCl to allow them to refold.
In vitro deamination kinetics
Deamination reactions were carried out under the conditions described above with the following modifications. The final volume of each reaction was 20 μL and concentrations of 10 nM RNA and 150 nM hADAR2 or hADAR2-D were used unless otherwise indicated. Reactions were quenched by adding 180 μL of 95 °C nuclease-free water followed by incubation at 95 °C for 1 minute. Deaminated RNA was purified by phenol-chloroform extraction and ethanol precipitation. The RNA solution was lyophilized to dryness and re-suspended in 200 μL nuclease free water. cDNA was generated from deaminated RNA via RT-PCR, Sanger sequenced and quantified using Chromas Lite (Technelysium) and ImageJ. The kobs (min−1) of each assay was calculated as described previously(36) using the ratio G/A+G as fraction product.
Results
ADAR2 deaminase activity is responsible for a slow growth effect in S. Cerevisiae
Spotting assays demonstrated that the S. cerevisiae strain INVSc1 exhibits a reduced growth rate when transfected with hADAR2 expression plasmid and grown in inducing media (i.e. in the presence of galactose) (Figure 2A). This effect is not observed for INVSc1 overexpressing the inactive hADAR2 mutant E396A or the empty parent vector (YEpTOP2PGAL1). Liquid culture growth assays demonstrated a negative correlation between the rate constant for deamination by various hADAR2 mutants in vitro (Figure 2B) and the growth rate of cells expressing those mutants (Figure 2C). This effect was not a result of differences in expression levels for these mutants as indicated by Western blotting analysis of whole cell lysates (Figure 2D). Thus, overexpression of human ADAR2 slows the growth of S. cerevisiae and this effect appears to be due to the deaminase activity of the enzyme.
Figure 2.

Yeast growth is slowed by expression of deaminase active human ADAR2. (A) Spotting analysis of INVSc1 transformed with hADAR2 wt or inactive hADAR2 mutant E396A and empty vector. (B) In vitro deamination rate constants of hADAR2 wt (kobs = 1 ± 0.2 min−1) and the mutants hADAR2 T375S R455S (SS, kobs = 0.062 ± 0.001 min−1), hADAR2 T375S R455G (SG, kobs = 0.035 ± 0.008 min−1), hADAR2 T375G R455S (GS, kobs = 0.016 ± 0.003 min−1) and hADAR2 E396A (kobs = 0 min−1).(63) Error represents standard error (n = 3) (C) Growth assay tracking cell count (estimated via OD600) over time. Yeast were transfected with hADAR2 variants and grown in galactose liquid media with shaking at 30 °C. Error represents standard error (n = 3). (D) Western blot analysis of whole cell lysates from cultures expressing hADAR2 mutants. Purified ADAR2 lanes contained 10 μL of 250 nM hADAR2 prepared as described previously(33) and negative control lanes (−con) contained whole cell lysate from INVSc1 cells transformed with the empty hADAR2 vector (YEpTOP2PGAL1) and GluR/B. Densitometric values were normalized to 3-GPK levels. Error represents standard error (n = 3).
ADAR2 targets select RNAs and sites in S. Cerevisiae
We hypothesized that this slow growth phenotype is a product of hADAR2 editing one or more RNA molecules in yeast. The effect was unlikely to be due to RNA binding by hADAR2 alone, since deaminase domain mutants with intact RNA-binding domains showed varying degrees of growth inhibition correlating with deaminase activity (Figure 2B, C). To identify transcripts in S. cerevisiae that are edited by hADAR2, transcriptomes of INVSc1 overexpressing wild type or inactive hADAR2 were sequenced using RNA-Seq. with an Illumina Genome Analyzer.(37) A site-by-site comparison was used to screen for A to G changes due to the presence of active hADAR2. Candidate sites were identified that had at least 50 reads of A in the control sample (deaminase inactive hADAR2 expressed) AND 50 reads of G in the wild type hADAR2 sample (Supplementary Table S1) where the % G was < 2% for the control and >10% for hADAR2 (i.e. at least a fivefold increase in G to A ratio). Of the 475,736 adenosine positions identified with at least 50 reads in the control sample, 10 sites satisfied the above mentioned criteria for a candidate editing site (0.002%). We also analyzed T to C changes for positions in the reference genome complementary to the RNA transcript using the same criteria for an additional seven sites out of the 477,215 T sites identified (0.001%) (Supplementary Table S2). Thus, the analysis led to the identification of 17 candidate sites for ADAR2-catalyzed adenosine deamination in S. cerevisiae transcripts (Table 1). These sites were found on eight transcripts with three in ORFs encoding proteins. Substituting a G for an A at all three of the sites in ORFs would result in an amino acid substitution in the protein encoded. Sanger sequencing was used to confirm A to G hits identified in the RNA-Seq. screen. Sequencing of ~300 bp RT-PCR products containing predicted edited sites amplified from yeast overexpressing active or inactive ADAR2 confirmed A to G changes in BDF2 mRNA, RL28 intronic RNA, U1 snRNA and U2 snRNA (Figure 3). Two additional editing sites were identified in the U1 snRNA during the Sanger sequencing follow-up analysis that were not found in the original RNA-Seq data set (Table 1). Interestingly, these adenosines are predicted to be in proximity to one of the RNA-Seq sites (A235, see below and Figure 4) in the U1 structure. Because Sanger sequencing may lack the sensitivity to detect some low-frequency single base substitutions(38, 39) we took total RNA derived from INVSc1, incubated it in the presence of 1.5 μM hADAR2 WT or hADAR E396A for 4 hours and sequenced their RT-PCR products. We were able to confirm two additional predicted editing sites within the HAC1 3′ UTR and 25S rRNA (Supplementary Figure 1).
Table 1.
