

Abstract
Cardiovascular diseases are a leading cause of mortality and morbidity worldwide, with hypertension being a major risk factor. Numerous studies support the contribution of reactive oxygen and nitrogen species in the pathogenesis of hypertension, as well as other pathologies associated with ischemia/reperfusion. However, the validation of oxidative stress-related biomarkers in these settings is still lacking and novel association of these biomarkers and other biomarkers such as endothelial progenitor cells, endothelial microparticles, and ischemia modified albumin, is just emerging. Oxidative stress has been suggested as a pathogenic factor and therapeutic target in early stages of essential hypertension. Systolic and diastolic blood pressure correlated positively with plasma F2-isoprostane levels and negatively with total antioxidant capacity of plasma in hypertensive and normotensive patients. Cardiac surgery with extracorporeal circulation causes an ischemia/reperfusion event associated with increased lipid peroxidation and protein carbonylation, two biomarkers associated with oxidative damage of cardiac tissue. An enhancement of the antioxidant defense system should contribute to ameliorating functional and structural abnormalities derived from this metabolic impairment. However, data have to be validated with the analysis of the appropriate oxidative stress and/or nitrosative stress biomarkers.
1. Introduction
Cardiovascular diseases are a leading cause of mortality and morbidity worldwide [1], with hypertension being a major risk factor [2]. These diseases affect more than 600 million people and it is estimated that there will be 29% of the world adult population suffering hypertension by 2025 [3]. The pathophysiology of cardiovascular diseases is complex due to the multiple biological pathways that have been implicated. Accordingly, the absolute risk of future cardiovascular events cannot be estimated from a single biomarker. These diseases are originated in the vascular endothelium, a target for the classical cardiovascular risk factors. Following endothelial activation oxidative stress has an important role in the development of atherosclerosis [4] and hypertension [5, 6], thereby contributing to the progression of the structural and functional cardiovascular damage. In cardiovascular disease related to ischemia/reperfusion injury, redox imbalance triggers the activity of a number of signaling pathways mediated by reactive oxygen species (ROSs) and reactive nitrogen species (RNSs) [7]. Consequently, in cardiac surgery with extracorporeal circulation, electrical and structural myocardial remodeling due to the excessive production of these reactive species may lead to the development of arrhythmias such as atrial fibrillation [8]. Furthermore, percutaneous transluminal coronary angioplasty following acute myocardial infarction results in heart reperfusion damage, thus enhancing the infarct size [9]. These cardiac abnormalities are associated with major changes in oxidative stress-related biomarkers. However, to date there is no available data allowing to have an optimal biomarker approach with the ability to help identify patients at risk for cardiovascular disease, to monitor the efficacy of treatments, and to develop new pharmacological tools. Further studies are still lacking to achieve the validation of oxidative stress-related biomarkers to implement strategies suitable to improve the prevention and reduction of cardiovascular risk. The aim of this review was to present a novel focus on the bases supporting the applicability of reliable biomarkers accounting for the antioxidant potential, as well as oxidative stress and nitrosative stress associated with clinical settings of cardiovascular diseases. Particularly, we analyze the biomarkers related to ischemia/reperfusion and essential hypertension, two examples of processes mediated by ROS and RNS. This novel strategy, together with other biomarkers, could contribute to the development of changes in health diagnostics and clinical research.
2. Oxidative Stress
The oxidation and reduction reactions in biological systems (redox reactions) represent the basis for numerous biochemical mechanisms of metabolic changes [10]. Oxidative stress constitutes a unifying mechanism of injury of many types of disease processes, and it occurs when there is an imbalance between the generation of reactive oxygen and nitrogen species and the antioxidant defense systems in the body, so that the latter become overwhelmed [11]. ROS are widely recognized as important mediators of cell growth, adhesion, differentiation, senescence, and apoptosis [12]; however, when these species are found in excess they might cause cell damage either directly or working as intermediates in diverse signaling pathways. RNS may have also a similar behavior.
In pathophysiological conditions, the main sources of ROS include the mitochondrial respiratory electron transport chain, xanthine oxidase (XO) activation through ischemia-reperfusion, the respiratory burst associated with neutrophil activation, and arachidonic acid metabolism. In the cellular metabolism, the oxygen molecule itself is reduced to water after forming, as successive intermediates, superoxide anion (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical. It is estimated that between 0.15 and 2% of cellular O2 consumption results in superoxide formation [13].
Xanthine oxidoreductase functions as either xanthine dehydrogenase (XDH) form, which transfers electron to NAD+, and generates NADH or XO form, which transfers electron to O2 and generates oxidative stress. Due to ischemia-induced cellular calcium overload converts XDH to XO, under reperfusion phase, enhanced XO can produce more ROS, such as superoxide, H2O2, and hydroxyl radicals [14].
Oxidants generated by cellular NADPH oxidases participate in many biological processes, serving both as critical elements of signaling pathways and important effector molecules [15–20]. The enzyme NADPH oxidase catalyzes the one electron reduction of O2 to generate O2•−, using NADPH as the source of electrons. There appear to be at least three isoforms of NADPH oxidase expressed in the vascular wall. Furthermore, they are distributed in nearly all cell types and tissues, and many cells express more than one NADPH oxidase protein isoform [21].
Nitric oxide (NO) synthases (NOS) are a family of enzymes that convert the amino acid L-arginine to L-citrulline and NO. Three main isoenzymes exist in mammals that are regulated by distinct genes: a constitutive neuronal NOS (nNOS or NOS-1), an endotoxin- and cytokine-inducible NOS (iNOS or NOS-2), and a constitutive endothelial NOS (eNOS or NOS-3) [22]. NOS-1 and NOS-3 are constitutively expressed and Ca+2/calmodulin dependent, while the inducible form (NOS-2) is induced by inflammatory stimuli and is Ca+2/calmodulin independent [23]. eNOS plays a major role in the regulation of vascular function. For instance, eNOS synthesizes NO by a two-step oxidation of the amino acid L-arginine, thereby leading to activation of guanylyl cyclase. The resulting second messenger cGMP in turn activates the cGMP-dependent kinase, which leads to decrease in intracellular Ca+2 concentrations, thereby causing vasorelaxation. However, it has become clear from studies with the purified enzyme that eNOS may become uncoupled in the absence of the NOS substrate L-arginine or the cofactor tetrahydrobiopterin. Uncoupled eNOS state results in the production of O2•− instead of NO [24, 25].
Myeloperoxidase is a neutrophil and monocyte enzyme that amplifies the reactivity of H2O2 through generation of hypochlorous acid, free radicals, and RNS. The myeloperoxidase enzyme (MPO) produces hypochlorous acid from H2O2 and chloride anion during the neutrophil's respiratory burst. It requires heme as a cofactor. In addition, it oxidizes tyrosine to tyrosyl radical using H2O2 as oxidizing agent [26]. Both hypochlorous acid and tyrosyl radical are cytotoxic and used by the neutrophil to kill bacteria and other pathogens.
In physiological conditions, cells have an antioxidant defense system to prevent ROS and RNS damage. Antioxidant substances are small molecules that can scavenge free radicals by accepting or donating an electron to eliminate the unpaired condition. Typically, this means that the antioxidant molecule becomes a free radical in the process of scavenging a ROS to a more stable and less reactive molecule. In most cases the scavenger molecule provides hydrogen radical that combines with the free radical. Consequently, a new radical that has an enhanced lifetime compared with the starting one is generated, for instance, due to a conjugated system [27]. However there are many mechanisms whereby antioxidants may act such as: (1) scavenging reactive oxygen species or their precursors, (2) inhibiting the formation of ROS, (3) attenuating the catalysis of ROS generation via binding to metal ions, (4) enhancing endogenous antioxidant generation, and (5) reducing apoptotic cell death by upregulating the anti-death gene Bcl-2 [28].
Enzymatic antioxidant defenses include superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT), among others. Nonenzymatic antioxidants include a variety of mostly exogenous biological molecules, such as ascorbic acid (vitamin C), α-tocopherol (vitamin E), reduced glutathione (GSH), carotenoids, flavonoids, polyphenols, and other exogenous antioxidants [29].
The first line of cellular defense against oxidative injury includes the antioxidant enzymes CAT, SOD, and GSH-Px [30]. The SOD converts superoxide anion to H2O2, which is a substrate for CAT and GSH-Px. Catalase metabolizes H2O2 to water and oxygen and GSH-Px reduces both H2O2 and organic hydroperoxides when reacting with GSH [31]. Reduced glutathione is present at high concentrations in all mammalian cells, especially in the renal cells, hepatocytes, and erythrocytes [32]. GSH protects protein thiol groups from nonenzymatic oxidation or can act as a cosubstrate of GSH-Px [33].
Exogenous antioxidants, such as vitamins C and E, exist at a number of locations, namely, on the cell membrane, intracellularly, and extracellularly. They react with ROS to either remove or inhibit them. The hydrophobic lipid inside of membranes requires a different spectrum of antioxidants. Vitamin E is thought to be the most important antioxidant found within lipid membranes in the body [34].
Polyunsaturated fatty acids (PUFAs) are prone to be oxidized by free radicals and other ROSs [35], which may be alleviated by dietary supplementation of antioxidants such as vitamin C, vitamin E, and carotenoids [36]. Nevertheless, the administration of repeated high doses of PUFA may render the organism less susceptible to lipid peroxidation due to adaptive intracellular mechanisms involving enhancement of the antioxidant response via Nrf2 activation [37]. Increasing extracellular concentrations of fatty acids and low-density lipoproteins induce iNOS expression in many cell types including pancreatic β-cells, vascular smooth muscle cells, and macrophages [38]. Similarly, feeding a high saturated-fat diet to rats increases iNOS activity in liver and colon [39] and stimulates free radical production and oxidative DNA damage in skeletal muscle mitochondria and the whole body [40, 41].
Cellular effects of ROS will rely on its cellular exposure levels [42]. Cell exposure to low to moderate ROS levels triggers a survival response and reinforces ROS scavengers of the antioxidant defence system to elicit a cytoprotective effect. The molecular mechanism believed to be responsible for this adaptive change involves enhanced antioxidant activity achieved by upregulating several housekeeping genes partly under the control of nuclear factor erythroid 2-related factor-2 (Nrf2) [43]. Nrf2 is normally sequestered in the cytosol by Kelch-like ECH-associated protein 1 (Keap1). Upon oxidative stimulation, ROS could either oxidise Nrf2 or covalently modify Keap1 thiol groups, leading to Nrf2 dissociation from Keap1 and subsequent nuclear translocation. In the nucleus, Nrf2 binds to antioxidant response elements (AREs) in target gene promoters [44], which increase the expression of antioxidant enzymes such as catalase, glutathione peroxidase, glutathione reductase, glutathione S-transferase, NADPH:quinone oxidoreductase 1, and heme oxygenase-1 [37].
Cell exposure to high levels of ROS induces nuclear factor kappaB (NF-κB) activation, which triggers a proinflammatory response characterized by increased levels of TNF-α, IL-1β, IL-6, and IL-8, increased expression of adhesion molecules, such as E-selectin, VCAM-1, and ICAM-1, and promotion of oxidative stress. NF-κB is one member of a ubiquitously expressed family of Rel-related transcription factors that serve as critical regulators of inflammatory related genes such as tumor necrosis factor (TNF) and IL-1 [45]. NF-κB mainly exists in the cytosol as a pre-formed trimeric complex that consists of the inhibitory protein I-kB and the P50/P65 protein dimer. ROS induces redox changes that result in phosphorylation of the I-kB subunit, thereby activating its proteolytic digestion. When the inhibitor subunit is dislodged from the P60/P65 heterodimer, NF-κB can translocate to the nucleus, bind DNA, and initiate transcription [42].
ROS can mediate cellular damage through direct interaction with cellular components. The redox reaction mediated by ROS determines an electron loss in a target molecule, which alters its structure and function. Virtually any cellular component can be a target of ROS interaction. Lipids, DNA, and proteins have been shown to interact with ROS and mediate cellular damage [46], given that it is not surprising that chronic and acute exposure to excessive levels of ROS can induce cell death processes (apoptosis, necrosis, and autophagy) [47–50].
