

Abstract
The dynamic nature of gene expression enables cellular programming, homeostasis, and environmental adaptation in living systems. Dissection of causal gene functions in cellular and organismal processes therefore necessitates approaches that enable spatially and temporally precise modulation of gene expression. Recently, a variety of microbial and plant-derived light-sensitive proteins have been engineered as optogenetic actuators, enabling high precision spatiotemporal control of many cellular functions1-11. However, versatile and robust technologies that enable optical modulation of transcription in the mammalian endogenous genome remain elusive. Here, we describe the development of Light-Inducible Transcriptional Effectors (LITEs), an optogenetic two-hybrid system integrating the customizable TALE DNA-binding domain12-14 with the light-sensitive cryptochrome 2 protein and its interacting partner CIB1 from Arabidopsis thaliana. LITEs do not require additional exogenous chemical co-factors, are easily customized to target many endogenous genomic loci, and can be activated within minutes with reversibility3,4,6,7,15. LITEs can be packaged into viral vectors and genetically targeted to probe specific cell populations. We have applied this system in primary mouse neurons, as well as in the brain of awake mice in vivo to mediate reversible modulation of mammalian endogenous gene expression as well as targeted epigenetic chromatin modifications. The LITE system establishes a novel mode of optogenetic control of endogenous cellular processes and enables direct testing of the causal roles of genetic and epigenetic regulation in normal biological processes and disease states.
The LITE system employs a modular design consisting of two independent components (Fig. 1a): The first component is the genomic anchor and includes a customizable DNA-binding domain, based on transcription activator-like effectors (TALEs)12,13 from Xanthamonas sp., fused to the light-sensitive cryptochrome 2 (CRY2) protein from Arabidopsis thaliana6,15 (TALE-CRY2). The second component includes CRY2’s interacting partner CIB16,15 fused to a desired effector domain (CIB1-effector). In the absence of light (inactive state), TALE-CRY2 binds the promoter region of the target gene while CIB1-effector remains free within the nuclear compartment. Illumination with blue light (peak ~450 nm) triggers a conformational change in CRY2 and subsequently recruits CIB1-effector (VP64 shown in Fig. 1a) to the target locus to mediate transcriptional modulation. This modular design allows each LITE component to be independently engineered, allowing the same genomic anchor to be combined with activating or repressing effectors16,17 to exert positive and negative transcriptional control over the same endogenous genomic locus. In principle, the genomic anchor may also be replaced with other DNA binding domains such as zinc finger proteins16 or RNA-guided DNA binding domains based on nucleolytically inactive mutants of Cas9 (Extended Data Fig. 1)18-22.
Figure 1. Design and Optimization of the LITE system.

(a) Schematic of the LITE system. Light stimulation induces dimerization of CRY2 and CIB1, recruiting the effector to the target promoter. (b) LITE architecture was optimized by fusing TALE and the transcriptional activator VP6414,16 to different truncations of CRY2 and CIB16 (n next to each bar). (c) Time course of light-dependent Neurog2 up-regulation and decay post-illumination (n = 3 biological replicas; *, p < 0.05; ***, p < 0.001). Cells were stimulated with 5 mW/cm2 light (460nm, 1 s pulses at 0.066 Hz). Mean ± s.e.m in all panels.
In order to identify the most effective architecture, we assessed the efficacy of different LITE designs by measuring blue light illumination induced transcriptional changes of the neural lineage-specifying transcription factor neurogenin 2 (Neurog2) (Fig. 1b). 3 out of 4 initial LITE pairings produced significant light-induced Neurog2 mRNA up-regulation in Neuro 2a cells (p < 0.001, Fig. 1b). Of these, TALE(Neurog2)-CRY2PHR::CIB1-VP64 yielded strongest light-mediated transcription activation as well as the highest induction ratio (light/no light mRNA levels). Therefore TALE-CRY2PHR::CIB1-VP64 was used in subsequent experiments. To ensure optimal function, we also systematically tuned light stimulation parameters (wavelength, Extended Data Fig. 2a; duty cycle, Extended Data Fig. 2b; light intensity, Extended Data Fig. 2c and d, and Supplementary Discussion23; and choice of activation domain, Extended Data Fig. 2e).