ADAR2 sites in S. cerevisiae strain INVSc1 from RNA-seq analysis
| Position | Edited Region | G/G+A | Transcript |
|---|---|---|---|
| ChrII: 278,998 | 5′-TCTATACTTTAACGTCAAGGA-3′ | 15% | GAL1 (5′-UTR) |
| ChrII: 278,999 | 5′-CTATACTTTAACGTCAAGGAG-3′ | 11% | GAL1 (5′-UTR) |
| ChrII: 680,804 | 5′-GCATCAAGAAACGGACTTGAT-3′ | 49% | U2 snRNA (A1054) |
| ChrII: 680,826 | 5′-AGGGATTTGAAGAATGCCGGC-3′ | 78% | U2 snRNA (A1032) |
| ChrII: 680,853 | 5′-CCTATGTCATAGAGGCGTTTT-3′ | 83% | U2 snRNA (A1004) |
| ChrII: 681,346 | 5′-GGGCGCTCTTATTGTTGATTT-3′ | 89% | U2 snRNA (A512) |
| ChrIV: 331,829 | 5′-ACTTGTCATTAGACGTTCAGT-3′ | 63% | BDF2 (coding R269G) |
| ChrVI: 75,228 | 5′-GAACTTGGCTATCCCTACCAA-3′ | 16% | HAC1 (coding I18V) |
| ChrVI: 75,995 | 5′-GGTTATTGGAAGCTTTCTTTT-3′ | 19% | HAC1 (intron) |
| ChrVI: 76,061 | 5′-CTTATTACTAAGAAATGGACG-3′ | 13% | HAC1 (intron) |
| ChrVI: 76,276 | 5′-GTGCTTACTCAACCGAAGAAG-3′ | 14% | HAC1 (3′UTR) |
| ChrVI: 76,404 | 5′-GGCTGCAAGGAGCAAGACTGG-3′ | 23% | HAC1 (3′UTR) |
| ChrVII: 311,091 | 5′-TGTGCAACCAATATGTCGTGT-3′ | 45% | RL28 (intron) |
| ChrXII: 453,079 | 5′-CGATCAACTTAGAACTGGTAC-3′ | 15% | 25 S rRNA (A2104) |
| ChrXIII: 634,284 | 5′-TTATTATTCGAGACCGCTTTG-3′ | 10% | HSC82 (coding E644G) |
| ChrXIV: 230,439 | 5′-TTTTTGGGTTAAACTGATTTT-3′ | 59% | U1 snRNA (A235) |
| ChrXIV:230,607 | 5′-TTCCAATAGTAGAAGGACGTT-3′ | 35% | U1 snRNA (A67) |
| ChrXIV: 230,471* | 5′-ATCATTGGTTAATCCCTTGAT-3′ | 59% | U1 snRNA (A203) |
| ChrXIV:230,470* | 5′-TCATTGGTTAATCCCTTGATT-3′ | 20% | U1 snRNA (A204) |
Site was not predicted in the whole transcriptome sequencing screen; it was observed while confirming candidate sites. G/G+A for these sites are observed values.
Figure 3.
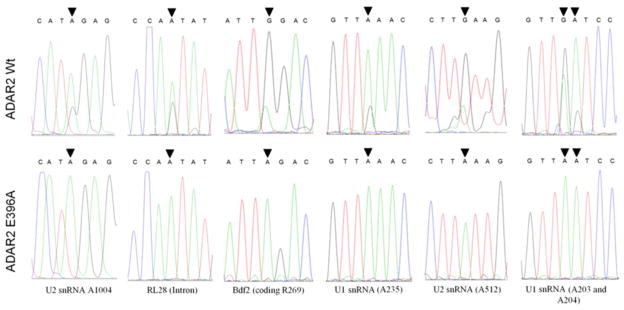
Confirmation of hADAR2 sites in yeast using Sanger sequencing. Sequencing traces for RT-PCR products amplified from total RNA extracted from INVSc1 transfected with expression plasmids for hADAR2 wt and hADAR E396A (inactive). Edited adenosines are indicated by a black arrow.
Figure 4.
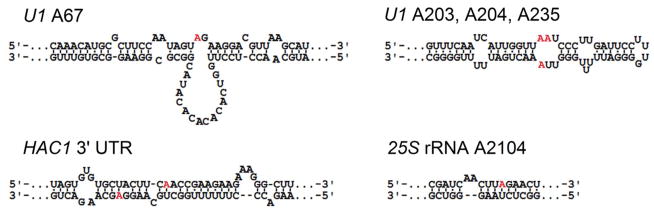
RNAs identified as hADAR2 editing substrates in INVSc1 with published secondary structures.(43–45) Edited adenosines are colored red.
ADAR2 binding and editing preferences are still not completely understood. The edited transcripts from yeast represent a novel set of ADAR2 substrates that could prove useful in further defining ADAR2 editing specificities. Factors that govern whether or not a given RNA molecule will be targeted by ADAR2 for editing include the transcript’s sequence and secondary structure.(40–42) For some of the ADAR2 sites discovered here, secondary structures have been reported for the RNAs near the target sites (U1 A67, U1 A235, HAC1 3′ UTR sites and 25S rRNA A2104) (Figure 4).(43–45) Regions containing the deamination sites for the remainder of targeted transcripts were analyzed using mFold (http://mfold.bioinfo.rpi.edu/) generating predicted secondary structures for these RNAs (Supplementary Figure 2). Generally, the targeted regions are predicted to be within or adjacent to base paired structure as expected for ADAR substrates. However, there is substantial variability in the predicted secondary structure context of the editing site adenosine itself.