Acute myocardial infarction (AMI) is a clinical model of oxidative stress by ischemia reperfusion. ROSs are major initiators of myocardial damage during ischemia/reperfusion. In pathophysiological conditions, AMI is initiated by myocardial ischemia due to coronary artery obstruction leading to an initial increase in ROS levels associated with a loss of antioxidant enzyme function, together with leakage of antioxidant enzymes into the extracellular fluid which in normal physiological conditions, kept oxygen free radical production under homeostatic control, and as a result, the unbalanced burst of free radicals on reperfusion easily overwhelms the available counteractive enzymes, so that control of ROS generation is lost [28]. ROSs including H2O2, O2•−, hydroxyl radical, and peroxynitrite have been shown to increase upon reperfusion of the heart following ischemia [51]. However, ROS seems to increase significantly after a few minutes of reperfusion, but its increase during ischemia alone is still controversial.
After an ischemic episode of the myocardium, left ventricle remodeling is known to occur; although its underlying mechanism is multifactorial, ROS and inflammatory cytokines may cause cardiodepressive reaction [52–54]. It is of interest to remark that ROSs also stimulate the production of inflammatory cytokines, and, in turn, inflammatory cytokines stimulate ROS formation. In chronic stage, ROS and inflammatory cytokines activate the matrix metalloproteinases [55, 56], thereby eliciting degradation of collagens which may cause a slippage in myofibrillar alignment causing left ventricular dilatation [57]. Oxidative stress may result in cellular defects including a depression in the sarcolemmal Ca2+-pump ATPase and (Na + K)-ATPase activities, changes leading to decreased Ca2+-efflux and increased Ca2+-influx, respectively [58]. The depression in Ca2+-regulatory mechanism by ROS ultimately results in intracellular Ca2+ ([Ca2+]i) overload and cell death. On the other hand, an increase in [Ca2+]i during ischemia induces the conversion of xanthine dehydrogenase to xanthine oxidase and subsequently results in generating superoxide radicals [59].
Oxidative stress can participate in the development of hypertension. Compelling experimental evidence indicates that ROSs play an important pathophysiological role in the development of hypertension. This is due, in large part, to O2•− excess and decreased NO bioavailability in the vasculature and kidneys, and to ROS-mediated cardiovascular remodeling [60–62]. In human hypertension, biomarkers of systemic oxidative stress are elevated [63]. Physiologically, ROSs are produced in a controlled manner at low concentrations and function as signaling molecules [64] to maintain vascular integrity by regulating endothelial function and vascular contraction-relaxation balance. Under pathological conditions, increased ROS bioactivity leads to endothelial dysfunction, an essential pathophysiological mechanism in the development of hypertension. Reactive oxygen species can inhibit the three major endothelium-dependent vasodilator pathways, that is, NO, prostacyclin, and endothelial derived hyperpolarizing factor (EDHF) [65]. Superoxide anions not only reduce the bioavailability of NO but also directly inhibit its main target, soluble guanylyl cyclase [66–68]. Furthermore, superoxide anions can react with NO leading to the formation of peroxynitrite (ONOO−) which uncouples NO synthase by oxidizing, on one hand, the Zn-thiolate complexes within the enzyme [69] and, on the other hand the essential cofactor tetrahydrobiopterin (BH4) [70]. When endothelial nitric oxide synthase (eNOS) becomes unstable and uncoupled, it leads to subsequent less NO production and more superoxide generation [71]. Additionally, ONOO− inhibits guanylyl cyclase, inactivates the prostacyclin synthase by tyrosine nitration, and further enhances oxidative stress by inhibiting superoxide dismutase [66–72].
Furthermore, increased ROS levels promote vascular smooth muscular cells growth, increased contractility, monocyte invasion, lipid peroxidation, inflammation, and increased deposition of extracellular matrix proteins, all of which are important factors in hypertensive vascular damage [73, 74]. A schema showing the cellular response to ROS is depicted in Figure 1.
Figure 1.
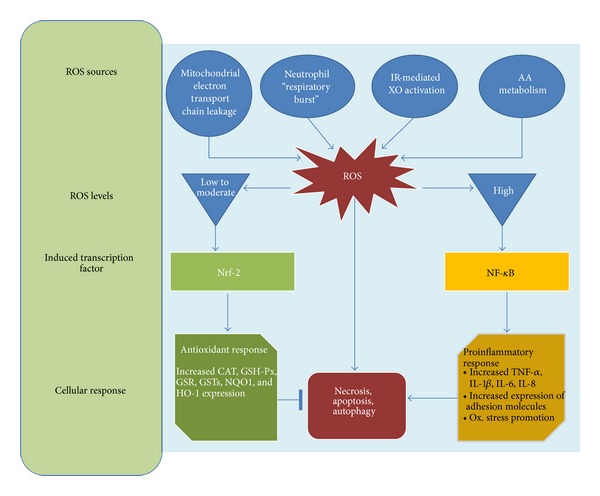
Cellular response to reactive oxygen species. Several pathophysiological conditions determine an increase on intracellular ROS levels. Transcription factor induction is dependent in the ROS levels. Low to moderate ROS levels induce Nrf-2 activation which increases the expression of antioxidant enzymes such as catalase, glutathione peroxidase, glutathione reductase, glutathione S-transferase, NADPH:quinone oxidoreductase 1, and heme oxygenase-1 while high levels of ROS induce NF-κB activation, which trigger a proinflammatory response characterized by increased levels of TNF-α, IL-1β, IL-6, and IL-8, increased expression of adhesion molecules, such as E-selectin, VCAM-1, and ICAM-1, and promotion of oxidative stress. AA: arachidonic acid; CAT: catalase; GSH-Px: glutathione peroxidase; GSR: glutathione reductase; glutathione S-transferase: GSTs; HO-1: heme oxygenase-1; NADPH:quinone oxidoreductase 1: NQO1; IL-1 β: Interleukin 1β; IL-6: Interleukin 6; IL-8: Interleukin 8; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf-2: nuclear factor- (erythroid-derived 2-) like 2.
3. Biomarkers
Epidemiologic literature that focuses on antioxidant status and chronic disease risk has in the past relied primarily upon biomarkers of exposure to antioxidant nutrients. However, this approach is in essence using exposure estimates to a select group of nutrients as a surrogate for estimating actual oxidative defense or oxidative stress status. Emphasis is now being placed on developing functional biomarkers of oxidative stress status, that is, biomarkers that integrate the effect of exposure to oxidants coupled with the full range of antioxidant protective mechanisms in vivo [75]. Many of such biomarkers are being studied including various measures of oxidation products of lipid, DNA, and protein (Table 1). Some of these biomarkers are now being applied in research of pathologies related to oxidative stress.
Table 1.
Biomarkers of oxidative stress.
| Type of biomarker | Location | Reference |
|---|---|---|
| Ferric reducing ability of plasma | Extracellular | Benzie and Strain, 1996 [96] |
| Carbonyls | Extracellular | Mohanty et al., 2010 [98] |
| Lipid peroxidation | ||
| Malondialdehyde | Extracellular | Ohkawa et al., 1979 [77] |
| F2-isoprostane | Extracellular | Collins et al., 1999 [202] |
| 4-Hydroxynonenal | Extracellular | Halliwell and Gutteridge, 2007 [89] |
| Plasma vitamins | ||
| Vitamin C | Extracellular | Roe and Kuether, 1943 [94] |
| Vitamin E | Extracellular | Teissier 1996 [93] |
| Antioxidant enzymes | ||
| Superoxide dismutase | Intracellular | Misra and Fridovich, 1972 [100] |
| Catalase | Intracellular | Aebi and Bergmeyer, 1974 [101] |
| Glutathione peroxidase | Intracellular | Flohé and Günzler, 1984 [102] |
| GSH/GSSG ratio in erythrocyte | Intracellular | Hissin and Hilf, 1976 [104] |
| Prooxidant enzymes | ||
| Xanthine oxidase | Intracellular | Haining and Legan 1967 [106] |
| NADPH oxidase | Intracellular | Nauseef, 2008 [17, 18] |
| Others | ||
| Endothelial microparticles | Extracellular | Burger and Touyz, 2012 [109] |
| Endothelial progenitor cells | Extracellular | Touyz and Schiffrin, 2012 [111] |
| Ischemia modified albumin | Extracellular | Sinha et al., 2003 [194] |
Lewandrowski et al. suggest the following features for an ideal cardiac biomarker, among others: (1) High concentration in the myocardium, (2) absence from nonmyocardial tissue, (3) high sensitivity and specificity in blood samples, (4) rapid release into the blood following myocardial injury, (5) favorable kinetics permitting detection of acute coronary syndromes and myocardial necrosis in days after the person seeks care, and (6) correlation between blood level of marker and extent of myocardial injury and prognosis [76].
3.1. Oxidative Stress-Related Biomarkers
Lipid peroxides have been estimated by measurement of thiobarbituric acid reactive substances in plasma by the method of Ohkawa et al. [77]. In this method, in the peroxidation process, peroxides are decomposed to aldehydes like malondialdehyde (MDA), which can be detected by thiobarbituric acid that gives an easily measurable pink color. All of these are termed thiobarbituric reactive species (TBARS) [78]. This method is one of the most widely used assays to assess peroxidation in the whole organism. The reaction of peroxynitrite with lipids leads to peroxidation and MDA and conjugated diene formation [79]. It is noteworthy also that MDA products may behave as toxic bifunctional electrophiles, due to reactivity with proteins, phospholipids, and DNA, generating stable products at the end of a series of reactions to form propane adducts [80]. Consequently, a change in the properties of the molecule, for example, in its charge profile, could result in modified cell-matrix interactions [81]. Lipid peroxides, normally present in lipoproteins or membranes, are known to further induce lipid peroxidation, inhibit the mitochondrial electron transport system, and to oxidize sulfhydryl groups on proteins, hence altering its function or otherwise disrupting signal transduction pathways [82].
The nonenzymatic peroxidation of arachidonic acid results in the formation of F2-isoprostanes, which are also products of lipid peroxidation. The levels of these compounds can serve as an index of lipid peroxidation in several diseases [83, 84]. Because of their mechanism of formation, specific structural features that distinguish them from other free radical-generated products and their chemical stability, they can provide a reliable index of lipid peroxidation in vivo in a variety of clinical settings associated with oxidative stress [85]. In addition, this determination provides a noninvasive method for dose selection to monitor the response to antioxidant treatment [86].
Another lipid peroxidation product is 4-hydroxynonenal (4-HNE), a highly cytotoxic and reactive α, β-aldehyde that is generated during various physiological and pathophysiological conditions based on the production of ROS [87, 88]. 4-HNE is also considered one of the most specific and sensitive measures of lipid autooxidation [89, 90]. HNE-4 can be measured by standard methods [91, 92].
Vitamin E can be measured by the method of HPLC method [93]. The determination of alpha-tocopherol in human serum is reported. UV-Detection is possible by using either one wavelength of 300 nm or after a lambda change mode with typical wavelengths for alpha-tocopherol (292 nm). According to short retention times (10 min) and rapid extraction the method is useful for clinical research and allows about 50 analyses per day and operator. Vitamin C (ascorbic acid) can be estimated by the method of Roe and Kuether [94]. This involves oxidation of ascorbic acid by copper followed by treatment with 2,4-dinitrophenylhydrazine that undergoes rearrangement to form a product with absorption maximum at 520 nm [95].
Total antioxidant activity is measured by the ferric reducing antioxidant power (FRAP) assay of Benzie and Strain [96]. FRAP is a test which measures the combined antioxidant effect of the nonenzymatic defenses in a biological fluid. At low pH, when a ferric-tripyridyltriazine (FeIII-TPTZ) complex reduces the ferrous (FeII) form, an intense blue color with an absorption maximum at 593 nm develops.
Another oxidative stress biomarker is the protein carbonylation. The direct damage of proteins during oxidative stress can give rise to the formation of protein carbonyls, which may serve as biomarkers for general oxidative stress, in addition to the data provided by lipid peroxidation. Most of the assays [97, 98] for the detection of protein CO groups involve derivatisation of the carbonyl group with 2,4-dinitrophenylhydrazine (DNPH), which leads to formation of a stable dinitrophenyl (DNP) hydrazone product. This then can be detected by various means, such as spectrophotometric assay, enzyme-linked immunosorbent assay (ELISA), and one-dimensional or two-dimensional electrophoresis followed by western blot immunoassay. At present, the measurement of protein CO groups after their derivatisation with DNPH is the most widely utilized measure of protein oxidation [99].
