

Summary
microRNAs (miRNAs) are typically generated as ~22-nucleotide double-stranded RNAs via processing of precursor hairpins by the RNase III enzyme Dicer, after which they are loaded into Argonaute (Ago) proteins to form RNA-induced silencing complex (RISC). However, the biogenesis of miR-451, an erythropoietic miRNA conserved in vertebrates, occurs independently of Dicer and instead requires cleavage of the 3′ arm of the pre-miR-451 precursor hairpin by Ago2. The 3′ end of the Ago2-cleaved pre-miR-451 intermediate is then trimmed to the mature length by an unknown nuclease. Here, using a classical chromatographic approach, we identified poly(A)-specific ribonuclease (PARN) as the enzyme responsible for the 3′-to-5′ exonucleolytic trimming of Ago2-cleaved pre-miR-451. Surprisingly, our data show that trimming of Ago2-cleaved pre-miRNAs is not essential for target silencing, indicating that RISC is functional with miRNAs longer than the mature length. Our findings define the maturation step in the miRNA biogenesis pathway that depends on Ago2-mediated cleavage.
Introduction
MicroRNAs (miRNAs) are endogenous small non-coding RNAs, which regulate gene expression in diverse biological processes. miRNAs and Argonaute (Ago) proteins form the effector complex, known as RNA-induced silencing complex (RISC), and silence their complementary target mRNAs. Among four Ago proteins (Ago1–4) in mammals, only Ago2 has the endonucleolytic ‘slicer’ activity (Liu et al., 2004; Meister et al., 2004), while all four are capable of repressing translation or promoting deadenylation and decay of target mRNAs. In the canonical biogenesis, miRNAs are produced as double-stranded RNAs via sequential cleavage of their precursors by two RNase III enzymes, Drosha and Dicer; primary miRNA (pri-miRNA) transcripts are first cropped by Drosha in the nucleus, and the resultant 60–70-nt precursor miRNA (pre-miRNA) hairpins are then diced by Dicer into ~22-nt miRNA/miRNA* duplexes in the cytoplasm, after which they are loaded into Ago proteins to form RISC (Carthew and Sontheimer, 2009; Ghildiyal and Zamore, 2009; Kim et al., 2009). However, several miRNA classes bypass the requirement of these RNase III enzymes. For example, intron-derived miRNAs called mirtrons (Okamura et al., 2007; Ruby et al., 2007), snoRNA-derived miRNAs (Brameier et al., 2011; Ender et al., 2008; Glazov et al., 2009; Taft et al., 2009), tRNA-derived miRNAs (Cole et al., 2009; Haussecker et al., 2010), RNaseZ-mediated (Bogerd et al., 2010) and Integrator complex-mediated viral miRNAs (Cazalla et al., 2011) are all produced in Drosha-independent manners. In contrast, the biogenesis of miR-451, a highly conserved erythropoietic miRNA in vertebrates, requires Drosha but bypasses Dicer (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). miR-451 is transcribed together with miR-144 from a bicistronic locus (Altuvia et al., 2005; Nelson et al., 2007) and processed by Drosha into an unusually short, 41–42-nt pre-miR-451 hairpin with a highly structured 17-nt stem region and 4-nt terminal loop. Pre-miR-451 is too short to be cleaved by Dicer, and is directly loaded into Ago proteins. Unlike canonical miRNA/miRNA* duplexes, maturation of miR-451 requires the slicer activity of Ago2; it has been shown that, in zebrafish, mice and humans, Ago2 cleaves the 3′ arm of pre-miR-451 by its slicer activity and yields a 30-nt intermediate RNA called Ago2-cleaved pre-miR-451 (ac-pre-miR451), whose 3′ end is then trimmed to generate mature, ~23-nt miR-451 (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). However, the ribonuclease for this trimming reaction remains unknown. In this study, by using a classical chromatographic approach, we identified poly(A)-specific ribonuclease (PARN) as the 3′ trimming enzyme required for maturation of Ago2-cleaved pre-miRNAs. Surprisingly, our in vitro and in vivo data show that trimming of miR-451 is not essential for efficiency and fidelity of target silencing, indicating that RISC is functional with guide RNAs longer than the mature length. Our results have uncovered the machinery responsible for trimming of Ago2-cleaved pre-miRNAs, providing a molecular mechanism for this unique form of Dicer-independent miRNA biogenesis.
Results
Recapitulation of pre-miR-451 maturation in vitro
To dissect the trimming mechanism of pre-miR-451, we developed an in vitro pre-miR-451 maturation assay. In this assay, 5′ 32P-radiolabeled pre-miR-451 (Figure 1A) was first loaded into FLAG-tagged Ago2 immobilized on beads. After washing, the Ago2:pre-miR-451 complex was incubated in K562 human leukemia cell lysate. Ago2-mediated nicking of the 3′ arm produced a 30-nt long, Ago2-cleaved pre-miR-451 (ac-pre-miR-451) (Figure 1B, time = 0), which was progressively trimmed into 23-nt mature miR-451 in K562 lysate (Figure 1B, time = 0.25–22). Trimming was recapitulated when the reaction was started with pre-nicked ac-pre-miR-451 (Figure 1A and C). As previously reported (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010), catalytic mutant Ago2 as well as slicer-deficient Ago1, 3, and 4 could load, but failed to nick and process pre-miR-451 (Figure S1A). In contrast, pre-nicked ac-pre-miR-451 loaded into these catalytically inactive Ago proteins was efficiently trimmed into mature miR-451 in K562 lysate (Figure S1B). Thus, the trimming reaction is not specific to Ago2 protein, but rather requires nicking by Ago2.
Figure 1. In vitro recapitulation of the miR-451 trimming reaction.
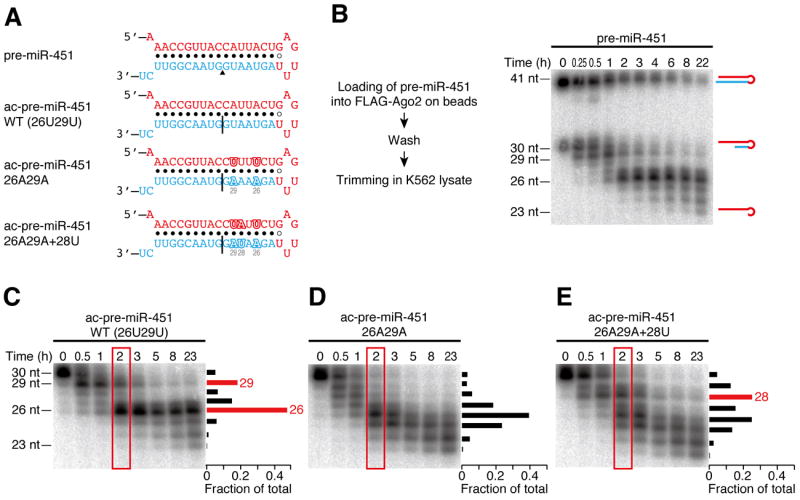
(A) The sequences and structures of pre-miR-451 and its derivatives used in B–E. The miR-451 guide strand is shown in red. A black triangle indicates the Ago2-cleavage site in pre-miR-451. Black lines show the nicks in ac-pre-miRNAs.
(B) Trimming of Ago2-loaded pre-miR-451 in vitro. The experimental scheme is shown on the left. Ago2-loaded pre-miR-451 was trimmed into mature miR-451 in K562 lysate. See also Figure S1.
(C–E) The trimming reaction prefers adenosine to uridine. (C) Trimming of wild-type ac-pre-miR-451 showed prominent accumulation of 29-nt and 26-nt intermediates bearing uridine at their 3′ ends. (D) Accumulation of the two intermediates was lost when the two uridines at positions 26 and 29 were substituted with adenosines. (E) A new 28-nt intermediate emerged by ectopic introduction of uridine at position 28. The signal intensities of the bands at 2 h (in red rectangles) are quantified and plotted on the right of each gel.