ADAR2 substrates in yeast reveal next-nearest neighbor preferences
The sequences surrounding the 19 different sites identified were also analyzed for preferred nucleotides (Figure 5A).(34) A preference for an A or U 5′ to the edited site and G immediately 3′ to the edited nucleotide was observed in the alignment, consistent with reported nearest neighbor preferences for hADAR2.(25, 41) However, there appeared to be additional preferences for a U two nucleotides to the 5′ of the editing site (5′ next-nearest neighbor) and for an A two nucleotides 3′ of the edited site (3′ next-nearest neighbor) in these yeast substrates. Prior reports on ADAR sequence preferences had not identified these next-nearest neighbor preferences for ADAR2.(25, 41, 42) Interestingly, two unrelated transcripts (BDF2 mRNA and 25S rRNA) have editing sites that fall within the five nucleotide sequence 5′-UUAGA-3′ that matches the consensus preferred flanking sequence. Four more transcripts have editing sites in locations that differ from the 5′-UUAGA-3′ sequence by only one base (U2 A1032, HAC1 A885, U1 A235 and U1 A67) (Figure 5B). The only invariant nucleotide in this group besides the editing site is the A two nucleotides to its 3′ side. This analysis was carried out using sites identified using our criteria of at least a fivefold increase in G to A ratio at the site from the presence of WT hADAR2. When a lower stringency threshold of a fourfold ratio was used (25 sites), a similar flanking sequence preference was observed whereas a three-fold ratio threshold (36 sites) showed only the known 5′-(U/A)AG-3′ preference (Supplementary Figure 3).
Figure 5.
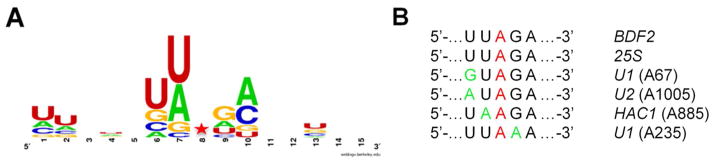
Analysis of yeast substrates reveals 3′ next nearest neighbor preference for adenosine. A WebLogo analysis of the hADAR2 edited transcripts in INVSc1 identified in the RNA-Seq. screen.(32) The sequence logo incorporates seven nucleotides 5′ and 3′ to the 19 edited sites from Table 1. B Alignment of six hADAR2 substrates found in S. cerevisiae closely related by sequence flanking the edited A. Red indicates editing site. Green indicates deviation from the sequence 5′-UUAGA-3′.
To test whether or not the presence of a 3′ next-nearest neighbor A could influence editing efficacy in an ADAR2 substrate, we mutated this A to G in the BDF2 sequence. The resulting plasmid (bdf2 A807G) was transformed into yeast also expressing wild type hADAR2. Total RNA was isolated from yeast expressing hADAR2 wt + BDF2, hADAR2 wt + bdf2 A807G and ADAR2 E396A + BDF2 following galactose induction for 48 hours. cDNA fragments for BDF2 containing the edited site were generated from the total RNA and subjected to Sanger sequencing (Figure 6A). Subsequent analysis revealed a reproducible approximately 20% decrease in editing efficiency in bdf2 A807G relative to BDF2 wt (Figure 6B). The editing efficiency at the A805 site appears lower here compared to that measured for the endogenous BDF2 message (Figure 3). However, this is likely a result of overexpression of the BDF2 transcript and the resulting effect on hADAR2 activity (see below). To further characterize the interaction between hADAR2 and the BDF2 RNA, we generated a DNA template that would allow for transcription of 200 nt surrounding the BDF2 A to I site (see Materials and Methods). In vitro RNA editing assays confirmed this RNA supports ADAR2-catalyzed deamination at the identified site (Figure 6, Supplementary Figure 4). Indeed, this is an excellent ADAR2 substrate given the observed rate of deamination under single turnover conditions (kobs = 0.77 ± 0.09 min−1).(31, 46) Furthermore, when the 3′ next-nearest neighbor A is mutated to G, C or U in this substrate, decreases in the hADAR2 deamination rate constant are observed, further highlighting the importance of adenosine at this location in the substrate RNA (Figure 6D). Interestingly, when an A was introduced to replace a 3′ next-nearest neighbor G in the well-characterized ADAR2 substrate GluR/B R/G site RNA, no statistically significant enhancement in rate was observed (Supplementary Figure 5), indicating a 3′ next-nearest neighbor A contributes to the reaction in some RNAs but not others (see below and Discussion).
Figure 6.

Importance of 3′ next-nearest neighbor A for BDF2 site deamination. A Sequencing trace for RT-PCR products of ADAR2 edited regions in BDF2 and bdf2 A807G. B Degree of A to G editing of BDF2 and bdf2 A807G in the presence of active hADAR2 wt in vivo. Graphs represent total area under G peaks divided by the total area of all the peaks at the edited site multiplied by 100%. BDF2 wt = 43 ± 3, bdf2 A807G = 24 ± 1. Error represents standard error (n = 3, **P < 0.01). C Predicted secondary structure near the BDF2 editing site showing mutations made to test importance of 3′ next nearest neighbor A. Edited adenosine colored red. D Single turnover rate constants for deamination of BDF2-derived and next-nearest neighbor mutant RNAs by hADAR2 in vitro. Bdf200 wt kobs = 0.77 ± 0.09, bdf200 nnnAG kobs = 0.22 ± 0.08, bdf200 nnnAC kobs = 0.4 ± 0.1, bdf200 nnnAU kobs = 0.45 ± 0.06. Error represents standard error (n = 3, *P < 0.05, **P < 0.01, nnnAC P = 0.075).