Although the interaction between CRY2 and CIB1 occurs on a sub-second timescale6, LITE-mediated transcriptional activation is likely dependent on many factors, including rate of transcription, mRNA processing, and transcript stability24,25. We found that LITE-mediated Neurog2 expression increased considerably as early as 30 min after initial stimulation and rose steadily until saturating at 12 h with approximately 20-fold up-regulation compared to GFP-transfected negative controls (Fig. 1c). Interestingly, Neurog2 transcript levels continued to increase for up to 30 min post-illumination, an effect that may have resulted from residual CRY2PHR-CIB1 dimerization or from previously recruited RNA polymerases. Thereafter, Neurog2 mRNA returned to baseline levels with a half-life of ~3 h. In contrast, a small-molecule inducible TALE system based on the plant hormone abcisic acid receptor26 exhibited slower on- and off-kinetics (Extended Data Fig. 3).
To apply LITE for neuronal applications, we developed an adeno-associated virus (AAV)-based vector (Figs. 2a, b) for the delivery of TALE genes and a simplified process for AAV production (Extended Data Fig. 4 and Online Methods). The ssDNA-based genome of AAV is less susceptible to recombination, providing an advantage over lentiviral vectors27. We evaluated a panel of 28 TALE activators targeting the mouse genome in primary neurons and found that most were able to up-regulate transcription in primary neurons (Fig. 2c). Moreover, in vivo expression of TALE(Grm2)-VP64 in the prefrontal cortex (PFC) (Fig. 2d and e) induced a 2.5-fold increase in Grm2 mRNA levels compared to GFP-only controls (Fig. 2f).
Figure 2. In vitro and in vivo AAV-mediated TALE delivery targeting endogenous loci in neurons.

(a) Schematic AAV vectors for TALE delivery. (b) Representative images of primary cortical neurons expressing TALE-VP64. (c) TALE-VP64 constructs targeting a variety of endogenous neuronal genes were screened for transcriptional activation in primary cortical neurons (*, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 3 biological replicas). (d) TALE-VP64 expression in PFC. (e) Higher magnification image of TALE-VP64-expressing neurons in PFC. (f) Grm2 mRNA up-regulation by TALE-VP64 in vivo in PFC (n = 4 animals). Mean ± s.e.m in all panels.
Similarly, we introduced LITEs into primary cortical neurons via co-delivery of two AAVs (Fig. 3a, b). We tested a Grm2-targeted LITE at 2 light pulsing frequencies with a reduced duty cycle of 0.8% to ensure neuron health (Extended Data Fig. 5a). Both stimulation conditions achieved a ~7-fold light-dependent increase in Grm2 mRNA levels (Fig. 3c). Further study verified that substantial target gene expression increases could be attained quickly (4-fold up-regulation of mRNA within 4 h; Fig. 3d). In addition, we observed significant up-regulation of mGluR2 protein after stimulation, confirming that LITE-mediated transcriptional changes are translated to the protein level (p < 0.01 vs GFP control, p < 0.05 vs no-light condition; Fig. 3e). To test the in vivo functionality of the LITE system, we stereotactically delivered a 1:1 mixture of high titer AAV vectors carrying TALE(Grm2)-CIB1 and CRY2PHR-VP64 into the PFC. We used a previously established fiber optic cannula system to deliver light to LITE-expressing neurons in vivo (Fig. 3f, g, and Extended Data Fig. 5b)28. After 12 h of stimulation, we observed a significant increase in Grm2 mRNA compared with unstimulated PFC (Fig. 3h, p ≤ 0.01). Taken together, these results confirm that LITEs enable optical control of endogenous gene expression in cultured neurons and in vivo.
Figure 3. LITE-mediated optogenetic modulation of endogenous transcription in primary neurons and in vivo.

(a) Schematic of AAV-LITE constructs. (b) Images of primary neurons expressing LITE constructs. (c) Light-induced activation of Grm2 in primary neurons after 24 h of stimulation (250 ms pulses at 0.033Hz or 500 ms pulses at 0.016Hz; 5mW/cm2; n = 4 biological replicas). (d) Up-regulation of Grm2 in primary cortical neurons after 4 h or 24 h of stimulation. Expression levels are shown relative to neurons transduced with GFP only (number of biological replica denoted within graph bars). (e) Light-mediated changes in mGluR2 protein levels (n = 7 biological replica). (f) Schematic of in vivo optogenetic stimulation setup. (g) Representative images of PFC neurons expressing both LITE components. (h) Light-induced activation of endogenous Grm2 expression using LITEs transduced into ILC. (**, p < 0.05; number of animals denoted within graph bars) (i) LITE2.0 significantly reduces the level of background activation in Neuro 2a cells (n = 3 biological replica). Mean ± s.e.m in all panels.