Trimming is an Mg2+-dependent 3′-to-5′ exonucleolytic reaction
Trimming may be catalyzed either endonucleolytically or exonucleolytically. We reasoned that a modification within the RNA region to be trimmed would block further exonucleolytic processing, but this would not affect endonucleolytic processing at unmodified positions. When we introduced a racemic phosphorothioate linkage between 28 and 29 of ac-pre-miR-451 or a 2′-O-methyl group at position 28, a 29-nt intermediate was accumulated and further trimming was suppressed in vitro (Figure S1C). Similarly, injection of a modified pre-miR-451 bearing a phosphorothioate linkage between positions 26 and 27 in zebrafish embryos resulted in accumulation of a 27-nt intermediate in vivo (Figure S1D). These results are consistent with 3′-5′ exonucleolytic trimming of ac-pre-miR-451 after Ago2-mediated cleavage.
Many nucleases utilize a metal ion as a cofactor for their activity. Trimming of pre-miR-451 was blocked in the presence of 5 mM EDTA but was recovered by the addition of 10 mM Mg2+ (Figure S1E). In contrast, 10 mM of EGTA, which chelates Ca2+, did not affect trimming (Figure S1E). Together, these data indicate that the pre-miR-451 trimming is an Mg2+-dependent 3′-to-5′ exonucleolytic reaction.
The trimming reaction shows a nucleotide preference
The kinetics of trimming in K562 lysate was not continuous; two predominant intermediates of 29-nt and 26-nt in length were accumulated before 23-nt mature miR-451 was produced (Figure 1B and C). This specific pattern was reminiscent of the size distribution of miR-451 in vivo (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). Interestingly, both intermediates have uridine at their 3′ ends, implying that the trimming reaction might prefer specific nucleotides. To test this idea, we substituted the two uridines at positions 26 and 29 of ac-pre-miR-451 with adenosines (Figure 1A, ac-pre-miR-451 26A29A), while the facing positions (12 and 15) were substituted to uridines to maintain the secondary structure. Compared to the wild-type ac-pre-miR-451, the marked accumulation of the two intermediates was lost in ac-pre-miR-451 26A29A (Figure 1D). Ectopic introduction of uridine at position 28 (Figure 1A, ac-pre-miR-451 26A29A+28U) resulted in the emergence of a new 28-nt intermediate (Figure 1E). Therefore, the trimming reaction prefers adenosine to uridine.
It has been suggested that, in vivo, Ago2-processed ac-pre-miR-451 is subject to oligouridylation at the 3′ end before it is trimmed (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). However, the impact of oligouridylation on the trimming remains unknown. Given that the trimming reaction disfavors uridines, oligouridylation of ac-per-miR-451 may negatively regulate the maturation process of miR-451. We prepared ac-pre-miR-451 derivatives with 1–3 uridines at the 3′ end (Figure S1F; ac-pre-miR-451+1U–3U), and monitored their trimming in K562 lysate. Contrary to our initial expectation, addition of 1–3 uridines did not detectably affect the kinetics of the trimming reaction (Figure S1F). We reason that the trimming exonuclease can process the dangling, single-stranded oligouridines at the 3′ end much more efficiently than the double-stranded stem region in ac-pre-miR-451. We concluded that oligouridylation of ac-pre-miR-451 is independent of the maturation of miR-451.
PARN, poly(A)-specific ribonuclease trims Ago2-cleaved pre-miR-451
We next attempted to identify the trimming enzyme using classical column chromatography (Figure 2A). After an eight-step purification, protein bands that showed close correlation with the trimming activity (Figure 2B) were analyzed by mass spectrometry. Throughout the purification steps, the trimming activity was eluted as single peaks, implying that there is one enzyme that is predominantly responsible for the activity. Among the mass spectrometry hits after the final chromatography, we found poly(A)-specific ribonuclease (PARN), a 73 kDa protein. PARN is widely conserved in many eukaryotes and is implicated in poly(A) tail length control and mRNA decay in both the nucleus and the cytoplasm (Goldstrohm and Wickens, 2008; Weill et al., 2012). Biochemical studies have shown that PARN has a preference for single-stranded poly(A) (Astrom et al., 1991; Astrom et al., 1992; Korner and Wahle, 1997; Korner et al., 1998; Martinez et al., 2000), and requires a divalent cation, particularly Mg2+, for its activity (Ren et al., 2002; Ren et al., 2004). These properties of PARN are consistent with the pre-miR-451 trimming reaction we observed (Figure 1 and S1).
Figure 2. Identification of PARN, poly(A)-specific ribonuclease, as the pre-miR-451 trimming enzyme.
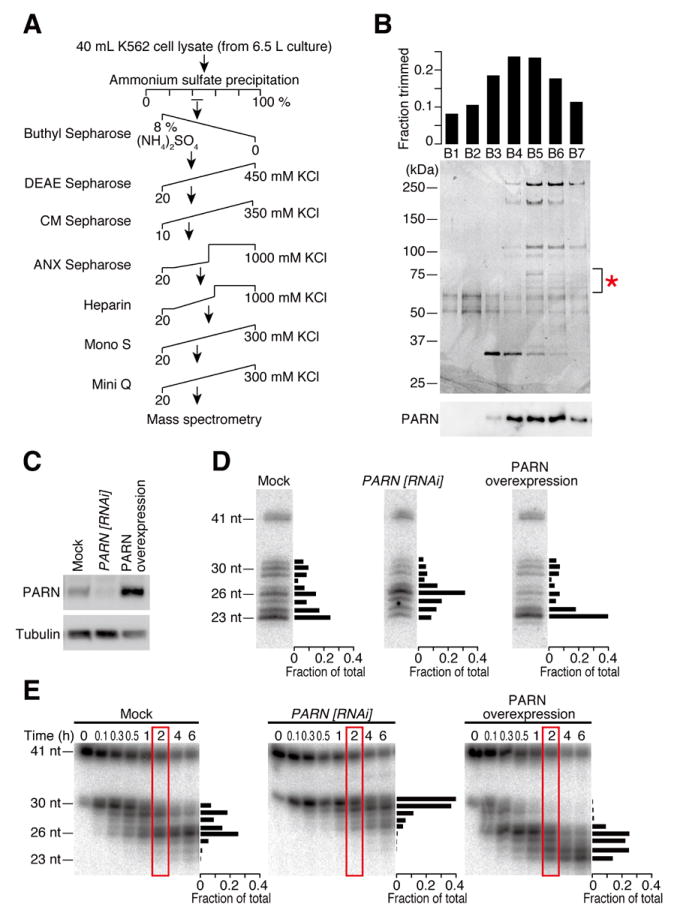
(A) Scheme of the chromatographic steps for purification of PARN.
(B) The profile of the final Mini Q purification. Each fraction was assayed for the trimming activity of ac-pre-miR-451 in vitro (top) and the proteins were separated by SDS-PAGE (middle). Protein bands that showed close correlation with the trimming activity were analyzed by mass spectrometry (indicated by *), among which PARN was found. Bottom, anti-PARN Western blotting.
(C) Anti-PARN Western blotting analysis of PARN knockdown or overexpression.
(D) Effect of PARN on pre-miR-451 trimming in HeLa S3 cells. Upon depletion or overexpression of PARN, a plasmid that expresses a miR-144/451 primary transcript was transfected and miR-451 was detected by Northern blotting. The signal intensities of the bands at 2 h are quantified and plotted on the right of each gel. PARN depletion impaired pre-miR-451 trimming while PARN overexpression promoted it. See also Figure S2 for miR-144 and endogenous let-7 controls.
(E) Effect of PARN on pre-miR-451 trimming in vitro. Lysates from PARN-depleted or PARN-overexpressing cells were tested for trimming. The signal intensities of the bands at 2 h (in red rectangles) are quantified and plotted on the right of each gel.