The BDF2-derived RNA is an excellent substrate for the isolated hADAR2 deaminase domain
We noted above that the 5′ and 3′ nearest neighbor preferences for ADAR2 had been previously reported.(37) These were defined by analyzing in vitro deamination efficiencies at different adenosine positions in perfectly matched duplex RNAs.(24) That approach did not reveal the next-nearest neighbor preferences described here. It may be the case that next-nearest neighbor effects are less important in determining editing efficiency at different sites when the edited adenosine resides in a perfectly matched duplex and are only apparent for more highly structured RNAs. Interestingly, our secondary structure analysis suggests the edited adenosine is in different structures for the sequence-related substrates identified here. For instance, the 25S substrate has the editing site near the middle of a perfectly matched six base pair stem whereas other substrates have the editing site in loops (e.g. U1 A67, U2 A1005, BDF2). This variability in secondary structure context for the editing sites suggested that the sequence preference may not be a function of the RNA-binding domain. Rather, the preferred sequence may constitute a contact surface for the catalytic domain, perhaps at a step in the reaction after initial binding and RNA conformational changes.(20) To test this idea, we carried out deamination reactions with the 200 nt BDF2-derived RNA and either hADAR2 or hADAR2-D, the isolated catalytic domain of human ADAR2 lacking the N-terminal dsRBDs (Figure 1). Remarkably, the BDF2 RNA is deaminated more rapidly by hADAR2-D than by the full-length protein at 150 nM enzyme concentration (Figure 7, A and B). Indeed, the single turnover reaction of hADAR2-D with the 200 nt BDF2-derived substrate RNA is too fast to be measured under these conditions using standard pipetting techniques (kobs > ~ 3 min.−1) (Figure 7B, Table 2). Efficient reaction of hADAR2-D with this substrate occurs at lower concentrations as well with nearly 50% editing observed after 10 minutes at 10 nM enzyme concentration (Figure 7C). This is in stark contrast with other substrates studied in our lab, including the GluR/B Q/R site(47) and the 5-HT2cR D site (see Figure 8) where ADAR2’s RNA-binding domain is required for efficient reaction. We also performed assays using hADAR2-D with 200 nt BDF2-dervied RNA where the 3′ next-nearest neighbor of the edited adenosine had been mutated from A to G (see above) and there was a reduction in its deamination rate relative to the original transcript (Table 2) as previously observed with hADAR2 (Figure 6D).
Figure 7.

Efficient deamination of the BDF2 site does not require ADAR2’s dsRBDs. A Sequencing traces for RT-PCR products of edited regions in BDF2 for two minute reactions containing 150 nM of either full length human ADAR2 (hADAR2) or the isolated catalytic domain (hADAR2-D); edited adenosines indicated by a star. B Plot of product vs time for reaction of either hADAR2 (squares, dashed line) or hADAR2-D (circles, solid line) with BDF2 RNA (n = 3). C % editing of BDF2 RNA at 10 minutes at different enzyme concentrations. Error represents standard error (n = 3).
Table 2.
Single Turnover Rate Constants for Deamination of BDF2-derived RNAsa
| enzyme | substrate | kobs, min−1b |
|---|---|---|
| hADAR2 | Bdf200 | 0.8 ± 0.2 |
| Bdf200 A → G | 0.2 ± 0.1 | |
| hADAR2-D | Bdf200 | >3 |
| Bdf200 A → G | 0.9 ± 0.1 |
Reactions were carried out with 150 nM enzyme and 10 nM RNA at 30 °C.
Data were fitted to the equation y = m1*(1−e(−m2*m0)), where m1 is the fitted reaction end point, m2 is the kobs or fitted rate constant (min−1), and y is the fraction product at time m0.
Figure 8.
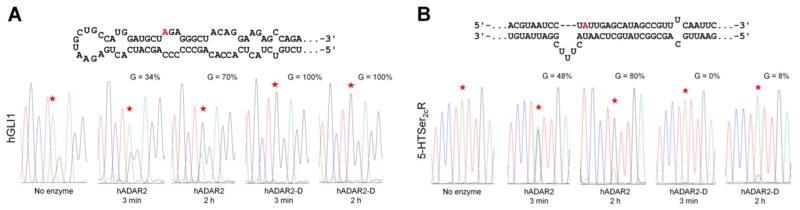
Human GLI1 (hGLI) RNA is efficiently edited by the hADAR2 catalytic domain (hADAR2-D). A Predicted secondary structure of the hGLI1 editing site(30) (edited adenosine colored red) and sequence traces of 150 nt hGLI1 derived RNA deamination products with hADAR2 and hADAR2-D (BOTTOM). B Predicted secondary structure of 5-HT2cR-derived RNA substrate (TOP) incorporating the D editing site (indicated by red color)(35) and sequence traces of 5-HT2cR RNA deamination products with hADAR2 and hADAR2-D (BOTTOM).
The human transcript of GLI1 is rapidly edited by hADAR2 catalytic domain
The high single turnover rate observed for BDF2 RNA with hADAR2-D indicated that the deaminase domain is capable of binding and editing the transcript independently of the dsRBDs. Indeed, the presence of the dsRBDs appeared to slow the deamination reaction (Figure 7B). We hypothesized that this may be because the region surrounding the BDF2 editing site is predisposed to bind and interact with the deaminase domain active site. This would suggest other hADAR2 substrates with a similar sequence and secondary structure may also be rapidly edited by hADAR2-D. The transcription factor glioma-associated oncogene 1 (GLI1) is an effector of sonic hedgehog signaling.(48, 49) It has been identified as a target of hADAR1 and hADAR2.(30) The region surrounding the edited adenosine in the hGLI transcript shares some similarities to BDF2 RNA; it is flanked by U and GA on its 5′ and 3′ sides, respectively, and is located opposite a C in a loop (Figure 8A). To determine whether hADAR2-D can also rapidly edit hGLI1 RNA we carried out deamination reactions with a 150 nt hGLI1-derived RNA substrate using both hADAR2 and hADAR2-D. We also performed deamination assays with 5-HT2cR RNA for comparison (Figure 8B). Like BDF2, hGLI1 RNA is rapidly deaminated by hADAR2-D. Quantitative editing of hGLI1 by hADAR2-D is observed after 3 minutes (Figure 8A) in contrast to the 5-HT2cR D-site; where only an 8% A to G change is observed after a 2 hour incubation with hADAR2-D (Figure 8B). Full-length hADAR2 also edits hGLI1 in vitro, though not as effectively as hADAR2-D (Figure 8A).