3.2. Antioxidant Potential Biomarkers
Superoxide Dismutase (SOD). The term superoxide dismutase characterizes a family of proteins differing in their structure and cofactors, among them being Mn-SOD and Cu-Zn-SOD. SOD activity enhances the spontaneous dismutation of superoxide radicals to H2O2 [46]. SOD can be measured utilizing the technique of Misra and Fridovich [100] based on inhibition of the formation of nicotine amide adenine dinucleotide, phenazine methosulfate, and amino blue tetrazolium formazan.
Catalase (CAT). The end product of the dismutation reaction −H2O2− can be removed by the activity of the enzyme catalase. CAT is an enzyme with a very high KM for its substrate and can remove H2O2 present in high concentrations [46]. The activity of CAT can be measured through the non-spontaneous decomposition of hydrogen peroxide, by the method described by Aebi [101].
Glutathione Peroxidase (GPx). The biochemical function of glutathione peroxidase is to reduce lipid hydroperoxides to their corresponding alcohols and to reduce free H2O2 to water. In contrast to catalase, peroxidase possesses high affinity for and can remove H2O2 even when it is present in low concentration [46]. Its activity can be estimated by the method described by Flohé and Günzler [102].
Thiol Index. GSH/GSSG ratio in erythrocyte. The GSH/GSSG index is a parameter of intracellular redox status. GSH is the major endogenous antioxidant produced by the cells, participating directly in the neutralization of free radicals and reactive oxygen compounds, as well as maintaining exogenous antioxidants such as vitamins C and E in their reduced forms [103]. GSH and GSSG levels can be measured in erythrocytes by the method described by Hissin and Hilf [104].
Xanthine Oxidase (XO). It is a key enzyme in reactive oxygen species formation, which plays a major role in cell oxidative stress. This enzyme catalyzes the oxidation of hypoxanthine to xanthine and can further catalyze the oxidation of xanthine to uric acid [105]. Xanthine oxidase can be estimated by the method of Haining and Legan [106].
NADPH Oxidase. The NADPH-oxidase complex utilizes electrons to produce superoxide radicals from the oxygen molecule [46]. NADPH oxidase may be measured through chemiluminescence [107] or electrochemistry [108] methods, among others. Approaches to quantitate oxygen consumption, extracellular release of O2•− or H2O2, and intracellular O2•− production provide reliable assessment of NADPH oxidase activity in a given population of cells [21].
3.3. Other Biomarkers of Cardiovascular Damage
Endothelial microparticles (EMPs) are nuclear fragments of cellular membrane shed from stressed or damaged cells. Typically defined as having a diameter of 0.1 to 1.0 mm, microparticles contain surface proteins and cytoplasmic material of the parental cells [109].
Flow cytometry has been largely used to quantify EMP levels in clinical samples, although this widely used methodology suffers from limitations according to MP probing, sizing, and counting in a standardized manner. For probing, EMP detection still presents the limitations of lacking fully specific antigens. CD144 was proposed as a marker for EMP detection. Others strategies have been developed as the combination of multicolor antibodies (CD31+/CD41−, CD31+/CD42b−, CD105+/CD45−) and monochrome composite markers (CD144+ CD105+, CD146+ CD105+) [110].
Endothelial progenitor cells have been a biomarker of cardiovascular damage. Of the various factors important in the protection of the endothelium are endothelial progenitor cells (EPCs). EPCs are bone marrow-derived cells capable of developing into mature endothelial cells [111]. EPCs are mobilized from the bone marrow into the peripheral blood in response to tissue ischemia or injury and through their differentiation improve blood flow and tissue repair [112]. The precise criteria for identifying EPCs by flow cytometry remain a contentious issue; however, the combined surface expression of CD133, CD34, and VEGFR2 is generally accepted as the most stringent criteria for EPCs [113, 114].
Ischemia modified albumin (IMA) has been another biomarker of cardiovascular damage. Under physiological conditions, transition metals can bind tightly to the exposed N-terminus of albumin [115]. In the presence of myocardial ischemia, structural changes take place in the N-terminus of the protein [116, 117] which reduce its binding capacity, possibly, in part, as a result of exposure to ROS. IMA rises in patients who develop myocardial ischemia during percutaneous coronary intervention (PCI) [84]. Serum IMA can be measured by the albumin cobalt binding (ACB) test. The first generation ACB test method has been validated and described in previous publications [118, 119].
4. Essential Hypertension
Hypertension has become an emerging epidemic and important worldwide public-health challenge, because it is one of the most common chronic conditions [120–123]. The importance of high blood pressure as a major cause of serious diseases has been recognized in most western countries for more than 50 years [124]. Essential or primary hypertension is a heterogeneous disorder in which secondary causes are not present. It accounts for 95% of all cases of hypertension [125], with different patients having different causal factors that lead to high blood pressure. The identification of population at risk is a challenge that requires laboratory blood tests; however, biomarkers to ensure regular monitoring of patients having essential hypertension are not available to date. Evidence has been provided that activation of the vascular endothelial cells in the presence of cardiovascular risk factors promotes oxidative stress and vascular inflammatory response, for either classic (smoking, aging, hypercholesterolemia, hypertension, hyperglycemia, and a family history of premature atherosclerotic disease) and novel (obesity, elevated C-reactive protein, and chronic systemic infection) risk factors, leading to acceleration of atherosclerotic vascular disease [126]. Therefore, appropriate biomarkers of these processes should give some clues for the management of early metabolic changes, otherwise leading to stages of permanent and progressive vascular lesions. Several studies have demonstrated excessive amounts of ROS in essential hypertensive patients and various animal models of hypertension [6, 46, 127–130].
4.1. Role of ROS in Vascular Homeostasis
The vasculature is a rich source of ROSs, which are produced in endothelial, adventitial, and smooth muscular cells. The regulation of vascular tone may be impaired by changes affecting the interaction between these vascular cells [131]. The endothelium senses mechanical stimuli, such as pressure and shear stress, as well as chemical stimuli induced by vasoactive substances. In response, it releases agents that regulate vasomotor function, trigger inflammatory processes, and affect hemostasis. Many crucial vasoactive endogenous compounds are produced by the endothelial cells to control the functions of vascular smooth muscle cells and of circulating blood cells [132]. These complex systems determine a fine equilibrium which regulates the vascular tone. Impairments in endothelium-dependent vasodilation lead to the so-called endothelial dysfunction. The endothelium modulates several biological processes through the balance between opposing mechanisms such as vasoconstriction/vasodilation, pro-coagulation/antithrombosis, cell proliferation/apoptosis, among others. This dynamic tissue layer constitutes a source and/or target of multiple growth factors and vasoactive mediators involved in regulating the physical and biochemical properties of the systemic vessels, as well as vascular contractility and cell growth. Concerning the regulation of vasomotor tone, the endothelium plays a protective role by generating vasorelaxing substances, such as NO, PGI2, and endothelium-derived hyperpolarizing factor (EDHF) [133], which oppose the vasoconstricting action. Vascular smooth muscle cells (VSMCs) are essential for good performance of the vasculature. They can alter the luminal diameter which enables blood vessels to maintain an appropriate blood pressure. VSMC are fit not only for short-term regulation of the vessel diameter, but also for long-term adaptation, via structural remodeling by changing cell number and connective tissue composition. The main function of vascular smooth muscle tonus is to regulate the caliber of the blood vessels in the body. Excessive vasoconstriction leads to hypertension, having physical effects on the vascular wall, such as tensile stress caused by pressure and shear stress caused by flow [134].
The vasoconstricting action of hormones, such as endothelin-1, angiotensin II, or urotensin II, among others, is mediated by ROS. Furthermore, ROS mediate many pathophysiological processes, such as growth, migration, apoptosis and secretion of inflammatory cytokines, as well as physiological processes, such as differentiation, by direct and indirect effects at multiple signaling levels. It was reported that H2O2 activates phospholipase C on VSMC through tyrosine phosphorylation and that this activation has a major role in rapid [Ca2+]i mobilization in this type of cells [135]. Ultimately, the physiological or pathophysiological effect of ROSs is dependent on their steady state concentration. Oxidative stress can lead to unbalanced vasoconstricting and vasodilating forces in favor of the first. Indeed, this impairment in vasomotor tone regulation belongs to a more complex phenomenon named endothelial dysfunction, characterized by a shift of the actions of the endothelium toward reduced vasodilation, a proinflammatory state, and prothrombotic setting [6].
A variety of enzymatic and nonenzymatic sources of ROS exist in blood vessels. Enzymatic sources of ROS include NADPH oxidases located on the cell membrane of polymorphonuclear cells, macrophages, and endothelial cells and cytochrome P450-dependent oxygenases. The proteolytic conversion of xanthine dehydrogenase to xanthine oxidase provides another enzymatic source of both superoxide and H2O2 (and therefore constitutes a source of the highly reactive hydroxyl radicals) and has been proposed to mediate deleterious processes in vivo. In addition to NADPH oxidase, the best characterized source of ROS, several other enzymes may contribute to ROS generation, including NO synthase, lipoxygenase, cyclooxygenases, xanthine oxidase, and cytochrome P450 enzymes. Also mitochondria could be considered a major source of ROS: in situations of metabolic perturbation, increased mitochondrial ROS generation might trigger endothelial dysfunction, possibly contributing to the development of hypertension [136].
4.2. Relationship between Oxidative Stress and Essential Hypertension
Systolic blood pressure and diastolic blood pressure correlated inversely with plasma antioxidant potential (e.g., FRAP) and directly with some oxidative stress-related parameters (e.g., F2-isoprostanes) [137]. On the other hand, other plasma blood pressure modulator levels, such as AT-II, ET-1, renin, aldosterone, and homocysteine, among others, showed no significant differences between normotensive and hypertensive subjects. These associations strongly suggest a role for oxidative stress as a modulator of blood pressure. These results are in agreement with previous studies reporting increased oxidative stress and decreased antioxidant capacity in patients with essential hypertension compared with normotensive subjects [138, 139]. These findings also suggest consideration of oxidative stress as a novel therapeutic target for the therapy of essential hypertension. F2-isoprostanes have been proposed as a new factor involved in the modulation of BP [140]. F2-isoprostanes have a direct correlation with blood pressure levels in normotensive and hypertensive subjects [137].
Long-term hypertension produces irreversible endothelial dysfunction due in part to self-sustaining ROS production [5, 141, 142]. In patients with already established endothelial damage, measurement of oxidative stress-related biomarkers will be of less use. In order to establish a functional interpretation of hypertension through oxidative stress-related biomarkers, the early phases of the disease should be studied. In the endothelial dysfunction phase, previous to the development of hypertension, the ROS fluctuations will appropriately reflect the balance between ROS production and their scavenging or depuration by the antioxidant defense system in the endothelium.
The role of the antioxidants vitamins C and E has emerged as a possible therapy for decreasing oxidative stress, thereby lowering BP. In fact, a concomitant decrease in BP and oxidative stress was shown following Vitamins C and E supplementation in patients with essential hypertension [143].
In contrast with these findings, the results of short-term trials of antioxidant supplements on blood pressure have been inconsistent [144, 145] and long-term clinical trials have failed to consistently support the antihypertensive effects of vitamins C and E in patients at high cardiovascular risk. This controversy may be explained by the patient inclusion and exclusion criteria, which did not consistently consider the influence of factors such as already established target organ damage, smoking status, physical activity, and intake of nonantihypertensive (e.g., statins) medication, which are potential confounders of oxidative stress status.
4.3. Antioxidant Therapy Effectiveness May Be Closely Dependant on Clinical Setting and Oxidative Status
Antioxidant therapy should be rather effective in the early stages of hypertension or atherosclerosis by preventing the oxidative-stress mediated “positive feedback loop” of progression from reversible endothelial dysfunction to atherosclerotic plaque formation [146]. Most patients participating in clinical trials are in the advanced stages of this progression (i.e., with atherosclerosis or cardiovascular disease) and the “positive feedback loop” will have generated irreversible structural changes such as vascular remodeling or atherosclerosis. Therefore the lack of benefit of antioxidant therapy in diminishing CV events may reflect incomplete information on the actual role of oxidative stress, which should be considered as a necessary component of the enrolled patients, as antioxidants may have no effect on the absence of marked oxidative stress and already established cardiovascular disease.