To confirm that PARN is the enzyme required for pre-miR-451 trimming, we depleted endogenous PARN by RNAi or overexpressed PARN in HeLa S3 cells (Figure 2C), and transfected a plasmid expressing the primary transcript of miR-144/miR-451 cluster. The ectopic expression of miR-144/miR-451 in HeLa cells has been previously used to study Dicer independence and structural requirements of miR-451 biogenesis (Yang et al., 2010). Northern analysis showed that depletion of PARN markedly decreased 23-nt mature miR-451, instead accumulating the 26-nt intermediate (Figure 2D, middle). In contrast, overexpression of PARN reduced the fraction of the intermediates and predominantly produced mature miR-451 (Figure 2D, right). The lengths of miR-144 and endogenous let-7 were unaffected by depletion or overexpression of PARN (Figure S2). Next, we prepared lysates from PARN-knockdown cells or PARN-overexpressing cells, and performed the in vitro trimming assay. Trimming was extremely slow in PARN-knockdown lysate, while 23-nt mature miR-451 was much more rapidly produced in PARN-overexpressing lysate when compared to the mock lysate (Figure 2E). Moreover, purified recombinant PARN from E. coli (Figure 3A) faithfully recapitulated trimming of Ago2-loaded pre-miR-451 (Figure 3B), with the same nucleotide specificity as observed in cell lysate (Figure 1C) and in cultured cells (Figure 2D). In contrast, mutating a catalytic residue of PARN (E30A, Figure 3A) blocked the trimming activity (Figure 3C). Much as in lysate, recombinant PARN could trim Ago2-cleaved ac-pre-miR-451 as well as pre-nicked ac-pre-miR-451 in slicer-deficient Ago proteins (Figure S3A and B). Based on these results, we concluded that PARN is the enzyme responsible for trimming and maturation of Ago2-cleaved pre-miR-451.
Figure 3. Recombinant PARN faithfully trims pre-miR-451.
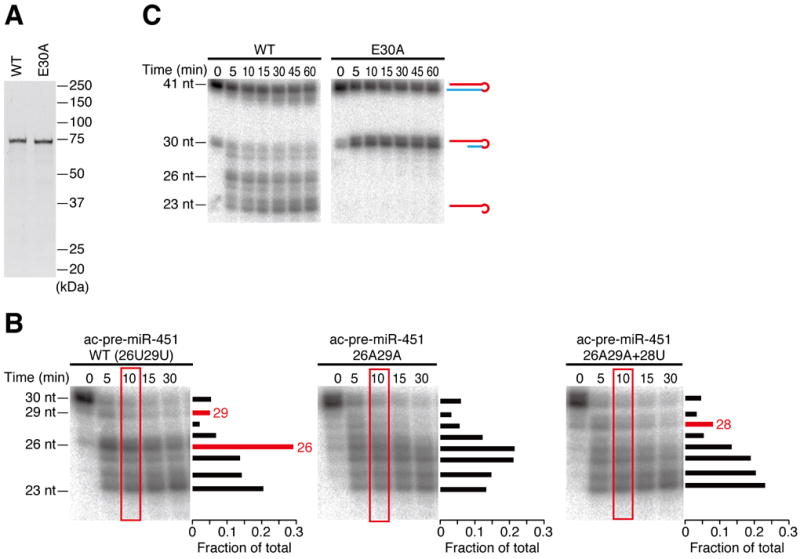
(A) Recombinant wild-type (WT) or catalytic mutant (E30A) PARN expressed and purified from E. coli.
(B) Recombinant PARN showed the same nucleotide specificity as in K562 lysate (Figure 1C–E). The signal intensities of the bands at 10 min (in red rectangles) are quantified and plotted on the right of each gel.
(C) Mutation of a catalytic residue (E30A) blocked the trimming activity. See also Figure S3.
Trimming of Ago2-cleaved pre-miRNA is dispensable for target silencing in vitro and in cultured cells
Given that miR-451 predominantly exists as 23–26-nt trimmed forms rather than the 30-nt untrimmed form in vivo (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010), it seemed plausible that trimming of pre-miR-451 is required for target silencing. To test this idea, we first compared the target cleavage efficiency in vitro with or without the trimming reaction. Ac-pre-miR-451 was loaded into FLAG-Ago2 and incubated in lysate for trimming or in buffer alone as a mock (Figure 4A). Contrary to expectation, cleavage of a target RNA perfectly complementary to miR-451 was comparable with or without trimming (Figure 4B), suggesting that trimming does not affect the target cleavage activity of Ago2-RISC. Next we sought to address the requirement of trimming by reporter assays in cultured cells. Because knockdown of PARN severely inhibited cell growth and reporter expression (data not shown), we introduced three successive phosphorothioate linkages at positions 27–30 in pre-miR-451 (pre-miR-451 3×PS) to block trimming in cells. We prepared Renilla luciferase reporters harboring three different types of target sites for miR-451 (Figure 4C): a single perfectly complementary, cleavable target site; two centrally mismatched, non-cleavable target sites; and the 3′ UTR sequence of CAB39, a previously identified endogenous mRNA bearing a seed-matching target site (Godlewski et al., 2010). These reporters and a control firefly luciferase reporter were co-transfected with pre-miR-451 or pre-miR-451 3×PS, and the luminescence was monitored after 24 h. At the same time, Ago2-bound small RNAs were immunopurified and the lengths of miR-451 were examined by Northern blotting. Whereas three phosphorothioate linkages hindered miR-451 maturation (Figure 4D), silencing efficiency of the reporter expression was unaltered by the phosphorothioate modification for any of the three reporter constructs (Figure 4E). These data together suggest that trimming of Ago2-cleaved pre-miR-451 is dispensable for target silencing in vitro and in cultured cells.
Figure 4. Trimming is dispensable for target silencing in vitro and in cultured cells.
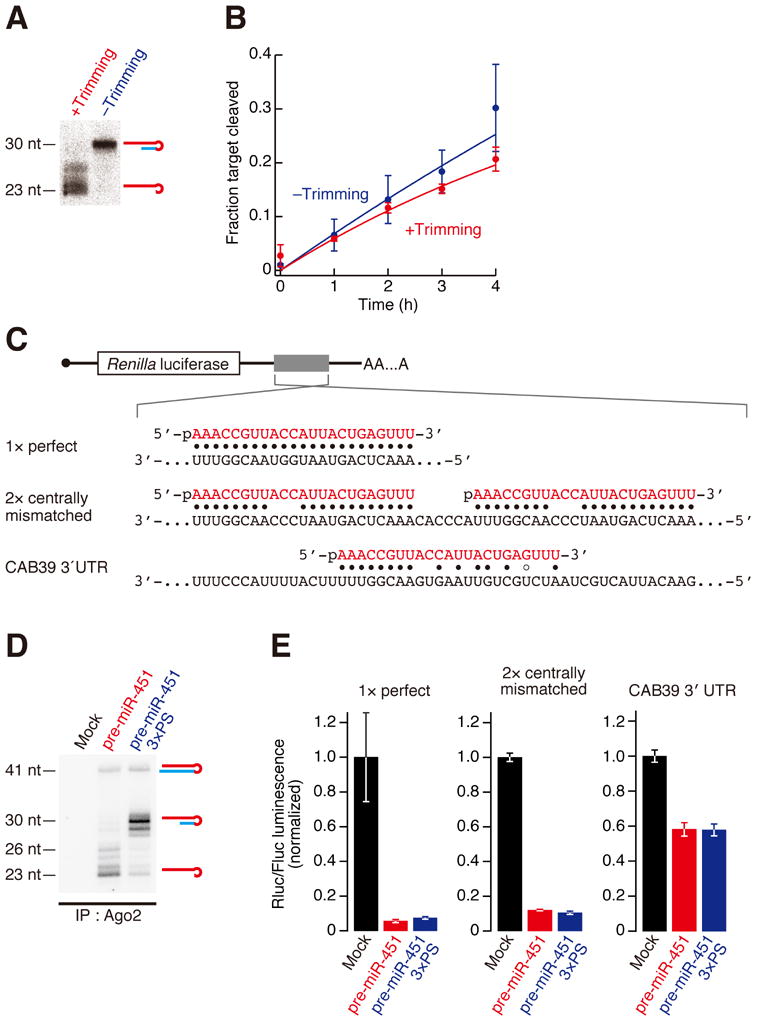
(A) FLAG-Ago2 loaded with ac-pre-miR-451 was incubated with PARN-overexpressing HeLa S3 lysate (+Trimming) or buffer alone (−Trimming) at 37 °C for 3 h. Trimming was confirmed by Northern blotting
(B) Target cleavage was comparable with or without trimming. The graph shows means and standard deviations from three independent cleavage reactions.