Overexpression of Bdf2p rescues growth in yeast expressing active ADAR2
Our RNA Seq. data indicated that both HAC1 and BDF2 mRNAs were modified by hADAR2; HAC1 was predicted to be edited at low % yields at multiple sites and BDF2 at a single site with > 60% yield, leading to a codon change. Hac1p is a well-characterized transcription factor involved in the unfolded protein response in yeast(50, 51) Bdf2p is a bromo domain protein also involved in transcriptional regulation.(52) To determine if either of these transcripts alter the hADAR2 effect on yeast growth, their sequences were inserted into the pYES3/CT expression vector downstream of the GAL1 promoter and co-transfected into yeast with an hADAR2 expression plasmid. Yeast strains were grown in liquid media and spotted on galactose-containing agar media to induce expression.
Yeast overexpressing the BDF2 transcript (but not HAC1) and wild type ADAR2 grew faster than when wild type ADAR2 is expressed alone (Supplementary Figure 6) and at a rate comparable to BDF2 + inactive ADAR2 E396A. Western blot analysis of the yeast lysates demonstrated the level of ADAR2 expression is not dependent on BDF2 expression (Supplementary Figure 6). This shows that overexpression of the BDF2 transcript can largely restore normal growth in yeast expressing active hADAR2. However, overexpression of wild type ADAR2 in a bdf2 knockout strain of S. cerevisiae also slowed yeast cell growth (data not shown) indicating that editing within the BDF2 mRNA is not solely responsible for the slow growth effect induced by ADAR2.
Discussion
The transcriptome sequencing approach described here successfully identified sites in S. cerevisiae transcripts that exhibited A to I changes in the presence of human ADAR2, providing us a novel set of editing substrates to analyze. A wide variety of sequences and secondary structures are present in yeast RNAs whereas background editing is nonexistent as ADARs are not naturally expressed in this organism. Whole transcriptome sequencing identified a relatively small number of sites/transcripts edited efficiently (Table 1). Thus hADAR2 appears to act selectively in S. cerevisiae and analysis of the preferred substrates revealed new information about ADAR2 selectivity (see below). One can envision using a similar approach to evaluate substrate specificity for other RNA editing enzymes as well (e.g. ADAR1, ADAR3, APOBECs, etc.).
We confirmed editing via Sanger sequencing at nine ADAR2 deamination sites in yeast RNAs (Figure 3, Supplementary Figure 1). Sites with low editing efficiency (<20%) in the RNA-Seq. data set may not be detectable using conventional Sanger sequencing.(38, 39) Yet three sites with high predicted editing efficiency for the RNA-Seq. data (U2 A1032, A1054 and U1 A67) did not sequence as G in Sanger sequencing. While it is possible these sites are false positives, the fact that they are within established hADAR2 substrate RNAs in yeast (i.e. editing has been confirmed at other adenosines in these RNAs) suggests they may indeed be ADAR2 deamination sites and other factors (e.g. subtle variation in yeast growth conditions, etc.) may be responsible for Sanger sequencing failing to confirm them.
ADARs will only bind and edit RNAs containing double helices of sufficient length.(22, 40, 53) This requirement is believed to be due to the presence of double stranded RNA binding domains (dsRBDs) in the ADAR structure.(22, 54) Therefore, the edited transcripts in yeast would be expected to fold into duplex structure near the targeted sites. Indeed, inspection of published secondary structures or structures predicted using mFold showed all the sites were either within or immediately adjacent to duplex structure (Figure 4, Supplementary Figure 2). The U2 snRNA is an interesting example. S. cerevisiae U2 is much larger than U2 snRNAs found in higher eukaryotes.(55) hADAR2 deaminates U2 at four different sites in the segment not found in these organisms. Secondary structure prediction suggests this region harbors an extremely long (> 200 bp) double helix with editing site A512 near the center (Supplementary Figure 2). Indeed, the high efficiency of the hADAR2 reaction at U2 A512 is likely related to its presence in this long double helical structure. This is reminiscent of the role of duplex structure present in RNAs containing Alu repeats in recruiting ADARs for efficient deamination recently described by Ohman.(56) Interestingly, deletion experiments carried out by Ares indicated this region is dispensable for S. cerevisiae U2 function in splicing.(57, 58)
The sequence analysis of the 19 ADAR2 sites identified in yeast shows a preference for a U or A 5′ to the edited site and G immediately 3′ to the edited nucleotide (Figure 5A). The U > A > C > G 5′ nearest neighbor and G > C > U = A 3′ nearest neighbor preferences have been reported for human ADAR2 and were determined by analyzing in vitro deamination efficiencies at different adenosine positions in perfectly matched duplex RNAs.(24, 41) Our results, obtained with a markedly different technique, are largely in agreement with these earlier studies. However, our analysis suggested an additional preference for the next-nearest neighbor nucleotide (two nucleotides from the edited site) not identified previously (Figure 5A). Mutation of the 3′ next nearest neighbor A to a G in the BDF2 mRNA substrate reduced editing efficiency in vivo and mutation to G, C or U reduced the single turnover deamination rate constant in vitro, supporting the notion that A is preferred at this position for this substrate (Figure 6B, D). Of note, the 5′-UUAGA-3′ sequence is found in at least four known ADAR editing sites in human transcripts, including in the 3′ UTR for the CaM kinase 1 mRNA.(11) However, the nature of the approach used for identification of those sites did not allow the identity of the responsible ADAR to be determined.(11)