In all animal studies with positive results, the vitamin E administration started before the constitution of histological atherosclerotic changes in the structure of the arterial wall [147]. Furthermore, the protective effect shown by vitamins C and E supplementation in the prevention of immune atherosclerotic lesions occurring after cardiac transplant also supports this assumption. [148, 149]. Correction of hypertension or prehypertension and other cardiovascular risk factors leading to endothelial dysfunction in their early stages may help reduce the burden of disease and mortality attributable to high blood pressure [150, 151].
Considerable evidence suggests that oxidative stress, which results in an excessive generation of ROS, plays a key role in the pathogenesis of hypertension. Antioxidant therapies have been evaluated in order to decrease ROSs production or increase their scavenging. In this line, polyphenols, widespread antioxidants in fruits, vegetables, and wine, have demonstrated their beneficial role in prevention and therapy of hypertension, by acting as free-radical scavengers, metal chelators, and in enzyme modulation and expression. Numerous studies have demonstrated the importance of naturally occurring dietary polyphenols in promoting cardiovascular health and emphasized the significant role that these compounds play in limiting the effects of cellular aging. Polyphenols such as resveratrol, epigallocatechin gallate (EGCG), and curcumin have been acknowledged for having beneficial effects on cardiovascular health, while some have also been shown to be protective in aging [152]. Polyphenols activate and enhance eNOS expression by several signaling pathways, increase glutathione levels, and inhibit ROS-producing enzymes such as NADPH and xanthine oxidases. Accordingly, antioxidant activity of phenolics in leaves of three red pepper (Capsicum annuum), mainly putrescine and anthocyanine derivatives, demonstrated a great potential for radical scavenging activity against oxidative stress [153]. In addition, some polyphenols can exert beneficial effects other than those related to their antioxidant properties. Thus, luteolin can reduce VSMCs' proliferation and migration [154], and effects also reported for resveratrol [155].
Also, polyphenols have the capacity to improve the endothelial control of vascular tone not only in several experimental models of cardiovascular diseases such as hypertension but also in healthy and diseased humans. Experiments with isolated arteries have shown that polyphenols cause nitric oxide (NO) mediated endothelium-dependent relaxations and increase the endothelial formation of NO [156], via the PI3-kinase/Akt pathway, leading to a rapid activation of endothelial NO synthase through its phosphorylation at Ser1177 [157].
It would then be concluded that the time window for an antioxidant therapy in essential hypertension is narrow, and as such should be focused on new onset and high-risk hypertension individuals having biomarkers consistent with a high ROS load.
As an example, in the Framingham Study, subjects who had isolated diastolic hypertension at baseline were generally male overweight or obese smokers and were at inordinately higher risk (23-fold) than those with optimal blood pressure to develop systo-diastolic hypertension [151]. As mentioned before, patients with these characteristics have shown favorable response to antihypertensive antioxidant therapy [139].
4.4. Cellular Biomarkers of Endothelial Health: EPCs and EMP
Endothelial progenitor cells (EPCs) are novel biomarkers for cardiovascular risk and show considerable potential as they reflect the body's capacity for endothelial repair [109]. EPCs have been validated as CV risk biomarkers for both low- and high-risk population [158–160]. Regarding essential hypertension, EPCs count in hypertension is decreased as is their overall function. Hypertension is associated with some specific EPCs function alterations, such as reduced in vitro migratory capacity [161] and increased senescence [162].
EPCs usefulness as a CV risk biomarker shows great promise but a consensus characterization of EPCs is still lacking [112], and further studies are needed to determine the usefulness of measuring EPC absolute number or functional capacity in different clinical settings [109]. Antioxidant agents, such as vitamin C, lower circulating EMP levels in patients with diabetes and dyslipidemia after myocardial infarction [110].
In turn, endothelial microparticles (EMPs) are considered as potential CV biomarkerss, which stems from the fact that they directly reflect endothelial damage [109]. Microparticles circulate in healthy individuals and their levels increase in cardiovascular and atherothrombotic diseases [163]. In subjects with high risk of coronary heart disease, baseline levels of EMP predicted outcome, independent of Framingham score and of C-reactive protein (CRP) and brain natriuretic peptide levels [110].
In patients with hypertension, plasma levels of EMP correlated with systolic and diastolic blood pressures [111].
5. Ischemia-Reperfusion Myocardial Damage
The main clinical model of ischemia-reperfusion injury is acute myocardial infarction (AMI), the leading cause of mortality worldwide. According to the WHO, in 2008 ischemic heart disease accounted for 7.25 million deaths (12.8%). Systemic thrombolysis and percutaneous coronary angioplasty (PCA) have been used to recover the myocardial perfusion, with the latter being the most successful, as it allows to re-establish the blood flow in the cardiac zones affected by the occlusion of a branch of the coronary artery. Nevertheless, as a consequence of this procedure, the ischemic zone is reperfused, giving rise to an ischemia-reperfusion event that generates increased production of ROS (oxidative stress) [164]. Therefore, this widely performed procedure leads to an enhancement of the previously established tissue damage (lethal reperfusion), as these reactive species attack biomolecules such as lipids, DNA, and proteins and trigger cell death pathways [165]. As ischemia-reperfusion injury remains a neglected therapeutic target [166], biomarkers of ischemia reperfusion could be used to measure the effectiveness of novel cardioprotective strategies to prevent lethal reperfusion.
A series of biochemical and metabolic changes in myocardial tissue occur due to deprivation of oxygen and nutrient supply during ischemia. Consequently, mitochondrial damage and ATP depletion impair myocardial contractile function [166]. Anaerobic glycolysis due to the absence of oxygen results in the accumulation of lactate and intracellular pH reduction (to <7.0). The latter activates the Na+-H+ ion exchanger, thus extruding protons from the cell in exchange for Na+ entry. Furthermore, the impaired function of (Na + K)-ATPase contributes to exacerbating the intracellular Na+ and Ca2+ overload [167]. At the onset of reperfusion, the level of tissue oxygenation increases, leading to a burst of ROS generation, thereby accounting for the syndrome of reperfusion injury [164]. Neutrophils are the primary source of ROS during reperfusion, although endothelial cells and cardiomyocytes can also generate these reactive species. Increased ROS production is mainly due to activation of XO in endothelial cells, mitochondrial electron transport chain reactions in cardiomyocytes, and NADPH oxidase in inflammatory cells [168].
Several biomarkers could be used to demonstrate the major role of oxidative stress in the pathophysiology of ischemia-reperfusion damage in myocardial infarction. Extracellular biomarkers of ischemia-reperfusion damage encompass products of lipid peroxidation, plasma antioxidant vitamin levels, total antioxidant capacity of plasma, protein carbonylation, and uric acid level. In addition, intracellular biomarkers such as activity of antioxidant enzymes, thiol index (GSH/GSSG ratio), carbonyl levels, and F2-isoprostane, among others, are suitable to estimate oxidative stress injury.
5.1. Lipid Peroxidation
Increased lipid peroxidation is thought to be a consequence of oxidative stress. It occurs due to an impairment altering the dynamic balance between prooxidant and antioxidant mechanisms. In ischemia, the ATP is drastically reduced and is converted to hypoxanthine and then to uric acid by xanthine oxidase upon reperfusion. During this process, enormous amounts of superoxide radicals are formed and can simulate Haber-Weiss reaction for further generation of ROS, thus initiating lipid peroxidation [169]. It has been shown that in AMI patients, the increase of oxidative stress markers, such as lipid peroxidation products, mirrors the decrease in low-density lipoprotein-cholesterol [170]. A reason for increased lipid peroxidation in plasma of AMI patients may be a poor enzymatic and nonenzymatic antioxidant defense system. Pasupathi et al. in a cohort of 100 patients with evidence of AMI, showed a significant rise in TBARS levels (P < 0.001), a lipid peroxidation biomarker indicative of elevated oxidative stress [171]. Furthermore, these authors showed an increased concentration of MDA in the circulation of total AMI patients, indicating increased lipid peroxidation. These results are in agreement with previous reports [172]. Nevertheless, lipid peroxidation might be increased due to the presence of systemic diseases including hyperlipidemia, diabetes mellitus, or renal disease, in addition to myocardial infarction [173], decreasing its specificity in this type of patients.
Also, F2-isoprostane, a representative isoprostane, has been reported to be a reliable marker for oxidant stress in vivo. In addition, it was reported that the level of immunoreactive 8-iso PGF(2 alpha) is increased in the great cardiac vein in patients with AMI [174]. The study showed transcardiac F2-isoprostane generation, being a reliable biomarker for the size of myocardium exposed to oxidant stress.
5.2. Plasma Antioxidant Vitamin Levels
In AMI patients, Pasupathi et al. showed lower levels of vitamins E and C compared with controls [171] This is in accordance with studies of Singh et al. [175], who demonstrated that there was a significant drop in vitamins C, E, A, and β-carotene, whereas lipid peroxides were significantly higher in AMI patients, compared with controls. This indicates severe damage to antioxidant system, which is unable to combat oxidative stress and inflammation [176].
5.3. Total Antioxidant Capacity (TAC)
This biomarker has been described as a critical tool for assessing redox status [177]. The TAC or related antioxidants may play an important role in protecting the organism from free-radicals-mediated damage [178]. The role that such compounds play in AMI development is important, since their presence may decrease the damage resulting from blood ROS during reperfusion.
5.4. Antioxidant Enzymes
Antioxidants constitute the foremost defense system that limit the toxicity associated with free radicals. It is known that plasma antioxidant capacity decreases and oxidative/antioxidative balance shift to the oxidative balance in patients with MI. SOD along with CAT and GPx, the preventive antioxidants, play a very important role in protection against lipid peroxidation [179]. Scott et al. showed that SOD, CAT, and GPx activities were significantly lower in MI and ischemic heart disease patients than in control subjects. Besides, decrease of SOD, CAT, and GPx activity was much more pronounced in smokers than in nonsmokers with AMI, thus making those individuals more vulnerable to oxidative stress. Free radical-scavenging enzymes such as SOD, CAT, and GPx are the first line of cellular defense against oxidative injury, decomposing O2•− and H2O2, otherwise interacting to form the more reactive hydroxyl radical. These enzymes protect the red cells against O2•− and H2O2-mediated lipid peroxidation [179]. Decreased activities of SOD and CAT in the erythrocyte in AMI patients have been reported [180, 181], likely due to inactivation by cross-linking or to exhaustion of the enzymes by increased peroxidation [182].
Glutathione peroxidase (GPx) catalyzes peroxide reduction utilizing GSH as the substrate and converting it to GSSG. Its activity has been reported decreased in erythrocytes of AMI patients [171]. Also, inactivation of GPx after endogenous exposure to products of lipid peroxidation [183] or of NO [184] has been reported. GSH is one of the most important endogenous intracellular antioxidants. It plays the role of a sulfhydryl (SH) group provider for direct scavenging reactions. GSH acts both as a substrate in the scavenging reaction catalyzed by GPx and as a scavenger of vitamins C and E radicals. A lowered serum concentration of reduced glutathione (GSH) has been considered the predictor of coronary restenosis after percutaneous coronary intervention, and its deficiency could lead to pronounced postreperfusion syndrome [185]. Decrease GSH concentration may also contribute to decreased GPx activity, becouse GSH Is one of its substrates [186]. In accordance with this it has been shown that plasma and erythrocyte GSH concentration were significantly decreased in AMI patients. It may be due to an increased utilization of GSH [180]. In a single center prospective cohort trial with a 15-month follow-up [187], the primary endpoint was major adverse cardiac events (MACE), defined either as re-PCI or coronary artery bypass grafting, MI, cardiac death, or cardiac rehospitalization, or a combination thereof. Results showed that GSH levels are significantly decreased in the whole MACE group. Authors acknowledge that this was a preliminary study with a relatively low number of subjects. Due to this limitation, they observed a relatively low number of MACEs during follow-up. Also they used preprocedural GSH concentrations as a discriminative variable, and we did not monitor the fluctuations in the GSH concentration over time, to use it as a harmonic (time-dependent) variable.