(C) Schematic representation of the Renilla luciferase reporters used in Figure 4E for cultured cells.
(D) Inhibition of trimming by three phosphorothioate linkages in HeLa S3 cells. Pre-miR-451 or pre-miR-451 3×PS was transfected and Ago2-bound RNA was immunopurified and analyzed by Northern blotting.
(E) Renilla luciferase reporter assay for 1× perfect target site, 2× centrally mismatched target sites or CAB39 3′ UTR containing an endogenous miR-451 target site by pre-miR-451 with or without 3×PS. Firefly luciferase served as a control. The Rluc/Fluc luminescence was normalized to the value of mock transfection. The mean values ± standard deviations from three independent experiments are shown. Trimming was dispensable for target silencing in cell culture.
Trimming of Ago2-cleaved pre-miRNA is dispensable for target silencing in vivo
We next asked if trimming is required for miRNA-mediated silencing in vivo using zebrafish embryos (Cifuentes et al., 2010). As in cultured cells, morpholino-mediated knockdown of PARN in zebrafish embryos caused severe defects in early development and reporter expression (data not shown). Therefore, we utilized phosphorothioate linkages to block trimming. We assayed the activity of a previously described pre-miR-430-derived hairpin RNA that mimics the secondary structure of pre-miR-451 (pre-miR-430Ago2 hairpin) (Figure 5A) (Cifuentes et al., 2010). Like pre-miR-451, this chimeric pre-miRNA is matured by Ago2-mediated cleavage and 3′-end trimming, but the trimming is strongly inhibited by the inclusion of three phosphorothioate linkages at positions 27–30 (pre-miR-430Ago2 hairpin 3×PS) (Figure 5B). When injected into embryos, pre-miR-430Ago2 hairpin 3×PS was able to repress a GFP reporter bearing three imperfect target sites for miR-430 (Figure 5C), with similar efficiencies as the unmodified pre-miR-430Ago2 hairpin (Figure 5D). This was also the case with a firefly luciferase reporter with three imperfect target sites (Figure 5C and E). These results suggest that trimming is not required for target silencing in zebrafish embryos. To further test the requirement of trimming for miRNA functionality, we utilized mutant zebrafish embryos deficient for maternal and zygotic Dicer activity (MZdicer). MZdicer embryos, in which miR-430-mediated clearance of several hundred maternal transcripts is disrupted, show defects during gastrulation and brain morphogenesis (Giraldez et al., 2005). These defects can be rescued by miR-430 duplex or Dicer-independent pre-miR-430Ago2 hairpin (Cifuentes et al., 2010). Strikingly, injecting the trimming-defective pre-miR-430Ago2 hairpin 3×PS rescued the brain morphogenesis defect in MZdicer, similarly to miR-430 duplex or pre-miR-430Ago2 hairpin (Figure 5F and S4).
Figure 5. Trimming is dispensable for target silencing in vivo.
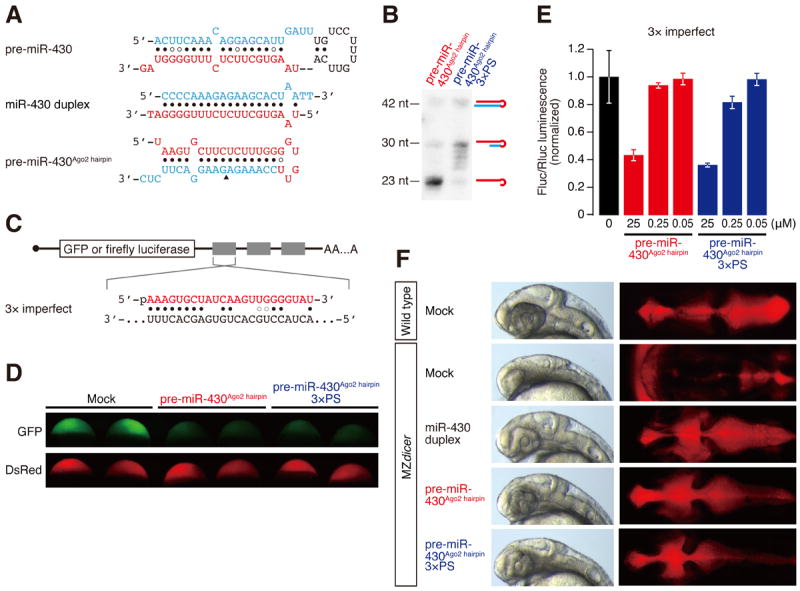
(A) The sequences and structures of pre-miR-430, miR-430 duplex and pre-miR-430Ago2 hairpin used in zebrafish experiments.
(B) Inhibition of trimming by three phosphorothioate linkages in zebrafish embryos. Pre-miR-430Ago2 hairpin with or without 3×PS was injected and RNAs were analyzed by Northern blotting.
(C) Schematic representation of the reporter mRNAs with 3× target sites used in D and E.
(D and E) Reporter assays for the 3× imperfect target sites by pre-miR-430Ago2 hairpin with or without 3×PS. (D) GFP reporter with DsRed control. (E) Firefly luciferase reporter with Renilla luciferase control. The Fluc/Rluc luminescence was normalized to the value of mock injection. The mean values ± standard deviations from three independent experiments are shown. Trimming was dispensable for reporter silencing in zebrafish embryos.
(F) Trimming is not required to rescue the brain morphologic defects in MZdicer zebrafish embryos. MZdicer embryos were injected miR-430 duplex, pre-miR-430Ago2 hairpin or pre-miR-430Ago2 hairpin 3×PS and analyzed at 30 hpf. See also Figure S4.
To gain quantitative insight in the broad consequences of trimming on miRNA activity and target selection, we analyzed the gene expression of the wild-type, MZdicer mutant and rescued MZdicer embryos by high-throughput sequencing (Table S1). miR-430 target mRNAs were significantly stabilized in MZdicer mutant compared to wild-type embryos at 6 hours post fertilization (hpf) (3.38 median fold change between MZdicer and wild type; P = 1.0 × 10-21, Wilcoxon signed-rank test) (Figure 6A). This expression difference was largely rescued upon miR-430 duplex injection (2.40 median fold change in duplex injected compared to uninjected MZdicer, P = 9.6 × 10-22) (Figure 6A). Despite similar capacity to rescue the developmental defects in MZdicer by 30 hpf (Figure 5F and S4), pre-miR-430Ago2 hairpin and pre-miR-430Ago2 hairpin 3×PS repressed miR-430 targets less efficiently than miR-430 duplex at 6 hpf, assayed by both the strength of the regulation (1.39 median fold change, P = 1.4 × 10-18; 1.27 median fold change, P = 2.3 × 10-20 respectively compared to MZdicer) (Figure 6A) and the number of target genes down-regulated >2-fold (Figure 6B). This difference in silencing activity could presumably be attributed to different processing kinetics between the miRNA duplex and the hairpin RNAs to accumulate active miRNA species. Importantly, while the number of miR-430 targets down-regulated >2-fold by pre-miR-430Ago2 hairpin was slightly higher than pre-miR-430Ago2 hairpin 3×PS (27 vs. 19; Figure 6B), the difference in the global distribution of targets was only minimal (1.05 median fold change, P = 4.6 × 10-4; Figure 6A), indicating that the lack of trimming does not affect overall silencing of canonical miR-430 targets. Next, we analyzed if the untrimmed 7-nt tail at the 3′ end of pre-miR-430Ago2 hairpin 3×PS may influence the fidelity of target mRNA selection by additional base paring. Out of 10,841 genes analyzed, there were only 17 and 3 genes that were >2-fold over-repressed by pre-miR-430Ago2 hairpin and pre-miR-430Ago2 hairpin 3×PS, respectively, compared to the levels observed in miR-430 duplex-injected embryos (Figure 6C). This result suggests that the additional nucleotides at the untrimmed 3′ end have a negligible effect on target mRNA selection. Taken together, we concluded that trimming of pre-miR-430Ago2 hairpin is dispensable for regulating miRNA targets and rescuing the physiological function of miR-430 in vivo.