Surprisingly, rapid deamination of the BDF2 or hGLI1 RNA substrate does not require hADAR2’s dsRBDs (Figure 7, Figure 8). This is unusual among the well-studied ADAR2 substrates where removal of the RNA-binding domain or disruption of dsRBD-RNA interactions typically reduces deamination rates.(24, 46) Editing sites have been described where the extent of editing observed appears to be enhanced by the deletion of dsRBDs from the ADAR protein.(24, 59) However, no substrate RNA had been previously reported where the rate of deamination by the isolated catalytic domain is faster than that of the full length ADAR under otherwise identical reaction conditions. These observations suggest the existence of two different types of ADAR substrate RNAs: those that require dsRBD interaction with duplex structure within the substrate RNA for efficient reaction and those that do not. The GluR/B Q/R editing site is likely one of the former since its local structural context (“CAG” triplet with A in an A:U pair) is suboptimal, yet it is an excellent substrate for hADAR2 in vivo. In addition, we have shown that dsRBD binding to a model GluR/B Q/R site substrate is necessary for efficient editing.(46) The 5-HT2cR D site also requires ADAR2’s dsRBD’s for efficient reaction (Figure 8B).(24) What, then, are the fundamental differences between the two types of ADAR substrates? Sites that react poorly with the isolated catalytic domain may have the editing site adenosine located in a suboptimal local structural environment for the reaction (Figure 9A). Efficient reaction at these sites may require increased residence time and positioning on the RNA that comes from dsRBD binding, they may require RNA distortion promoted by dsRBD binding or they could require dsRBD-induced changes in the editing site adenosine pKa. We know from earlier work that ADAR2’s RNA-binding domain alone can induce conformational changes in a substrate RNA,(60) which may facilitate reaction at certain sites. On the other hand, substrates that do not require dsRBD binding may be pre-organized in an RNA conformation found on the reaction pathway, perhaps one with an unstacked adenosine and flanking nucleotides optimal for catalytic domain interactions (Figure 9B). These sites may also have optimal flanking sequence for tuning the pKa of the reactive adenosine.(61) One might expect that ADAR-induced conformational changes in the RNA would be minimal for substrates of this type. Such a hypothesis could be tested using 2-aminopurine (2-AP) modified RNAs and measuring ADAR-induced changes in 2-AP fluorescence. We are currently investigating the extent to which the BDF2 RNA can be reduced in size while maintaining efficient deamination by hADAR2-D such that synthetic variants, including those containing 2-AP, could be used to study catalytic domain-RNA interactions.
Figure 9.
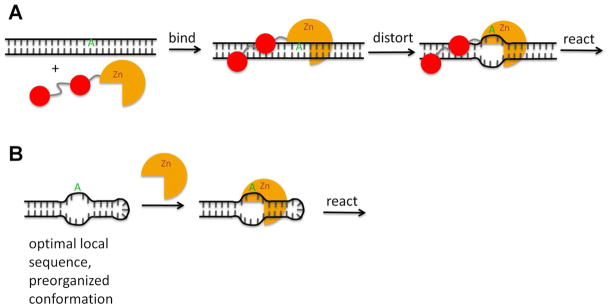
Two types of ADAR2 substrates with differing requirements for dsRBD binding. A ADAR2 binds to dsRNA via its dsRBDs. Local dsRNA structure is distorted facilitating base-flipping and deamination of the edited adenosine by the catalytic domain. B RNA structure around the editing site resembles the intermediate complex formed when ADAR2 distorts local dsRNA and the sequences flanking the edited adenosine are optimal for deaminase domain binding. Such sites do not require dsRBD binding.
Experiments involving overexpression of target transcripts along with ADAR2 demonstrated that BDF2 transcript overexpression is capable of suppressing ADAR2’s effect on yeast growth. Since the growth effect is still observed in a BDF2 knockout strain, editing of other transcripts must also contribute to the effect. However, suppression of the effect indicates BDF2 overexpression inhibits ADAR2 activity, likely by providing an RNA ligand that sequesters the protein and reducing editing efficiency at other sites. These results suggest one could carry out selections in yeast for molecules capable of suppressing the ADAR2-induced slow growth effect to discover inhibitors of human ADAR2.
Supplementary Material
Acknowledgments
We thank Charles Nicolet of the Sequencing Core and Dawei Lin and Nikhil Joshi in the Bioinformatics Core at the UC Davis Genome Center for their assistance in acquiring and analyzing transcriptome datasets and Daniel Melters for his advice and assistance with the bioinformatics analysis.
This study was funded by a grant from the National Institutes of Health (R01-GM061115).
Abbreviations
- A
adenosine
- 2-AP
2-aminopurine
- Bdf
bromodomain factor
- BSA
bovine serum albumin
- CNS
central nervous system
- CM
complete media
- C
cytidine
- dsRBD
double stranded RNA binding domain
- EDTA
ethylenediaminetetraacetic acid
- G
guanosine
- Glu R B
glutamate receptor B subunit
- hADAR2
human adenosine deaminase acting on RNA 2
- hADAR2-D
human adenosine deaminase acting on RNA 2 deaminase domain
- hGLI1
human glioma-associated oncogene 1
- I
inosine
- LB
Luria-Bertani
- NMR
nuclear magnetic resonance
- OD
optical density
- ORF
open reading frame
- 3-PGK
phosphoglycerate kinase
- RT-PCR
reverse transcription polymerase chain reaction
- TLC
thin-layer chromatography
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- 5-HT2cR
serotonin receptor 2c subtype
- U
uridine
Footnotes
Notes
The authors declare no competing financial interest.