High plasma MPO is reported to be a risk factor for early adverse cardiac events in patients with chest pain [188] or acute coronary syndromes [189] and to be associated with endothelial dysfunction [190, 191]. In myocardial infarction a retrospective study concluded that levels of MPO were independently predictive of 5-year survival and might provide additional prognostic information if used in combination with the established markers, left ventricular ejection fraction and NT-pro-BNP [192].
Ischemia modified albumin (IMA) is a recently developed biomarker of transient myocardial ischemia [193]. Circulating IMA is increased in patients with myocardial ischemia, after percutaneous coronary intervention [194] or in acute coronary syndromes [195]. The test has recently been licensed by the US Food and Drug Administration for diagnostic use in suspected myocardial ischemia. IMA is serum albumin in which the N-terminus has been chemically modified. It has been proposed that ROSs such as superoxide (•O2−) and hydroxyl (•OH) radicals generated during myocardial ischemia-reperfusion modify the N-terminus of serum albumin resulting in IMA formation. In vivo modifications of the albumin N-terminus are proposed to be related to the ROS production during myocardial ischemia reperfusion. For example, an increase in IMA was observed in patients minutes after transient occlusion and reperfusion during coronary angioplasty [195]. Roy et al. in 2006 showed for the first time that IMA formation is directly related to ROS in vitro, generating and providing novel information on the nature of the species contributing to IMA formation, namely •OH [196].
Polyphenols induce cardiovascular protective effects, thanks to their antioxidant, vasodilator, anti-inflammatory, anti-fibrotic, antiapoptotic and metabolic properties. Polyphenol-dependent cardioprotection occurs mainly through NO and via several powerful prosurvival cellular cascades that involve metabolic intermediates [197]. It has been reported that the polyphenol luteolin protects against myocardial injury and enhances cellular antioxidant defense capacity through the activation of Akt and ERK signal pathways that leads to Nrf2 activation and subsequently heme oxygenase-1 induction [198] In addition, it preserves cardiac function, reduces infarct size and cardiomyocyte apoptotic rate after ischemia/reperfusion injury in diabetic rats [199]. These authors also demonstrated that luteolin has an anti-apoptotic role in an experimental model of ischemia/reperfusion in rats [199], being a potential candidate for preventing and treating cardiovascular diseases [200].
Nevertheless, common oxidative stress-related biomarkers for both pathologies could be the diminution of TAC of plasma and increased lipid peroxidation products, such as F2-isoprostanes and MDA in percutaneous coronary angioplasty following coronary infarction [139, 201].
6. Concluding Remarks and Perspectives
Increasing body of evidence accounts for the validity of using biomarkers to assess the factors involved in pathogenesis and clinical outcome of human diseases. Several biomarkers could be used to demonstrate the major role of oxidative stress in cardiovascular pathologies, such as those derived from essential hypertension and ischemia reperfusion damage. Indeed, the applicability of oxidative stress-related biomarkers arises from the role of ROS in the pathophysiology of these clinical settings. The role of ROS in the development of essential hypertension arises from both decreased vasodilation due to a diminished bioavailability of NO and prostacyclin together with an increased vasoconstriction by their cellular effect on VSMC, thus accounting for blood pressure elevation. The contribution of ROS to myocardial oxidative damage caused by infarction is due to direct and cellular mediated effects of ROS, such as necrosis and apoptosis, respectively.
Extracellular oxidative stress-related biomarkers include total antioxidant capacity of plasma, plasma antioxidant vitamin levels, lipid peroxidation product levels, protein carbonylation, and uric acid level. Intracellular oxidative stress related biomarkers include the activity of antioxidant enzymes, prooxidant enzymes, and thiol index.
Cardiac biomarker testing in the clinical laboratory continues to evolve based on the availability of improved technologies and the results of clinical trials among patients with acute coronary syndromes. Although still at its infancy, the use of cellular biomarkers of endothelial dysfunction has the potential to yield powerful mechanistic insight into endothelial dysfunction. They provide a direct window to the active state of the endothelium or to endothelial repair. There is ample consensus that these biomarkers provide a direct window to the active state of the endothelium or to endothelial repair and for these reasons, they represent special value as biomarkers in these clinical settings. In the same fashion, ischemia modified albumin value lies in how it specifically denotes the presence of ischemia in the organism.
In summary, the applicability of oxidative stress-related biomarkers to study the pathogenesis and clinical outcome of essential hypertension and ischemia reperfusion cardiac pathologies arises from the role of ROS in cardiovascular diseases. Lipid peroxidation biomarkers such as plasma F2-isoprostane levels have been widely studied in the assessment of in vivo oxidative stress-related human disease. However, novel biomarkers are emerging to account for additional pathological processes. Thus, increased inflammatory and oxidative stress biomarkers are associated with endothelial progenitor cell dysfunction, which is reversed upon treatment with anti-inflammatory or antioxidant drugs. Therefore, inflammatory and redox signaling pathways that interfere with EPC bioactivity need to be further investigated.
Acknowledgment
This study was supported by the Fondo Nacional de Investigación Científica y Tecnológica (FONDECYT), Grant 1120594.
Abbreviations
- AMI:
Acute myocardial infarction
- ARE:
Antioxidant response elements
- BP:
Blood pressure
- ACB:
Albumin cobalt binding
- CAT:
Catalase
- CABG:
Coronary artery bypass grafting
- CO:
Carbon monoxide
- CV:
Cardiovascular
- CRP:
C-reactive protein
- DNPH:
2,4-dinitrophenylhydrazine
- DNP:
Stable dinitrophenyl
- ELISA:
Enzyme-linked immunosorbent assay
- EDHF:
Endothelium-derived hyperpolarizing factor
- FRAP:
Ferric reducing antioxidant power
- EMP:
Endothelial microparticles
- EPCs:
Endothelial progenitor cells
- FeIII-TPTZ:
Ferric-tripyridyltriazine
- GSH:
Reduced glutathione
- GSH-Px:
Glutathione peroxidase
- H2O2:
Hydrogen peroxide
- OH:
Hydroxyl
- 4-HNE:
4-hydroxynonenal
- IMA:
Ischemia modified albumin
- MACE:
Major adverse cardiac events
- MPO:
Myeloperoxidase
- MDA:
malondialdehyde
- MI:
Myocardial infarction
- NAD+:
Nicotinamide adenine dinucleotide
- NADH:
Nicotinamide adenine dinucleotide dehydrogenase
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NO:
Nitric oxide
- NOS:
Nitric oxide synthases
- Nrf2:
Nuclear factor erythroid 2-related factor-2
- nNOS:
A constitutive neuronal NOS
- iNOS:
Cytokine-inducible NOS
- eNOS:
Constitutive endothelial NOS
- O2•−:
Superoxide anion
- •OH:
Hydroxyl radical
- PCI:
Percutaneous coronary intervention
- PUFA:
Polyunsaturated fatty acids
- ROS:
Reactive oxygen species.
- RNS:
Reactive nitrogen species
- SOD:
Superoxide dismutase
- SH:
Sulfhydryl
- TBARS:
Thiobarbituric reactive species
- TNF:
Tumor necrosis factor
- VSMC:
Vascular smooth muscle cells
- XO:
Xanthine oxidase
- XDH:
Xanthine dehydrogenase.
References
- 1.Quam L, Smith R, Yach D. Rising to the global challenge of the chronic disease epidemic. The Lancet. 2006;368(9543):1221–1223. doi: 10.1016/S0140-6736(06)69422-1. [DOI] [PubMed] [Google Scholar]
- 2.Gu D, Reynolds K, Wu X, et al. Prevalence, awareness, treatment, and control of hypertension in China. Hypertension. 2002;40(6):920–927. doi: 10.1161/01.hyp.0000040263.94619.d5. [DOI] [PubMed] [Google Scholar]
- 3.Mittal BV, Singh AK. Hypertension in the developing world: challenges and opportunities. American Journal of Kidney Diseases. 2010;55(3):590–598. doi: 10.1053/j.ajkd.2009.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Peluso I, Morabito G, Urban L, Ioannone F, Serafini M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocrine Metabolic&Immune Disorders Drug Targets. 2012;12(4):351–360. doi: 10.2174/187153012803832602. [DOI] [PubMed] [Google Scholar]
- 5.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigo R, González J, Paoletto F. The role of oxidative stress in the pathophysiology of hypertension. Hypertension Research. 2011;34:431–440. doi: 10.1038/hr.2010.264. [DOI] [PubMed] [Google Scholar]
- 7.Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxidative Medicine and Cellular Longevity. 2009;2(5):259–269. doi: 10.4161/oxim.2.5.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigo R, Cereceda M, Castillo R, et al. Prevention of atrial fibrillation following cardiac surgery: basis for a novel therapeutic strategy based on non-hypoxic myocardial preconditioning. Pharmacology and Therapeutics. 2008;118(1):104–127. doi: 10.1016/j.pharmthera.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Rodrigo R, Prieto JC, Castillo R. Cardioprotection against ischaemia/reperfusion by vitamins C and E plus n-3 fatty acids: molecular mechanisms and potential clinical applications. Clinical Science. 2013;124:1–15. doi: 10.1042/CS20110663. [DOI] [PubMed] [Google Scholar]
- 10.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Juránek I, Bezek Š. Controversy of free radical hypothesis: reactive oxygen species—cause or consequence of tissue injury. General Physiology and Biophysics. 2005;24(3):263–278. [PubMed] [Google Scholar]
- 12.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 13.Tahara EB, Navarete FDT, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radical Biology and Medicine. 2009;46(9):1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Tsuda H, Kawada N, Kaimori J-Y, et al. Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochemical Biophysical Research Communication. 2012;427(2):266–272. doi: 10.1016/j.bbrc.2012.09.032. [DOI] [PubMed] [Google Scholar]
- 15.Takac I, Schröder K, Brandes RP. The nox family of NADPH oxidases: friend or foe of the vascular system? Current Hypertension Reports. 2012;14(1):70–78. doi: 10.1007/s11906-011-0238-3. [DOI] [PubMed] [Google Scholar]
- 16.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. The FEBS Journal. 2008;275(15):3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 17.Nauseef WM. Biological roles for the NOX family NADPH oxidases. The Journal of Biological Chemistry. 2008;283(25):16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nauseef WM. Nox enzymes in immune cells. Seminars in Immunopathology. 2008;30(3):195–208. doi: 10.1007/s00281-008-0117-4. [DOI] [PubMed] [Google Scholar]
- 19.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bedard K, Lardy B, Krause K-H. NOX family NADPH oxidases: not just in mammals. Biochimie. 2007;89(9):1107–1112. doi: 10.1016/j.biochi.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Nauseff WM. Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. doi: 10.1016/j.bbagen.2013.04.040. Biochimica et Biophysica Acta. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montesanto A, Crocco P, Tallaro F, et al. Common polymorphisms in nitric oxide synthase (NOS) genes influence quality of aging and longevity in humans. Biogerontology. 2013;14(2):177–186. doi: 10.1007/s10522-013-9421-z. [DOI] [PubMed] [Google Scholar]
- 23.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. European Heart Journal. 2012;33(7):829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vásquez-Vivar J, Kalyanaraman B, Martásek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y, Zweier JL. Direct measurement of nitric oxide generation from nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(23):12705–12710. doi: 10.1073/pnas.94.23.12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinecke JW, Li W, Francis GA, Goldstein JA. Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of proteins. Journal of Clinical Investigation. 1993;91(6):2866–2872. doi: 10.1172/JCI116531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spiteller G. Are lipid peroxidation processes induced by changes in the cell wall structure and how are these processes connected with diseases? Medical Hypotheses. 2003;60(1):69–83. doi: 10.1016/s0306-9877(02)00333-x. [DOI] [PubMed] [Google Scholar]