Figure 6. Trimming does not affect efficiency and fidelity of endogenous target silencing.
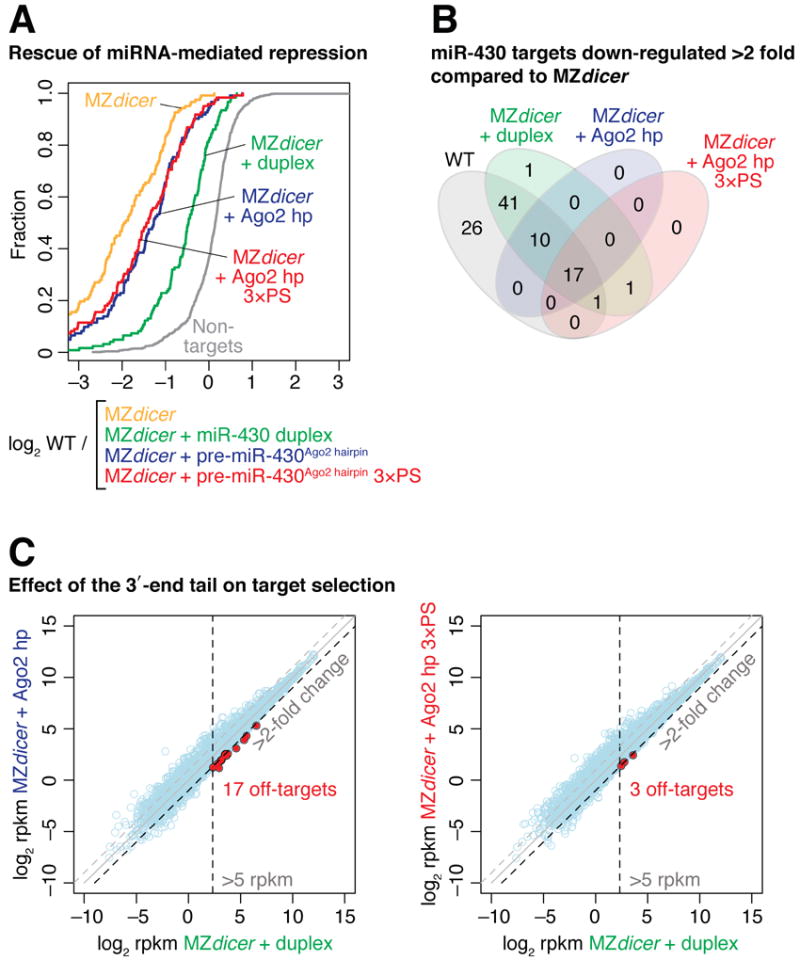
(A) pre-miR-430Ago2 hairpin is able to repress endogenous target genes independently of trimming. Cumulative distributions of fold changes between wild-type embryos and uninjected or injected MZdicer embryos for 122 validated miR-430 targets are shown. The cumulative distribution of non-target genes in wild type / MZdicer is plotted as a reference (grey line). See also Table S1.
(B) Venn diagram showing the number of miR-430 targets that are repressed in wild-type embryos or in MZdicer embryos injected with miR-430 duplex, pre-miR-430Ago2 hairpin or pre-miR-430Ago2 hairpin 3×PS compared to uninjected MZdicer embryos.
(C) Trimming has a negligible effect on target selection. Genes that are expressed >5 rpkm and over-repressed >2-fold by pre-miR-430Ago2 hairpin or pre-miR-430Ago2 hairpin 3×PS compared to miR-430 duplex are shown in red.
Discussion
Here, by using classical column chromatography, we identified PARN, a Mg2+-dependent poly(A)-specific 3′-to-5′ exonuclease, as the enzyme responsible for 3′-end trimming and maturation of Ago2-cleaved pre-miR-451 (Figure 7). Knockdown of PARN inhibited the trimming reaction, while its overexpression enhanced miR-451 maturation (Figure 2C–E). The biochemical characteristics of PARN including its nucleotide preference (Figure 3) were fully consistent with the properties of the pre-miR-451 maturation process, observed in vitro (Figure 1), in cultured cells (Yang et al., 2010), in zebrafish (Cifuentes et al., 2010) and in mice (Cheloufi et al., 2010). Our findings define the maturation step in the Ago2-dependent miRNA biogenesis pathway conserved in vertebrates (Figure 7).
Figure 7. A model for biogenesis and function of Ago2-cleaved pre-miRNAs.
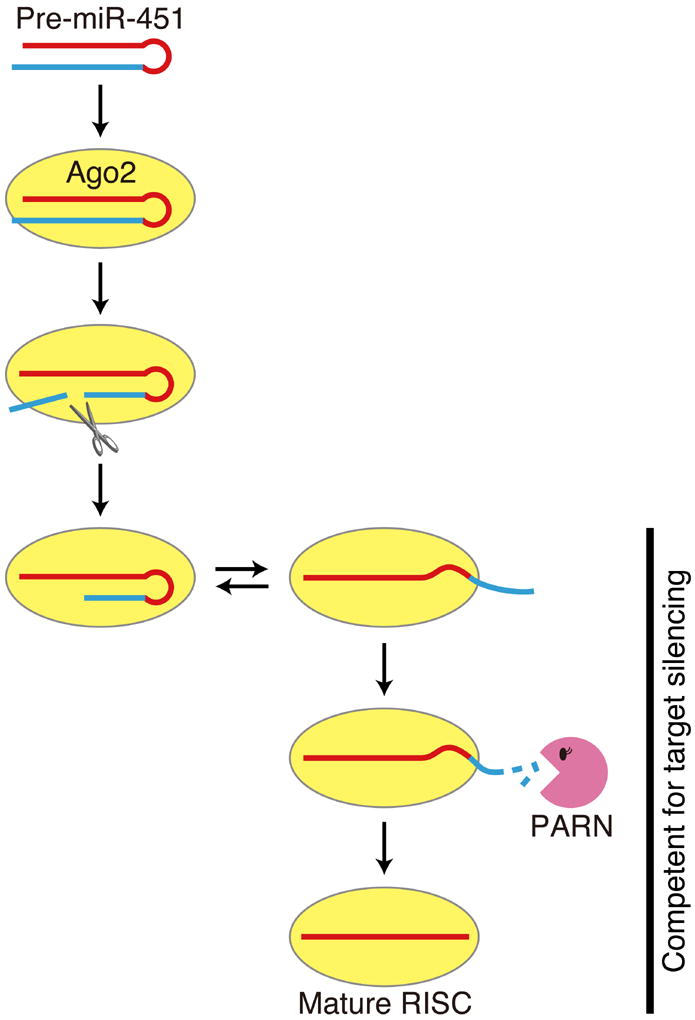
Surprisingly, our in vitro and in vivo results (Figure 4-6) together show that 3′ trimming of Ago2-cleaved pre-miRNAs is not essential for target silencing (Figure 7). Although our data do not exclude the possibility that trimming plays a role in modulating the activity of miR-451 (e.g., long-term turnover of RISC), pre-miR-430Ago2 hairpin was able to rescue the morphologic defects and restore the genome-wide regulation of target mRNAs in MZdicer zebrafish embryos regardless of the 3′ trimming, achieving physiological function in vivo (Figure 5F and 6).