Tables of predicted editing sites in yeast transcriptome read counts for A to G (Table S1) and C to T (Table S2) changes, sequence traces of predicted hADAR2 editing sites in yeast confirmed via Sanger sequencing (Figure S1), predicted secondary structures of S. cerevisiae RNA targeted for editing by hADAR2 (Figure S2), extended WebLogo analysis of predicted ADAR2 editing site regions in S. cerevisiae (Figure S3), fitted time courses of BDF2 RNA and 3′ next nearest neighbor variants (Bdf200) deamination reactions by hADAR2 (Figure S4), in vitro hADAR2 deamination rates of GluR/B and GluR/B G2292A, C2335U (Figure S5), yeast spotting assays with supporting western blot data demonstrating rescue of cell growth in yeast expressing hADAR2 by BDF2 overexpression (Figure S6). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Schaub M, Keller W. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie. 2002;84:791–803. doi: 10.1016/s0300-9084(02)01446-3. [DOI] [PubMed] [Google Scholar]
- 2.Grosjean H. Fine-tuning of RNA functions by modification and editing. In: Grosjean H, editor. Topics in Current Genetics. Springer Berlin Heidelberg; New York: 2005. pp. 1–22. [Google Scholar]
- 3.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 4.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 5.Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology. 2001;24:478–491. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34:349–356. doi: 10.1016/s0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 7.Iwamoto K, Kato T. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci Lett. 2003;346:169–172. doi: 10.1016/s0304-3940(03)00608-6. [DOI] [PubMed] [Google Scholar]
- 8.Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427:801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- 9.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 10.Doe CM, Relkovic D, Garfield AS, Dalley JW, Theobald DE, Humby T, Wilkinson LS, Isles AR. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum Mol Genet. 2009;18:2140–2148. doi: 10.1093/hmg/ddp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM. Genome-Wide Identification of Human RNA Editing Sites by Parallel DNA Capturing and Sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 12.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xing Q, Wang M, Chen X, Qian X, Qin W, Gao J, Wu S, Gao R, Feng G, He L. Identification of a novel ADAR mutation in a Chinese family with dyschromatosis symmetrica hereditaria (DSH) Arch Dermatol Res. 2005;297:139–142. doi: 10.1007/s00403-005-0589-1. [DOI] [PubMed] [Google Scholar]
- 15.Patterson JP, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 17.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem. 1996;271:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 18.Hoopengardner B. Adenosine-to-inosine RNA editing: perspectives and predictions. Mini-Rev Med Chem. 2006;6:1213–1216. doi: 10.2174/138955706778742812. [DOI] [PubMed] [Google Scholar]
- 19.Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O’Connell MA. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011;30:4211–4222. doi: 10.1038/emboj.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman RA, Macbeth MR, Beal PA. ADAR Proteins: Structure and Catalytic Mechanism. Curr Top Microbiol Immunol. 2012;353:1–33. doi: 10.1007/82_2011_144. [DOI] [PubMed] [Google Scholar]
- 21.Doyle M, Jantsch MF. New and old roles of the double-stranded RNA-binding domain. J Struct Biol. 2002;140:147–153. doi: 10.1016/s1047-8477(02)00544-0. [DOI] [PubMed] [Google Scholar]
- 22.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bass BL, Hurst SR, Singer JD. Binding properties of newly identified Xenopus proteins containing dsRNA-binding motifs. Curr Biol. 1994;4:301–314. doi: 10.1016/s0960-9822(00)00069-5. [DOI] [PubMed] [Google Scholar]
- 24.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macbeth MR, Schubert HL, VanDemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pokharel S, Beal PA. High-throughput screening for functional adenosine to inosine RNA editing systems. ACS Chem Biol. 2006;1:761–765. doi: 10.1021/cb6003838. [DOI] [PubMed] [Google Scholar]
- 27.Eifler T, Chan D, Beal PA. A Screening Protocol for Identification of Functional Mutants of RNA Editing Adenosine Deaminases. Curr Protoc Chem Biol. 2012;4:357–369. doi: 10.1002/9780470559277.ch120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuttan A, Bass BL. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci U S A. 2012;109:E3295–E3304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garncarz W, Tariq A, Handl C, Pusch O, Jantsch MF. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol. 2013;10:192–204. doi: 10.4161/rna.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimokawa T, Rahman MF, Tostar U, Sonkoly E, Stahle M, Pivarcsi A, Palaniswamy R, Zaphiropoulos PG. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013;10:321–333. doi: 10.4161/rna.23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephens OM, Yi-Brunozzi HY, Beal PA. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000;39:12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 32.Printen JA, Sprague GF. Protein-Protein Interactions in the Yeast Pheromone Response Pathway - Ste5p Interacts with All Members of the Map Kinase Cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macbeth MR, Bass BL. Large-scale overexpression and purification of ADARs from Saccharomyces cerevisiae for biophysical and biochemical studies. Methods Enzymol. 2007;424:319–331. doi: 10.1016/S0076-6879(07)24015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schirle NT, Goodman RA, Krishnamurthy M, Beal PA. Selective inhibition of ADAR2-catalyzed editing of the serotonin 2c receptor pre-mRNA by a helix-threading peptide. Org Biomol Chem. 2010;8:4898–4904. doi: 10.1039/c0ob00309c. [DOI] [PubMed] [Google Scholar]