- 28.Venardos KM, Kaye DM. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Current Medicinal Chemistry. 14(14):1539–1549. doi: 10.2174/092986707780831078. [DOI] [PubMed] [Google Scholar]
- 29.Gasparetto C, Malinverno A, Culacciati D, et al. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. International Journal of Immunopathology and Pharmacology. 2005;18(3):487–496. doi: 10.1177/039463200501800308. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigo R. Prevention of postoperative atrial fibrillation: novel and safe strategy based on the modulation of the antioxidant system. Frontiers in Physiology. 2012;3, article 93 doi: 10.3389/fphys.2012.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreoli SP. Reactive oxygen molecules, oxidant injury and renal disease. Pediatric Nephrology. 1991;5(6):733–742. doi: 10.1007/BF00857888. [DOI] [PubMed] [Google Scholar]
- 32.Sehirli AO, Sener G, Satiroglu H, Ayanoğlu-Dülger G. Protective effect of N-acetylcysteine on renal ischemia/reperfusion injury in the rat. Journal of Nephrology. 2003;16(1):75–80. [PubMed] [Google Scholar]
- 33.Meister A, Anderson ME. Glutathione. Annual Review of Biochemistry. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 34.Byers T, Perry G. Dietary carotenes, vitamin C, and vitamin E as protective antioxidants in human cancers. Annual Review of Nutrition. 1992;12:139–159. doi: 10.1146/annurev.nu.12.070192.001035. [DOI] [PubMed] [Google Scholar]
- 35.Hennig B, Toborek M, McClain CJ. High-energy diets, fatty acids and endothelial cell function: implications for atherosclerosis. Journal of the American College of Nutrition. 2001;20(2):97–105. doi: 10.1080/07315724.2001.10719021. [DOI] [PubMed] [Google Scholar]
- 36.Evans P, Halliwell B. Micronutrients: oxidant/antioxidant status. British Journal of Nutrition. 2001;85(2):S67–S74. [PubMed] [Google Scholar]
- 37.Zhu H, Jia Z, Misra BR, et al. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovascular Toxicology. 2008;8(2):71–85. doi: 10.1007/s12012-008-9016-0. [DOI] [PubMed] [Google Scholar]
- 38.Wu G, Meininger CJ. Regulation of nitric oxide synthesis by dietary factors. Annual Review of Nutrition. 2002;22:61–86. doi: 10.1146/annurev.nutr.22.110901.145329. [DOI] [PubMed] [Google Scholar]
- 39.Wan G, Ohnomi S, Kato N. Increased hepatic activity of inducible nitric oxide synthase in rats fed on a high-fat diet. Bioscience, Biotechnology and Biochemistry. 2000;64(3):555–561. doi: 10.1271/bbb.64.555. [DOI] [PubMed] [Google Scholar]
- 40.Haegele AD, Briggs SP, Thompson HJ. Antioxidant status and dietary lipid unsaturation modulate oxidative DNA damage. Free Radical Biology and Medicine. 1994;16(1):111–115. doi: 10.1016/0891-5849(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 41.Sreekumar R, Unnikrishnan J, Fu A, et al. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. American Journal of Physiology—Endocrinology and Metabolism. 2002;282(5):E1055–E1061. doi: 10.1152/ajpendo.00554.2001. [DOI] [PubMed] [Google Scholar]
- 42.Gloire G, Legrand-Poels S, Piette J. NF-κB activation by reactive oxygen species: fifteen years later. Biochemical Pharmacology. 2006;72(11):1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 43.de Vries HE, Witte M, Hondius D, et al. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radical Biology and Medicine. 2008;45(10):1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxidants and Redox Signaling. 2005;7(3-4):385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 45.Baldwin AS., Jr. Series introduction: the transcription factor NF-κB and human disease. The Journal of Clinical Investigation. 2001;107(1):3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicologic Pathology. 2002;30(6):620–650. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- 47.von Harsdorf R, Li P-F, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99(22):2934–2941. doi: 10.1161/01.cir.99.22.2934. [DOI] [PubMed] [Google Scholar]
- 48.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxidants and Redox Signaling. 2011;14(11):2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai D-F, Rabinovitch P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy. 2011;7(8):917–918. doi: 10.4161/auto.7.8.15813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18(54):7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 51.Eaton P, Clements-Jewery H. Peroxynitrite: in vivo cardioprotectant or arrhythmogen? British Journal of Pharmacology. 2008;155(7):972–973. doi: 10.1038/bjp.2008.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovascular Research. 2002;53(1):31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 53.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circulation Research. 2000;87(3):241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 54.Suematsu N, Tsutsui H, Wen J, et al. Oxidative stress mediates tumor necrosis factor-α-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107(10):1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 55.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. American Journal of Physiology—Cell Physiology. 2001;280(1):C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 56.Deten A, Hölzl A, Leicht M, Barth W, Zimmer H-G. Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats. Journal of Molecular and Cellular Cardiology. 2001;33(6):1191–1207. doi: 10.1006/jmcc.2001.1383. [DOI] [PubMed] [Google Scholar]
- 57.Rohde LE, Ducharme A, Arroyo LH, et al. Matrix metalloproteinase inhibition attenuates early left ventricular injury. Cardiovascular Research. 2000;47:446–456. [Google Scholar]
- 58.Dixon IM, Hata T, Dhalla NS. Sarcolemmal Na(+)-K(+)-ATPase activity in congestive heart failure due to myocardial infarction. American Journal of Physiology—Cell Physiology. 1992;262(3):C664–C671. doi: 10.1152/ajpcell.1992.262.3.C664. [DOI] [PubMed] [Google Scholar]
- 59.Sasaki M, Joh T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. Journal of Clinical Biochemistry and Nutrition. 2007;40(1):1–12. doi: 10.3164/jcbn.40.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Touyz RM. Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Review of Cardiovascular Therapy. 2003;1(1):91–106. doi: 10.1586/14779072.1.1.91. [DOI] [PubMed] [Google Scholar]
- 61.Chabrashvili T, Tojo A, Onozato ML, et al. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39(2 I):269–274. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 62.Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109(19):2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- 63.Redón J, Oliva MR, Tormos C, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41(5):1096–1101. doi: 10.1161/01.HYP.0000068370.21009.38. [DOI] [PubMed] [Google Scholar]
- 64.Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clinical and Experimental Pharmacology and Physiology. 2003;30(11):860–866. doi: 10.1046/j.1440-1681.2003.03930.x. [DOI] [PubMed] [Google Scholar]
- 65.Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (the wiggers award lecture) The American Jounral of Physiology Heart Circulatory Physiology. 2006;291:H985–H1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- 66.Münzel T, Daiber A, Ullrich V, Mülsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the CGMP-dependent protein kinase. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(8):1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- 67.Price DT, Vita JA, Keaney JF., Jr. Redox control of vascular nitric oxide bioavailability. Antioxidants and Redox Signaling. 2000;2(4):919–935. doi: 10.1089/ars.2000.2.4-919. [DOI] [PubMed] [Google Scholar]
- 68.Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. American Journal of Physiology. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822. [DOI] [PubMed] [Google Scholar]
- 69.Zou M-H, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. Journal of Clinical Investigation. 2002;109(6):817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen W, Druhan LJ, Chen C-A, et al. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. 2010;49(14):3129–3137. doi: 10.1021/bi9016632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. American Journal of Physiology—Endocrinology and Metabolism. 2012;302(5):E481–E495. doi: 10.1152/ajpendo.00540.2011. [DOI] [PubMed] [Google Scholar]
- 72.Zou M-H, Cohen RA, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11(2):89–97. doi: 10.1080/10623320490482619. [DOI] [PubMed] [Google Scholar]
- 73.Diep QN, Amiri F, Touyz RM, et al. PPARα activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension. 2002;40(6):866–871. doi: 10.1161/01.hyp.0000037969.41360.cc. [DOI] [PubMed] [Google Scholar]
- 74.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42(6):1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- 75.Mayne ST. Antioxidant nutrients and chronic disease: use of biomarkers of exposure and oxidative stress status in epidemiologic research. The Journal of Nutrition. 2003;133(3):933S–940S. doi: 10.1093/jn/133.3.933S. [DOI] [PubMed] [Google Scholar]
- 76.Lewandrowski K, Chen A, Januzzi J. Cardiac markers for myocardial infarction. a brief review. The American Journal of Clinical Pathology. 2002;118:S93–99. doi: 10.1309/3EK7-YVV9-228C-E1XT. [DOI] [PubMed] [Google Scholar]
- 77.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Analytical Biochemistry. 1979;95(2):351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 78.Esterbauer H. Estimation of peroxidative damage. a critical review. Pathologie Biologie. 1996;44(1):25–28. [PubMed] [Google Scholar]
- 79.Var A, Kuşcu NK, Koyuncu F, et al. Atherogenic profile in preeclampsia. Archives of Gynecology and Obstetrics. 2003;268(1):45–47. doi: 10.1007/s00404-002-0317-4. [DOI] [PubMed] [Google Scholar]
- 80.Blair IA. Lipid hydroperoxide-mediated DNA damage. Experimental Gerontology. 2001;36(9):1473–1481. doi: 10.1016/s0531-5565(01)00133-4. [DOI] [PubMed] [Google Scholar]
- 81.Slatter DA, Bolton CH, Bailey AJ. The importance of lipid-derived malondialdehyde in diabetes mellitus. Diabetologia. 2000;43(5):550–557. doi: 10.1007/s001250051342. [DOI] [PubMed] [Google Scholar]
- 82.Ischiropoulos H, Zhu L, Chen J, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Archives of Biochemistry and Biophysics. 1992;298(2):431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 83.Hubel CA, Lyall F, Weissfeld L, Gandley RE, Roberts JM. Small low-density lipoproteins and vascular cell adhesion molecule-1 are increased in association with hyperlipidemia in preeclampsia. Metabolism. 1998;47(10):1281–1288. doi: 10.1016/s0026-0495(98)90337-7. [DOI] [PubMed] [Google Scholar]
- 84.Barden A, Beilin LJ, Ritchie J, Croft KD, Walters BN, Michael CA. Plasma and urinary 8-iso-prostane as an indicator of lipid peroxidation in pre-eclampsia and normal pregnancy. Clinical Science. 1996;91(6):711–718. doi: 10.1042/cs0910711. [DOI] [PubMed] [Google Scholar]
- 85.Cracowski J-L, Devillier P, Durand T, Stanke-Labesque F, Bessard G. Vascular biology of the isoprostanes. Journal of Vascular Research. 2001;38(2):93–103. doi: 10.1159/000051036. [DOI] [PubMed] [Google Scholar]
- 86.Praticò D, Lawson JA, Rokach J, Fitzgerald GA. The isoprostanes in biology and medicine. Trends in Endocrinology and Metabolism. 2001;12(6):243–247. doi: 10.1016/s1043-2760(01)00411-8. [DOI] [PubMed] [Google Scholar]
- 87.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24(4-5):149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 88.Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24(4-5):167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 89.Halliwell B, Gutteridge JMC. Free Radical in Biology and Medicine. 4th edition. New York, NY, USA: Oxford University Press; 2007. [Google Scholar]
- 90.Zarkovic K. 4-Hydroxynonenal and neurodegenerative diseases. Molecular Aspects of Medicine. 2003;24(4-5):293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 91.EsterbauermH H, Cheesman KH. [42] Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods in Enzymology. 1990;186:407–421. doi: 10.1016/0076-6879(90)86134-h. [DOI] [PubMed] [Google Scholar]
- 92.Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radical Biology and Medicine. 1990;9(6):515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 93.Teissier E, Walters-Laporte E, Duhem C, Luc G, Fruchart JC, Duriez P. Rapid quantification of alpha-tocopherol in plasma and low- and high-density lipoproteins. Clinical Chemistry. 1996;42(1):430–435. [PubMed] [Google Scholar]