Recent reports have shown that a 3′-to-5′ exonuclease, Nibbler, trims the 3′ ends of miRNAs in Drosophila (Han et al., 2011; Liu et al., 2011). For a subset of fly miRNAs, Dicer-1 processing generates miRNA/miRNA* duplexes that are slightly longer than the mature lengths. These intermediate duplexes are loaded into Ago1, the miRNA* strands are ejected, and then Nibbler shapes the 3′ ends of the Ago1-bound miRNA strands by trimming a few nucleotides (Han et al., 2011). On the other hand, in the mirtron pathway, the RNA exosome including Rrp6, another 3′-to-5′ exonuclease, is required for the removal of the 3′ tail of the pre-miRNA-like hairpin after splicing and debranching (Flynt et al., 2010). In mammals, yet another 3′-to-5′ exonuclease DIS3L2 acts to degrade pre-let-7 when its 3′ end is oligouridylated via LIN-28 binding (Chang et al., 2013). Future studies will be needed to understand why so many different exonucleases act at different steps in the diverse miRNA biogenesis pathways.
Experimental Procedures
General methods
Preparation of lysis buffer (30 mM HEPES-KOH [pH 7.4], 100 mM KOAc, 2 mM Mg(OAc)2), 2× urea loading buffer and 2× PK buffer, and RNA radiolabeling with T4 polynucleotide kinase has been previously described in detail (Haley and Zamore, 2004).
Cell culture
K562 cells were cultured in RPMI 1640 medium and HeLa S3 cells and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin, at 37 °C in 5% CO2.
Cell lysate preparation
Cells were washed in PBS (pH 7.4) and collected by centrifugation. The cell pellets were resuspended in 2 pellet volume of lysis buffer containing 5 mM DTT and 1× Complete EDTA-free protease inhibitor tablets (Roche), and homogenized by Douncing. For preparation of K562 cell lysate for column chromatography, we used low-salt lysis buffer (30 mM HEPES-KOH [pH 7.4], 20 mM KCl, 2 mM Mg(OAc)2) containing 5 mM DTT and 1× Complete EDTA-free protease inhibitor tablets. Lysate was clarified by centrifugation at 17,000 × g for 20 min at 4 °C. The supernatant was flash frozen in liquid nitrogen and stored at −80 °C.
In vitro trimming assay
Synthetic RNA oligonucleotides were purchased from Gene Design and IDT. Pre-miR-451 and the series of its derivatives were denatured for 2 min at 95 °C and annealed for ~1 h at room temperature. Typically for 1 reaction, 10 μl of lysate from HEK 293T cells expressing FLAG-tagged Ago2 was incubated with 5 μl of Dynabeads protein G paramagnetic beads (Life Technologies) and 0.5 μl of anti-FLAG M2 antibody (Sigma) for 1 h at 4 °C. Beads were then incubated with 10 nM 5′ 32P-radiolabeled pre-miRNAs in lysis buffer for 15 min at 25 °C and then washed three times with wash buffer (2 M NaCl, 10% glycerol and 0.1% Triton in lysis buffer) and rinsed three times with lysis buffer. Immunopurified Ago2 complexes containing radiolabeled pre-miRNAs were incubated with 40 μl of lysate or 30 μl of ~800 nM recombinant PARN. RNAs were extracted from the precipitate by Proteinase K treatment in 2× PK buffer, concentrated by ethanol precipitation, and separated by electrophoresis on 15% denaturing PAGE. The radioactive bands were detected by FLA-7000 image analyzer (Fujifilm Life Sciences) and quantified by ImageGauge software (Fujifilm Life Sciences).
Purification of PARN by column chromatography
All purification steps were performed at 4 °C. Forty mL lysate from 6.5 L culture of K562 cells was precipitated with 40% ammonium sulfate. After centrifugation at 8,000 × g for 30 min, the precipitate was resuspended in 20 ml of buffer A (30 mM HEPES-KOH [pH 6.5], 20 mM KCl, 2 mM Mg(OAc)2, 5% glycerol, 0.1% NP-40, 5 mM DTT) with 8% (NH4)2SO4, and the precipitate was removed by centrifugation at 8,000 × g for 30 min. After filtration, the supernatant was loaded onto a 20 ml HiPrep Buthyl column and was eluted by a 8–0% linear gradient of (NH4)2SO4 in buffer A. The fractions were assayed for in vitro pre-miR-451 trimming and the peak activity was detected at ~5% (NH4)2SO4. The active fractions were pooled and dialyzed with buffer A, and loaded onto a 5 ml HiTrap DEAE column and eluted with a liner gradient of 20–450 mM KCl in buffer B (30 mM HEPES-KOH [pH 6.5], 2 mM Mg(OAc)2, 5% glycerol, 0.1% NP-40, 5 mM DTT). The fractions of the peak activity were diluted with dilution buffer (30 mM HEPES-KOH [pH 6.5], 2 mM Mg(OAc)2, 5% glycerol, 0.1% NP-40, 5 mM DTT) to adjust the KCl concentration to 20 mM. Subsequently, the active sample was applied to a 1 mL HiTrap CM column and eluted with a liner gradient of 10–350 mM KCl in buffer B. Further purification was similarly performed by using HiTrap ANX column (20–1000 mM KCl), HiTrap Heparin column (20–1000 mM KCl), Mono S column (20–300 mM KCl), and Mini Q column (20–300 mM KCl). The active fractions of the last Mini Q purification were precipitated by trichloroacetic acid and the proteins were separated by SDS-PAGE. All columns were purchased from GE Healthcare Life Sciences.
Mass spectrometry Analysis
Proteins separated by SDS-PAGE were stained with GelCode Blue Safe (Pierce) and the bands of interest were cut out from the gel, followed by in-gel digestion with trypsin. The peptide mixtures were analyzed by a ion trap mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific) equipped with a nanospray ion source, and a splitless nano HPLC system (DiNa, KYA Technologies) with ODS capillary column (100 × 0.1 mm, 3 μm particle size, KYA Technologies). The tryptic digests were separated in 2% acetonitrile and 0.1% formic acid in water, using linear gradient from 0% to 80% of a solvent consisting of 70% acetonitrile and 0.1% formic acid for 30 min at a flow rate of 300 nl/min. Proteins were identified from the protein database of Homo Sapiens by employing MASCOT (Matrix Science) as a search engine for peptide mass fingerprinting.
Plasmid construction
To construct pCAGEN-FLAG-Ago1–4, full-length Ago1–4 sequences were amplified from pIRES-neo-FLAG-HA-Ago1-4 (Meister et al., 2004) by PCR using the following sets of the 5′ primer containing an EcoRI site and a FLAG-tag sequence and the 3′ primer containing a NotI site, and inserted into the EcoRI-NotI sites of the pCAGEN vector: 5′ -GAGTGAATTCATGGACTACAAGGACGACGATGACAAGATGGAAGCGGGA CCCTCGGG-3′ and 5′-CACTGCGGCCGCTCAAGCGAAGTACATGGTGC-3′ for Ago1, 5′ -GAGTGAATTCATGGACTACAAGGACGACGATGACAAGATGTACTCGGGAG CCGGCCC-3′ and 5′-CACTGCGGCCGCTCAAGCAAAGTACATGGTGC-3′ for Ago2, 5′ -GAGTGAATTCATGGACTACAAGGACGACGATGACAAGATGGAAATCGGCT CCGCAGG-3′ and 5′-CACTGCGGCCGCTTAAGCGAAGTACATTGTGC-3′ for Ago3, 5′ -GAGTGAATTCATGGACTACAAGGACGACGATGACAAGATGGAGGCGCTGG GACCCGGA-3′ and 5′-CACTGCGGCCGCTCAGGCAAAATACATCGTGTG-3′ for Ago4.