- 36.Maydanovych O, Easterwood LM, Cui T, Veliz EA, Pokharel S, Beal PA. Probing adenosine-to-inosine editing reactions using RNA-containing nucleoside analogs. Methods Enzymol. 2007;424:369–386. doi: 10.1016/S0076-6879(07)24017-0. [DOI] [PubMed] [Google Scholar]
- 37.Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y, Garraway LA, LaFramboise T, Lee JC, Shah K, O’Neill K, Sasaki H, Lindeman N, Wong KK, Borras AM, Gutmann EJ, Dragnev KH, DeBiasi R, Chen TH, Glatt KA, Greulich H, Desany B, Lubeski CK, Brockman W, Alvarez P, Hutchison SK, Leamon JH, Ronan MT, Turenchalk GS, Egholm M, Sellers WR, Rothberg JM, Meyerson M. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med. 2006;12:852–855. doi: 10.1038/nm1437. [DOI] [PubMed] [Google Scholar]
- 39.Jancik S, Drabek J, Berkovcova J, Xu YZ, Stankova M, Klein J, Kolek V, Skarda J, Tichy T, Grygarkova I, Radzioch D, Hajduch M. A comparison of Direct sequencing, Pyrosequencing, High resolution melting analysis, TheraScreen DxS, and the K-ras StripAssay for detecting KRAS mutations in non small cell lung carcinomas. J Exp Clin Cancer Res. 2012;31:79. doi: 10.1186/1756-9966-31-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann KA, Bass BL. The importance of internal loops within RNA substrates of ADAR1. J Mol Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 42.Dawson TR, Sansam CL, Emeson RB. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J Biol Chem. 2004;279:4941–4951. doi: 10.1074/jbc.M310068200. [DOI] [PubMed] [Google Scholar]
- 43.Kretzner L, Krol A, Rosbash M. Saccharomyces-Cerevisiae U1 Small Nuclear-RNA Secondary Structure Contains Both Universal and Yeast-Specific Domains. Proc Natl Acad Sci U S A. 1990;87:851–855. doi: 10.1073/pnas.87.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeeninga RE, Van Delft Y, de Graaff-Vincent M, Dirks-Mulder A, Venema J, Raue HA. Variable regions V13 and V3 of Saccharomyces cerevisiae contain structural features essential for normal biogenesis and stability of 5.8S and 25S rRNA. RNA. 1997;3:476–488. [PMC free article] [PubMed] [Google Scholar]
- 45.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephens OM, Haudenschild BL, Beal PA. The binding selectivity of ADAR2’s dsRBMs contributes to RNA-editing selectivity. Chem Biol. 2004;11:1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Dabiri GA, Lai F, Drakas RA, Nishikura K. Editing of the GLuR-B ion channel RNA in vitro by recombinant double-stranded RNA adenosine deaminase. EMBO J. 1996;15:34–45. [PMC free article] [PubMed] [Google Scholar]
- 48.Choy SW, Cheng SH. Hedgehog signaling. Vitam Horm. 2012;88:1–23. doi: 10.1016/B978-0-12-394622-5.00001-8. [DOI] [PubMed] [Google Scholar]
- 49.Nilsson M, Unden AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, Toftgard R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nojima H, Leem SH, Araki H, Sakai A, Nakashima N, Kanaoka Y, Ono Y. Hac1: a novel yeast bZIP protein binding to the CRE motif is a multicopy suppressor for cdc10 mutant of Schizosaccharomyces pombe. Nucleic Acids Res. 1994;22:5279–5288. doi: 10.1093/nar/22.24.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shamu CE. Splicing: HACking into the unfolded-protein response. Curr Biol. 1998;8:R121–R123. doi: 10.1016/s0960-9822(98)70986-8. [DOI] [PubMed] [Google Scholar]
- 52.Sawa C, Nedea E, Krogan N, Wada T, Handa H, Greenblatt J, Buratowski S. Bromodomain factor 1 (Bdf1) is phosphorylated by protein kinase CK2. Mol Cell Biol. 2004;24:4734–4742. doi: 10.1128/MCB.24.11.4734-4742.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 54.Chang KY, Ramos A. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J. 2005;272:2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x. [DOI] [PubMed] [Google Scholar]
- 55.Shuster EO, Guthrie C. Human U2 snRNA can function in pre-mRNA splicing in yeast. Nature. 1990;345:270–273. doi: 10.1038/345270a0. [DOI] [PubMed] [Google Scholar]
- 56.Daniel C, Veno MT, Ekdahl Y, Kjems J, Ohman M. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res. 2012;40:9876–9886. doi: 10.1093/nar/gks691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ares M, Jr, Igel AH. Mutations define essential and nonessential U2 RNA structures. Mol Biol Rep. 1990;14:131–132. doi: 10.1007/BF00360444. [DOI] [PubMed] [Google Scholar]
- 58.Miraglia L, Seiwert S, Igel AH, Ares M., Jr Limited functional equivalence of phylogenetic variation in small nuclear RNA: yeast U2 RNA with altered branchpoint complementarity inhibits splicing and produces a dominant lethal phenotype. Proc Natl Acad Sci U S A. 1991;88:7061–7065. doi: 10.1073/pnas.88.16.7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Macbeth MR, Lingam AT, Bass BL. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10:1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yi-Brunozzi HY, Stephens OM, Beal PA. Conformational changes that occur during an RNA-editing adenosine deamination reaction. J Biol Chem. 2001;276:37827–37833. doi: 10.1074/jbc.M106299200. [DOI] [PubMed] [Google Scholar]
- 61.Wilcox JL, Bevilacqua PC. A simple fluorescence method for pK(a) determination in RNA and DNA reveals highly shifted pK(a)’s. J Am Chem Soc. 2013;135:7390–7393. doi: 10.1021/ja3125299. [DOI] [PubMed] [Google Scholar]
- 62.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 63.Pokharel S, Jayalath P, Maydanovych O, Goodman RA, Wang SC, Tantillo DJ, Beal PA. Matching active site and substrate structures for an RNA editing reaction. J Am Chem Soc. 2009;131:11882–11891. doi: 10.1021/ja9034076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.