- 94.Roe HJ, Kuether CA. Detection of ascorbic acid in whole blood and urine through the 2, 4-dinitrophenyl-hydrazine derivative of dehydro ascorbic acid. The Journal of Biological of Chemistry. 1943;147:399–407. [Google Scholar]
- 95.Chung WY, Chung JKO, Szeto YT, Tomlinson B, Benzie IFF. Plasma ascorbic acid: measurement, stability and clinical utility revisited. Clinical Biochemistry. 2001;34(8):623–627. doi: 10.1016/s0009-9120(01)00270-3. [DOI] [PubMed] [Google Scholar]
- 96.Benzie IFF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Analytical Biochemistry. 1996;239(1):70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- 97.Buss H, Chan TP, Sluis KB, Domigan NM, Winterbourn CC. Protein carbonyl measurement by a sensitive ELISA method. Free Radical Biology & Medicine. 1997;23:361–366. doi: 10.1016/s0891-5849(97)00104-4. [DOI] [PubMed] [Google Scholar]
- 98.Mohanty JG, Bhamidipaty S, Evans MK, Rifkind JM. A fluorimetric semi-microplate format assay of protein carbonyls in blood plasma. Analytical Biochemistry. 2010;400(2):289–294. doi: 10.1016/j.ab.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clinica Chimica Acta. 2003;329(1-2):23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 100.Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. The Journal of Biological Chemistry. 1972;247(10):3170–3175. [PubMed] [Google Scholar]
- 101.Aebi M. Catalase. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Vol. 2. NY, USA: Verlag Chemie-Academic Press; 1974. [Google Scholar]
- 102.Flohé L, Günzler WA. Assays of glutathione peroxidase. Methods in Enzymology. 1984;105:114–120. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 103.Scholz RW, Graham KS, Gumpricht E, Reddy CC. Mechanism of interaction of vitamin E and glutathione in the protection against membrane lipid peroxidation. Annals of the New York Academy of Sciences. 1989;570:514–517. [Google Scholar]
- 104.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Analytical Biochemistry. 1976;74(1):214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 105.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radical Biology and Medicine. 2002;33(6):774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 106.Haining JL, Legan JS. Fluorometric assay for xanthine oxidase. Analytical Biochemistry. 1967;21(3):337–343. doi: 10.1016/0003-2697(67)90308-9. [DOI] [PubMed] [Google Scholar]
- 107.Yamazaki T, Kawai C, Yamauchi A, Kuribayashi F. A highly sensitive chemiluminescence assay for superoxide detection and chronic granulomatous disease diagnosis. Tropical Medicine and Health. 2011;39(2):41–45. doi: 10.2149/tmh.2011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ashkenazi A, Abu-Rabeah K, Marks RS. Electrochemistry and chemiluminescence techniques compared in the detection of NADPH oxidase activity in phagocyte cells. Talanta. 2009;77(4):1460–1465. doi: 10.1016/j.talanta.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 109.Burger D, Touyz RM. Cellular biomarkers of endothelial health: microparticles, endothelial progenitor cells, and circulating endothelial cells. Journal of the American Society of Hypertension. 2012;6(2):85–99. doi: 10.1016/j.jash.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 110.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(1):27–33. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 111.Touyz RM, Schiffrin E. Arterial hypertension. In: Hill J, editor. Muscle Fundamental Biology and Mechanisms of Disease. Vol. 2. San Diego, Calif, USA: Elsevier; 2012. pp. 1311–1319. [Google Scholar]
- 112.Lin C-P, Lin F-Y, Huang P-H, et al. Endothelial progenitor cell dysfunction in cardiovascular diseases: role of reactive oxygen species and inflammation. Biomed Research International. 2013;2013:10 pages. doi: 10.1155/2013/845037.845037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sen S, McDonald SP, Coates PTH, Bonder CS. Endothelial progenitor cells: novel biomarker and promising cell therapy for cardiovascular disease. Clinical Science. 2011;120(7):263–283. doi: 10.1042/CS20100429. [DOI] [PubMed] [Google Scholar]
- 114.Peichev M, Naiyer AJ, Pereira D, et al. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood. 2000;95(3):952–958. [PubMed] [Google Scholar]
- 115.Sadler PJ, Tucker A, Viles JH. Involvement of a lysine residue in the N-terminal Ni2+ and Cu2+ binding site of serum albumins. Comparison with Co2+, Cd2+ and Al3+ European Journal of Biochemistry. 1994;220(1):193–200. doi: 10.1111/j.1432-1033.1994.tb18614.x. [DOI] [PubMed] [Google Scholar]
- 116.Chan B, Dodsworth N, Woodrow J, Tucker A, Harris R. Site-specific N-terminal auto-degradation of human serum albumin. European Journal of Biochemistry. 1995;227(1-2):524–528. doi: 10.1111/j.1432-1033.1995.tb20419.x. [DOI] [PubMed] [Google Scholar]
- 117.Bar-Or D, Curtis G, Rao N, Bampos N, Lau E. Characterization of the Co2+ and Ni2+ binding amino-acid residues of the N-terminus of human albumin: an insight into the mechanism of a new assay for myocardial ischemia. European Journal of Biochemistry. 2001;268(1):42–47. doi: 10.1046/j.1432-1327.2001.01846.x. [DOI] [PubMed] [Google Scholar]
- 118.Bar-Or D, Winkler JV, VanBenthuysen K, Harris L, Lau E, Hetzel FW. Reduced albumin-cobalt binding with transient myocardial ischemia after elective percutaneous transluminal coronary angioplasty: a preliminary comparison to creatine kinase-MB, myoglobin, and troponin I. American Heart Journal. 2001;141(6):985–991. doi: 10.1067/mhj.2001.114800. [DOI] [PubMed] [Google Scholar]
- 119.Christenson RH, Duh SH, Sanhai WR, et al. Characteristics of an albumin cobalt binding test for assessment of acute coronary syndrome patients: a multicenter study. Clinical Chemistry. 2001;47(3):464–470. [PubMed] [Google Scholar]
- 120.Alderman MH, Ogihara T. Global challenge for overcoming high blood pressure: fukuoka statement, 19 October 2006. Journal of Hypertension. 2007;25(3):p. 727. doi: 10.1097/HJH.0b013e3280961a1f. [DOI] [PubMed] [Google Scholar]
- 121.Ezzati M, Lopez AD, Rodgers A, Hoorn SV, Murray CJ, Comparative Risk Assessment Collaborating Group Selected major risk factors and global and regional burden of disease. The Lancet. 2002;360(9343):1347–1360. doi: 10.1016/S0140-6736(02)11403-6. [DOI] [PubMed] [Google Scholar]
- 122.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. The Lancet. 2005;365(9455):217–223. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 123.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. The Lancet. 2006;367(9524):1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 124.Perkovic V, Huxley R, Wu Y, Prabhakaran D, MacMahon S. The burden of blood pressure-related disease: a neglected priority for global health. Hypertension. 2007;50(6):991–997. doi: 10.1161/HYPERTENSIONAHA.107.095497. [DOI] [PubMed] [Google Scholar]
- 125.Carretero OA, Oparil S. Essential hypertension—part I: definition and etiology. Circulation. 2000;101(3):329–335. doi: 10.1161/01.cir.101.3.329. [DOI] [PubMed] [Google Scholar]
- 126.Hadi HAR, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vascular health and risk management. 2005;1(3):183–198. [PMC free article] [PubMed] [Google Scholar]
- 127.Lacy F, Kailasam MT, O’Connor DT, Schmid-Schönbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension. 2000;36(5):878–884. doi: 10.1161/01.hyp.36.5.878. [DOI] [PubMed] [Google Scholar]
- 128.Stojiljkovic MP, Lopes HF, Zhang D, Morrow JD, Goodfriend TL, Egan BM. Increasing plasma fatty acids elevates F2-isoprostanes in humans: implications for the cardiovascular risk factor cluster. Journal of Hypertension. 2002;20(6):1215–1221. doi: 10.1097/00004872-200206000-00036. [DOI] [PubMed] [Google Scholar]
- 129.Tanito M, Nakamura H, Kwon Y-W, et al. Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxidants and Redox Signaling. 2004;6(1):89–97. doi: 10.1089/152308604771978381. [DOI] [PubMed] [Google Scholar]
- 130.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochemistry and Cell Biology. 2004;122(4):339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 131.Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxidants and Redox Signaling. 2011;15(6):1517–1530. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cines DB, Pollak ES, Buck CA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91(10):3527–3561. [PubMed] [Google Scholar]
- 133.Félétou M, Köhler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Current Hypertension Reports. 2010;12(4):267–275. doi: 10.1007/s11906-010-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. Endothelium-dependent hyperpolarization, bringing the concepts together. Trends in Pharmacological Sciences. 2002;23(8):374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 135.Mayet J, Hughes A. Cardiac and vascular pathophysiology in hypertension. Heart. 2003;89(9):1104–1109. doi: 10.1136/heart.89.9.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.González-Pacheco FR, Caramelo C, Castilla MÁ, et al. Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: role of phospholipse C gamma. Nephrology Dialysis Transplantation. 2002;17(3):392–398. doi: 10.1093/ndt/17.3.392. [DOI] [PubMed] [Google Scholar]
- 137.Rodrigo R, Bächler JP, Araya J, Prat H, Passalacqua W. Relationship between (Na + K)-ATPase activity, lipid peroxidation and fatty acid profile in erythrocytes of hypertensive and normotensive subjects. Molecular and Cellular Biochemistry. 2007;303(1-2):73–81. doi: 10.1007/s11010-007-9457-y. [DOI] [PubMed] [Google Scholar]
- 138.Russo C, Olivieri O, Girelli D, et al. Anti-oxidant status and lipid peroxidation in patients with essential hypertension. Journal of Hypertension. 1998;16(9):1267–1271. doi: 10.1097/00004872-199816090-00007. [DOI] [PubMed] [Google Scholar]
- 139.Rodrigo R, Prat H, Passalacqua W, Araya J, Guichard C, Bächler JP. Relationship between oxidative stress and essential hypertension. Hypertension Research. 2007;30(12):1159–1167. doi: 10.1291/hypres.30.1159. [DOI] [PubMed] [Google Scholar]
- 140.Gross M, Steffes M, Jacobs DR, Jr., et al. Plasma F2-isoprostanes and coronary artery calcification: the CARDIA study. Clinical Chemistry. 2005;51(1):125–131. doi: 10.1373/clinchem.2004.037630. [DOI] [PubMed] [Google Scholar]
- 141.Panza JA, Casino PR, Kilcoyne CM, Quyyumi AA. Role of endothelium-derived nitric oxide in the abnormal endothelium- dependent vascular relaxation of patients with essential hypertension. Circulation. 1993;87(5):1468–1474. doi: 10.1161/01.cir.87.5.1468. [DOI] [PubMed] [Google Scholar]
- 142.Vaziri ND. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Current Opinion in Nephrology and Hypertension. 2004;13(1):93–99. doi: 10.1097/00041552-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 143.Rodrigo R, Prat H, Passalacqua W, Araya J, Bächler JP. Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clinical Science. 2008;114(9-10):625–634. doi: 10.1042/CS20070343. [DOI] [PubMed] [Google Scholar]
- 144.Juraschek SP, Guallar E, Appel LJ, Miller ER., III Effects of vitamin c supplementation on blood pressure: a meta-analysis of randomized controlled trials. American Journal of Clinical Nutrition. 2012;95(5):1079–1088. doi: 10.3945/ajcn.111.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.John JH, Ziebland S, Yudkin P, Roe LS, Neil HAW. Effects of fruit and vegetable consumption on plasma antioxidant concentrations and blood pressure: a randomised controlled trial. The Lancet. 2002;359(9322):1969–1974. doi: 10.1016/s0140-6736(02)98858-6. [DOI] [PubMed] [Google Scholar]
- 146.Widlansky ME, Gokce N, Keaney JF, Jr., Vita JA. The clinical implications of endothelial dysfunction. Journal of the American College of Cardiology. 2003;42(7):1149–1160. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 147.Munteanu A, Zingg J-M, Azzi A. Anti-atherosclerotic effects of vitamin E-myth or reality? Journal of Cellular and Molecular Medicine. 2004;8(1):59–76. doi: 10.1111/j.1582-4934.2004.tb00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu L, Meydani M. Combined vitamin C and E supplementation retards early progression of arteriosclerosis in heart transplant patients. Nutrition Reviews. 2002;60(11):368–371. doi: 10.1301/00296640260385810. [DOI] [PubMed] [Google Scholar]
- 149.Fang JC, Kinlay S, Beltrame J, et al. Effect of vitamins C and E on progression of transplant-associated arteriosclerosis: a randomised trial. The Lancet. 2002;359(9312):1108–1113. doi: 10.1016/S0140-6736(02)08154-0. [DOI] [PubMed] [Google Scholar]