Expression of FLAG -tagged Ago1-4 proteins in HEK 293T cells
HEK 293T cells were transfected with pCAGEN-FLAG-Ago1–4 plasmid constructs using X-treme GENE9 Transfection Reagent (Roche). On the day before transfection, exponentially growing cells were plated into 15-cm dishes at a density of 2 × 106 cells in antibiotic-free medium. On the following day, the cells were transfected with 8 μg per dish of plasmid DNAs and harvested after 36 h.
Knockdown and overexpression of PARN in HeLa S3 cells
For PARN knockdown, HeLa S3 cells were plated into 10-cm dishes at a density of 1.6 × 106 cells in antibiotic-free medium and immediately transfected with 50 nM siRNA mixture composed of siRNA duplex 1 (5′-GGAGAAAACAGGAAGAGAAdTdT-3′ and 5′- UUCUCUUCCUGUUUUCUCCdTdT-3′) and siRNA duplex 2 (5′- UCAUCUCCAUGGCCAAUUAdTdT-3′ and 5′- UAAUUGGCCAUGGAGAUGAdTdT-3′) using Lipofectamine 2000 (Life Technologies). The cells were harvested after 72 h. For PARN overexpression, exponentially growing cells were first plated into 10-cm dishes at a density of 1 × 106 cells in antibiotic-free medium. On the following day, the cells were transfected with 12 μg per dish of pDESR12.2 plasmid harboring the PARN coding region by using Lipofectamine 2000 (Life Technologies), and harvested after 48 h.
Preparation of recombinant PARN
Full-length PARN containing an NdeI site at the 5′ end (5′-GTAGAGTCATATGATGGAGATAATCAGGAGCAATTTTAA-3′) and a XhoI site at the 3′ end (5′-ACTCTACCTCGAGTTACCATGTGTCAGGAACTTCAAAGAG-3′) was amplified by PCR from pDESR12.2-PARN and was inserted into the NdeI-XhoI sites of pCold I vector (TaKaRa). His-tagged PARN protein was expressed in the E. coli strain Rosetta 2(DE3) (MERCK). After growth to OD600 of ~0.5 at 37 °C, cells were induced with 1 mM IPTG and grown overnight at 15 °C. After cell lysis, His-tagged PARN was purified using His-trap HP column (GE Healthcare). The catalytic mutation (E30A) was introduced by PCR using 5′ -TCGCCATCGATGGGGCGTTTTCAGGAATCAG-3′ and 5′-CTGATTCCTGAAAACGCCCCATCGATGGCGA-3′.
In vitro target cleavage assay
To create the miR-451 target, DNA fragments containing the complementary target site were amplified from pGL3-Basic (Promega) by PCR using the primers (5′-GCGTAATACGACTCACTATAGTCACATCTCATCTACCTCC-3′ and 5′ -CCCATTTAGGTGACACTATAGATTTACATCGCGTTGAGTGTAGAACGGTTGT ATAAAAGGAAACCGTTACCATTACTGAGTTTAAGAGAGGAGTTCATG-3′). The target was transcribed from the corresponding DNA fragment using T7-Scribe Standard RNA IVT Kit (Cell Script). The transcripts were cap-radiolabeled with [α-32P] GTP using ScriptCap m7G Capping System (Epicentre), and gel-purified to ensure the integrity. For target cleavage, 10 nM ac-pre-miR-451 was loaded into FLAG-Ago2 on beads and incubated in lysate from HeLa S3 cells overexpressing PARN for trimming or buffer alone for 3 h at 37 °C, before 0.5 nM cap-radiolabeled target was added and further incubated at 37 °C.
miRNA reporter assay
The miR-451 reporter plasmids bearing 1× perfect or 2× centrally mismatched target sites or the 3′ UTR sequence of CAB39 were constructed by inserting the corresponding DNA fragments into the XhoI-NotI sites of psiCHECK-2 (Promega). Twenty nM of pre-miR-451 with or without 3× PS and 33 pg/ml of each reporter plasmid were co-transfected into HeLa S3 cells using Lipofectamine 2000 (Life Technologies), and the reporter assay was performed 24 h after transfection using the Dual-Luciferase Reporter Assay System (Promega). The miR-430 reporter plasmids were assembled by ligation of a DNA fragment containing three imperfect target sites (5′-ACTACCTGCACTGTGAGCACTTT-3′) for miR-430b into the XhoI-XbaI sites of pCS2+/GFP or pCS2+/Luciferase. Reporter constructs were linearized with NotI and in vitro transcribed with mMessage Machine SP6 (Ambion). For luciferase assay in vivo (Figure 5E) embryos were injected with 1 nl of Fluc reporter mRNA and Rluc control mRNA at 0.2 ng/μl each, 1 nl of 0.1 μg/μl mouse Ago2 mRNA, 1 nl of 0.2 μg/μl α-amanitin and 1 nl of titrating concentrations of pre-miR-430Ago2 hairpin with or without 3× PS (Figure 5E). Groups of 8 embryos were collected per triplicate and flash frozen in liquid nitrogen 6 h after injection. Reporter expression was quantified with the Dual-Luciferase Reporter Assay System (Promega) in a Veritas microplate luminometer (Turner BioSystems). The embryos for the GFP reporter assay (Figure 5D) were injected with 1 nl of GFP reporter mRNA and DsRed control mRNA at 0.1 ng/μl each, 1 nl of 0.1 μg/μl mouse Ago2 mRNA, 1 nl of 0.2 μg/μl α-amanitin and 1 nl of the indicated pre-miR-430 hairpin or duplex at 25 μM. Embryos were imaged for GFP and DsRed expression at 7 h after injection using a Zeiss Discovery microscope and photographed with a Zeiss Axiocam digital camera. Images were processed with Zeiss AxioVision 3.0.6.
Western blot analysis
Anti-FLAG (1:5000, Sigma), anti-PARN (1:4000, Bethyl) and anti-Tubulin (1:3000, Sigma) primary antibodies were used for detection. Chemiluminescence was induced by Luminata Forte Western HRP Substrate (Millipore) and images were acquired by a LAS-3000 imaging system and quantified using ImageGauge software (Fujifilm Life Sciences).
Northern blot analysis
Small RNAs were isolated using mirVana (Ambion) (Figure 2D). For Figure 4D, endogenous Ago2 was immunoprecipitated by anti-human Ago2 antibody (Wako) and the bound RNAs were extracted from the precipitate by Proteinase K treatment in 2× PK buffer. RNA was separated by 15% denaturing PAGE and transferred to Hybond-N+ membrane (GE Healthcare) using a semi-dry Trans-Blot SD (BioRad) at 10 V (0.15 mA) for 35 min. The membrane was UV crosslinked and hybridized with 5′ 32P-radiolabeled RNA probes for overnight at 65 °C. Small RNAs from zebrafish embryos were isolated by TRIzol (Life Technologies) at 7 h post-injection. After separation by 15% denaturing PAGE, small RNAs were transferred to a Zeta-Probe Blotting membrane (BioRad) using a semi-dry Trans-Blot SD (BioRad) at 20 V (0.68 A) for 35 min. The membrane was blotted with 5′ 32P-radiolabeled starfire probe from IDT overnight at 30 °C. The RNA probe sequences are 5′-AAACUCAGUAAUGGUAACGGUUU-3′ for miR-451, 5′-AGUACAUCAUCUAUACUGUA-3′ for miR-144, and 5′-UAUACAACCUACUACCUCUCU-3′ for let-7. The sequences of the StarFire DNA oligonucleotides are 5′-ATACCCCAACTTGATAGCACTTT-3′ for miR-430 and 5′-AACTCAGTAATGGTAACGGTTT-3′ for miR-451.