- 150.Sierra C, de la Sierra A. Early detection and management of the high-risk patient with elevated blood pressure. Vascular Health and Risk Management. 2008;4(2):289–296. doi: 10.2147/vhrm.s930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schiffrin EL. The vascular phenotypes in hypertension: relation with the natural history of hypertension. Journal of the American Society of Hypertension. 2007;1(1):56–67. doi: 10.1016/j.jash.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 152.Khurana S, Venkataraman K, Hollingsworth A, Piche M, Tai TC. Polyphenols: benefits to the cardiovascular system in health and in aging. Nutrients. 2013;5(10):3779–3827. doi: 10.3390/nu5103779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim W, Kim EO, Kang K, et al. Antioxidant activity of phenolics in leaves of three red pepper (Capsicum annuum) cultivars. Journal of Agricultural Food and Chemistry. 2013 doi: 10.1021/jf403006c. [DOI] [PubMed] [Google Scholar]
- 154.Jiang D, Li D, Wu W. Inhibitory effects and mechanisms of luteolin on proliferation and migration of vascular smooth muscle cells. Nutrients. 2013;5:1648–1659. doi: 10.3390/nu5051648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shi Y, Hou X, Zhang X, Wang Y, Chen Y, Zou J. Inhibition of oxidized phospholipid-induced vascular smooth muscle cell proliferation by resveratrol is associated with reducing Cx43 phosphorylation. Journal and Agricultural Food Chemistry. 2013 doi: 10.1021/jf4036723. [DOI] [PubMed] [Google Scholar]
- 156.Schini-Kerth VB, Auger C, Kim J-H, Etienne-Selloum N, Chataigneau T. Nutritional improvement of the endothelial control of vascular tone by polyphenols: role of NO and EDHF. Pflügers Archiv. 2010;459(6):853–862. doi: 10.1007/s00424-010-0806-4. [DOI] [PubMed] [Google Scholar]
- 157.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt- dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 158.Schmidt-Lucke C, Rössig L, Fichtlscherer S, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111(22):2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 159.Werner N, Kosiol S, Schiegl T, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. The New England Journal of Medicine. 2005;353(10):999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 160.Hill JM, Zalos G, Halcox JPJ, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. The New England Journal of Medicine. 2003;348(7):593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 161.Vasa M, Fichtlscherer S, Aicher A, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circulation Research. 2001;89(1):E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 162.Zhou Z, Peng J, Wang C-J, et al. Accelerated senescence of endothelial progenitor cells in hypertension is related to the reduction of calcitonin gene-related peptide. Journal of Hypertension. 2010;28(5):931–939. doi: 10.1097/HJH.0b013e3283399326. [DOI] [PubMed] [Google Scholar]
- 163.Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006;48(2):180–186. doi: 10.1161/01.HYP.0000231507.00962.b5. [DOI] [PubMed] [Google Scholar]
- 164.Maxwell SRJ. Anti-oxidant therapy: does it have a role in the treatment of human disease? Expert Opinion on Investigational Drugs. 1997;6(3):211–236. doi: 10.1517/13543784.6.3.211. [DOI] [PubMed] [Google Scholar]
- 165.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovascular Research. 2009;81(3):457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 166.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. Journal of Clinical Investigation. 2013;123(1):92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Avkiran M, Marber MS. Na+/H+ exchange inhibitors for cardioprotective therapy: progress, problems and prospects. Journal of the American College of Cardiology. 2002;39(5):747–753. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- 168.Duilio C, Ambrosio G, Kuppusamy P, Dipaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. American Journal of Physiology—Heart and Circulatory Physiology. 2001;280(6):H2649–H2657. doi: 10.1152/ajpheart.2001.280.6.H2649. [DOI] [PubMed] [Google Scholar]
- 169.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovascular Research. 2004;61(3):461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 170.Chamblee BB, Timm TC, Hunsaker LA, Vander Jagt DL. Relationship of oxidative stress indices to decreased LDL-cholesterol after acute myocardial infarction. Clinical Biochemistry. 2000;33(5):423–426. doi: 10.1016/s0009-9120(00)00153-3. [DOI] [PubMed] [Google Scholar]
- 171.Pasupathi P, Rao YY, Farook J, Saravanan G, Bakthavathsalam G. Oxidative stress and cardiac biomarkers in patients with acute myocardial infarction. European Journal of Scientific Research. 2009;27(2):275–285. [Google Scholar]
- 172.Senthil S, Veerappan RM, Ramakrishna Rao M, Pugalendi KV. Oxidative stress and antioxidants in patients with cardiogenic shock complicating acute myocardial infarction. Clinica Chimica Acta. 2004;348(1-2):131–137. doi: 10.1016/j.cccn.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 173.Kasap S, Gönenç A, Şener DE, Hisar I. Serum cardiac markers in patients with acute myocardial infarction: oxidative stress, C-reactive protein and N-terminal probrain natriuretic peptide. Journal of Clinical Biochemistry and Nutrition. 2007;41(1):50–57. doi: 10.3164/jcbn.2007007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kijima Y, Hashimura K, Matsu-Ura Y, et al. Transcardiac 8-iso-prostaglandin F2α generation from acute myocardial infarction heart: insight into abrupt reperfusion and oxidant stress. Prostaglandins Leukotrienes and Essential Fatty Acids. 2001;64(3):161–166. doi: 10.1054/plef.2001.0256. [DOI] [PubMed] [Google Scholar]
- 175.Singh RB, Niaz MA, Sharma JP, Kumar R, Bishnoi I, Begom R. Plasma levels of antioxidant vitamins and oxidative stress in patients with acute myocardial infarction. Acta Cardiologica. 1994;49(5):441–452. [PubMed] [Google Scholar]
- 176.Kurtul N, Pence S, Akarsu E, Kocoglu H, Aksoy Y, Aksoy H. Adenosine deaminase activity in the serum of type 2 diabetic patients. Acta Medica. 2004;47:33–35. [PubMed] [Google Scholar]
- 177.Ghiselli A, Serafini M, Natella F, Scaccini C. Total antioxidant capacity as a tool to assess redox status: critical view and experimental data. Free Radical Biology and Medicine. 2000;29(11):1106–1114. doi: 10.1016/s0891-5849(00)00394-4. [DOI] [PubMed] [Google Scholar]
- 178.Patra RC, Swarup D, Dwivedi SK. Antioxidant effects of α tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology. 2001;162(2):81–88. doi: 10.1016/s0300-483x(01)00345-6. [DOI] [PubMed] [Google Scholar]
- 179.Scott MD, Lubin BH, Zuo L, Kuypers FA. Erythrocyte defense against hydrogen peroxide: preeminent importance of catalase. Journal of Laboratory and Clinical Medicine. 1991;118(1):7–16. [PubMed] [Google Scholar]
- 180.Anbarasi K, Vani G, Balakrishna K, Devi CSS. Effect of bacoside a on brain antioxidant status in cigarette smoke exposed rats. Life Sciences. 2006;78(12):1378–1384. doi: 10.1016/j.lfs.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 181.Kotrikadze N, Alibegashvili M, Zibribadze M, et al. Activity and content of antioxidant enzymes in prostate tumors. Experimental Oncology. 2008;30(3):244–247. [PubMed] [Google Scholar]
- 182.Salo DC, Pacifici RE, Lin SW, Giulivi C, Davies KJA. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. The Journal of Biological Chemistry. 1990;265(20):11919–11927. [PubMed] [Google Scholar]
- 183.Asahi M, Fujii J, Suzuki K, et al. Inactivation of glutathione peroxidase by nitric oxide. Implication for cytotoxicity. The Journal of Biological Chemistry. 1995;270(36):21035–21039. doi: 10.1074/jbc.270.36.21035. [DOI] [PubMed] [Google Scholar]
- 184.Seo HG, Takata I, Nakamura M, et al. Induction of nitric oxide synthase and concomitant suppression of superoxide dismutases in experimental colitis in rats. Archives of Biochemistry and Biophysics. 1995;324(1):41–47. doi: 10.1006/abbi.1995.9932. [DOI] [PubMed] [Google Scholar]
- 185.Serdar Z, Aslan K, Dirican M, Sarandöl E, Yeşilbursa D, Serdar A. Lipid and protein oxidation and antioxidant status in patients with angiographically proven coronary artery disease. Clinical Biochemistry. 2006;39(8):794–803. doi: 10.1016/j.clinbiochem.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 186.Ścibior D, Skrzycki M, Podsiad M, Czeczot H. Glutathione level and glutathione-dependent enzyme activities in blood serum of patients with gastrointestinal tract tumors. Clinical Biochemistry. 2008;41(10-11):852–858. doi: 10.1016/j.clinbiochem.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 187.Pietruszyńsk R, Markuszewski L, Masiarek K, Makowski M, Retelewska W, Watala C. Role of preprocedural glutathione concentrations in the prediction of major adverse cardiac events in patients with acute coronary syndrome treated with percutaneous coronary intervention. Polskie Archiwum Medycyny Wewnętrznej. 2013;123:228–237. doi: 10.20452/pamw.1728. [DOI] [PubMed] [Google Scholar]
- 188.Brennan M-L, Penn MS, Van Lente F, et al. Prognostic value of Myeloperoxidase in patients with chest pain. The New England Journal of Medicine. 2003;349(17):1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 189.Baldus S, Heeschen C, Meinertz T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108(12):1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 190.Vita JA, Brennan M-L, Gokce N, et al. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110(9):1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Baldus S, Rudolph V, Roiss M, et al. Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation. 2006;113(15):1871–1878. doi: 10.1161/CIRCULATIONAHA.105.590083. [DOI] [PubMed] [Google Scholar]
- 192.Mocatta TJ, Pilbrow AP, Cameron VA, et al. Plasma Concentrations of Myeloperoxidase predict mortality after myocardial infarction. Journal of the American College of Cardiology. 2007;49(20):1993–2000. doi: 10.1016/j.jacc.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 193.Morrow DA, de Lemos JA, Sabatine MS, Antman EM. The search for a biomarker of cardiac ischemia. Clinical Chemistry. 2003;49(4):537–539. doi: 10.1373/49.4.537. [DOI] [PubMed] [Google Scholar]
- 194.Sinha MK, Gaze DC, Tippins JR, Collinson PO, Kaski JC. Ischemia modified albumin is a sensitive marker of myocardial ischemia after percutaneous coronary intervention. Circulation. 2003;107(19):2403–2405. doi: 10.1161/01.CIR.0000072764.18315.6B. [DOI] [PubMed] [Google Scholar]
- 195.Sinha MK, Roy D, Gaze DC, Collinson PO, Kaski J-C. Role of “ischemia modified albumin”, a new biochemical marker of myocardial ischaemia, in the early diagnosis of acute coronary syndromes. Emergency Medicine Journal. 2004;21(1):29–34. doi: 10.1136/emj.2003.006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Roy D, Quiles J, Gaze DC, Collinson P, Kaski JC, Baxter GF. Role of reactive oxygen species on the formation of the novel diagnostic marker ischaemia modified albumin. Heart. 2006;92(1):113–114. doi: 10.1136/hrt.2004.049643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lecour S, Lamont KT. Natural polyphenols and cardioprotection. Mini Reviews in Medicinal Chemistry. 2011;11(14):1191–1199. doi: 10.2174/13895575111091191. [DOI] [PubMed] [Google Scholar]
- 198.Sun G-B, Sun X, Wang M, et al. Oxidative stress suppression by luteolin-induced heme oxygenase-1 expression. Toxicology and Applied Pharmacology. 2012;265(2):229–240. doi: 10.1016/j.taap.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 199.Sun D, Huang J, Zhang Z, et al. Luteolin limits infarct size and improves cardiac function after myocardium ischemia/reperfusion injury in diabetic rats. PLoS ONE. 2012;7(3) doi: 10.1371/journal.pone.0033491.e33491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xu T, Li D, Jiang D. Targeting cell signaling and apoptotic pathways by luteolin: cardioprotective role in rat cardiomyocytes following ischemia/reperfusion. Nutrients. 2012;4(12):2008–2019. doi: 10.3390/nu4122008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Iuliano L, Praticò D, Greco C, et al. Angioplasty increases coronary sinus F2-isoprostane formation: evidence in vivo oxidative stress during PTCA. Journal of the American College of Cardiology. 2001;37(1):76–80. doi: 10.1016/s0735-1097(00)01040-8. [DOI] [PubMed] [Google Scholar]
- 202.Collins CE, Quaggiotto P, Wood L, O’Loughlin EV, Henry RL, Garg ML. Elevated plasma levels of F2α isoprostane in cystic fibrosis. Lipids. 1999;34(6):551–556. doi: 10.1007/s11745-999-0397-1. [DOI] [PubMed] [Google Scholar]