MZdicer rescue and brain ventricle labeling
MZdicer mutant embryos (dicerhu896 allele) were injected at 1-cell stage with 1000 pl of 25 μM stock solution of the indicated hairpin or duplex miRNA. At 30 hpf, embryos were injected in the brain ventricle with Texas Red dextran 70,000 MW (1:20 dilution of a 500 μg/μl stock, Molecular Probes) and imaged as described previously (Giraldez et al., 2005). Different focal planes were merged into a single image using Adobe Photoshop CS2 software.
RNA-seq sample preparation and sequencing analysis
MZdicer embryos were injected with 1 nl of a 25 μM stock solution of miR-430 duplex, pre-miR-430Ago2 hairpin or pre-miR-430Ago2 hairpin 3×PS at one-cell stage. 15 embryos per sample were collected at shield stage (~6 hpf) together with wild type and MZdicer uninjected embryos. Total RNA was extracted using the TRIzol method and submitted for library cloning and sequencing at the Yale Center for Genome Analysis. RNA-seq libraries were prepared using standard Illumina protocol and sequenced in multiplex in the HiSeq 2000 sequencing system. Raw reads were processed with TopHat 2.0.8 and mapped to the Zv9 Danio rerio genomic sequence and ENSEMBL 70 splicing junctions. Expression depth as reads per kilobase of gene model per million CDS-aligning reads (rpkm) was calculated for each gene using the uniquely mapping reads allowing up to one mismatch. Known miR-430 targets were identified in (Giraldez et al., 2005) and represent 122 experimentally validated miR-430 targets by microarray. The background set of 881 non-target genes is defined as in (Bazzini et al., 2012). Briefly, it consists only of genes with 3′ UTRs ≥ 300-nt and mismatches of ≥ 6-nt to the miR-430 seed in either the CDS or the 3′ UTR. To analyze the off-targeting of pre-miR-430Ago2 hairpin and pre-miR-430Ago2 hairpin 3×PS, we quantified the repression of a set of 10,841 genes, defined as the genes with more than 5 rpkm at 2 hpf in wild type, a sample described in (Bazzini et al., 2012).
Supplementary Material
Highlights.
PARN is responsible for trimming of Ago2-cleaved pre-miR-451.
Trimming of Ago2-cleaved pre-miRNAs is dispensable for target silencing.
Acknowledgments
We thank Charles Vejnar for mapping high-throughput sequencing results, Jesse Reinhart for mass spectrometry analysis, and Ian MacRae for providing recombinant human Ago2 used for additional experiments not presented here. We also thank Hervé Seitz and the members of our laboratories for discussions and critical comments on the manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas (“Functional machinery for non-coding RNAs,” Grant Number 21115002) from the Ministry of Education, Culture, Sports, Science and Technology of Japan to Y.T., the March of Dimes, the Pew Scholar, and NIH grants 5R01GM101108-02, 5R01GM102251-02, 5R21HD073768-02 to A.J.G., a NIH grant K99HD071968-01 to D.C., and postdoctoral fellowships from the Japan Society for the Promotion of Science to M.Y. and N.I.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom J, Astrom A, Virtanen A. In vitro deadenylation of mammalian mRNA by a HeLa cell 3’ exonuclease. EMBO J. 1991;10:3067–071. doi: 10.1002/j.1460-2075.1991.tb07858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom J, Astrom A, Virtanen A. Properties of a HeLa cell 3’ exonuclease specific for degrading poly(A) tails of mammalian mRNA. J Biol Chem. 1992;267:18154–8159. [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–37. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Karnowski HW, Cai X, Shin J, Pohlers M, Cullen BR. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Mol Cell. 2010;37:135–42. doi: 10.1016/j.molcel.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameier M, Herwig A, Reinhardt R, Walter L, Gruber J. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 2011;39:675–86. doi: 10.1093/nar/gkq776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Xie M, Steitz JA. A primate herpesvirus uses the integrator complex to generate viral microRNAs. Mol Cell. 2011;43:982–92. doi: 10.1016/j.molcel.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Triboulet R, Thornton JE, Gregory RI. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013;497:244–48. doi: 10.1038/nature12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–89. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, Hutvagner G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA. 2009;15:2147–160. doi: 10.1261/rna.1738409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. A human snoRNA with microRNA-like functions. Mol Cell. 2008;32:519–28. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Flynt AS, Greimann JC, Chung WJ, Lima CD, Lai EC. MicroRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol Cell. 2010;38:900–07. doi: 10.1016/j.molcel.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–08. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–38. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- Glazov EA, Kongsuwan K, Assavalapsakul W, Horwood PF, Mitter N, Mahony TJ. Repertoire of bovine miRNA and miRNA-like small regulatory RNAs expressed upon viral infection. PLoS One. 2009;4:e6349. doi: 10.1371/journal.pone.0006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010;9:2742–748. [PubMed] [Google Scholar]
- Goldstrohm AC, Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol. 2008;9:337–44. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nat Struct Mol Biol. 2004;11:599–06. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- Han BW, Hung JH, Weng Z, Zamore PD, Ameres SL. The 3’-to-5’ exoribonuclease Nibbler shapes the 3’ ends of microRNAs bound to Drosophila Argonaute1. Curr Biol. 2011;21:1878–887. doi: 10.1016/j.cub.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–95. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–39. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Korner CG, Wahle E. Poly(A) tail shortening by a mammalian poly(A)-specific 3’-exoribonuclease. J Biol Chem. 1997;272:10448–0456. doi: 10.1074/jbc.272.16.10448. [DOI] [PubMed] [Google Scholar]
- Korner CG, Wormington M, Muckenthaler M, Schneider S, Dehlin E, Wahle E. The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J. 1998;17:5427–437. doi: 10.1093/emboj/17.18.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Liu N, Abe M, Sabin LR, Hendriks GJ, Naqvi AS, Yu Z, Cherry S, Bonini NM. The exoribonuclease Nibbler controls 3’ end processing of microRNAs in Drosophila. Curr Biol. 2011;21:1888–893. doi: 10.1016/j.cub.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Ren YG, Thuresson AC, Hellman U, Astrom J, Virtanen A. A 54-kDa fragment of the Poly(A)-specific ribonuclease is an oligomeric, processive, and cap-interacting Poly(A)-specific 3’ exonuclease. J Biol Chem. 2000;275:24222–4230. doi: 10.1074/jbc.M001705200. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–97. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Nelson PT, De Planell-Saguer M, Lamprinaki S, Kiriakidou M, Zhang P, O’Doherty U, Mourelatos Z. A novel monoclonal antibody against human Argonaute proteins reveals unexpected characteristics of miRNAs in human blood cells. RNA. 2007;13:1787–792. doi: 10.1261/rna.646007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–00. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren YG, Kirsebom LA, Virtanen A. Coordination of divalent metal ions in the active site of poly(A)-specific ribonuclease. J Biol Chem. 2004;279:48702–8706. doi: 10.1074/jbc.M403858200. [DOI] [PubMed] [Google Scholar]
- Ren YG, Martinez J, Virtanen A. Identification of the active site of poly(A)-specific ribonuclease by site-directed mutagenesis and Fe(2+)-mediated cleavage. J Biol Chem. 2002;277:5982–987. doi: 10.1074/jbc.M111515200. [DOI] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–6. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. Small RNAs derived from snoRNAs. RNA. 2009;15:1233–240. doi: 10.1261/rna.1528909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava FA, Mendez R. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol. 2012;19:577–85. doi: 10.1038/nsmb.2311. [DOI] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 2010;107:15163–5168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.