Given persistent baseline up-regulation in vivo, we undertook further rounds of optimization to reduce background activity and improve the gene induction ratio. We observed that TALE(Grm2)-CIB1 alone produced similar levels of up-regulation as background activation, yet CRY2PHR-VP64 alone did not significantly affect transcription (Extended Data Fig. 5c). Therefore we rationalized that LITE-dependent background transcriptional activation arises mainly from TALE-CIB1.
The subsequent comprehensive screen to reduce baseline TALE-CIB1-mediated up-regulation focuses on two strategies: First, although CIB1 is a plant transcription factor, it may have intrinsic activity in mammalian cells29. To address this, we deleted three CIB1 regions conserved amongst basic helix-loop-helix transcription factors of higher plants (Extended Data Fig. 6). Second, to prevent TALE-CIB1 from binding the target locus in absence of light, we engineered TALE-CIB1 to localize in the cytoplasm pending light-induced dimerization with the NLS-containing CRY2PHR-VP64 (Extended Data Fig. 7a and b). To test both strategies independently or in combination, we evaluated 73 distinct LITE architectures and identified 12 effector-targeting domain pairs (denoted by the “+” column in Extended Data Fig. 6) with both improved light-induction efficiency and reduced background (fold mRNA increase in the no-light condition compared with the original LITE; p < 0.05). One architecture successfully incorporating both strategies, designated LITE2.0, demonstrated the strongest light induction (light/no-light = 20.4) and resulted in greater than 6-fold reduction of background activation compared with the original design (Fig. 3i). Another – LITE1.9.1 – produced minimal background activation (1.06) while maintaining four-fold light induction (Extended Data Fig. 7c).
Finally, we sought to expand the range of transcriptional processes modulatable by TALE and LITE. We hypothesized that TALE-mediated targeting of histone effectors to endogenous loci could induce specific epigenetic modifications, which would enable the interrogation of epigenetic as well as transcriptional dynamics (Fig. 4a). We fused CRY2PHR with SID4X (Fig. 4b and Extended Data Fig. 8) and observed light-mediated transcription repression of Grm2 in neurons (Fig. 4c, d) accompanied by ~2-fold reduction in H3K9 acetylation at the targeted Grm2 promoter (Fig. 4d). In order to expand the diversity of histone residue targets for locus-specific histone modification, we next derived a set of 32 repressive histone effector domains (Supplementary Tables 1-5). Selected from across a wide phylogenetic spectrum, the domains include histone deacetylases (HDACs), methyltransferases (HMTs), acetyltransferase (HAT) inhibitors, as well as HDAC and HMT recruiting proteins. Preference was given to proteins and functional truncations of small size to facilitate efficient AAV packaging. The resulting epigenetic mark-modifying TALE-histone effector fusion constructs (epiTALEs) were evaluated in primary neurons and Neuro 2a cells for their ability to repress Grm2 and Neurog2 transcription, respectively (Fig. 4e, f and Extended Data Fig. 9). In primary neurons, 23 out of 24 epiTALEs successfully repressed transcription of Grm2 (p < 0.05). Similarly, epiTALE expression in Neuro 2a cells led to decreased Neurog2 expression for 20 of the 32 histone effector domains tested (Extended Data Fig. 9; p < 0.05). We then expressed a subset of promising epiTALEs in primary neurons and Neuro 2a cells and quantified the relative histone residue mark levels at the target locus using ChIP-RT-qPCR (Fig. 4g, h and Extended Data Fig. 10). In primary neurons and Neuro 2a cells, levels of H3K9me1, H4K20me3, H3K27me3, H3K9ac, and H4K8ac were altered by epiTALEs consisting of, respectively, KYP (A. thaliana), TgSET8 (T. gondii), NUE and PHF19 (C. trachomatis and H. sapiens), Sin3a, Sirt3 and NcoR, (all H. sapiens) and HDAC8, RPD3, and Sir2a (X laevis, S cerevisiae, P falciparum). These domains provide a ready source of epigenetic effectors for LITE-mediated control of specific epigenetic modifications.
Figure 4. TALE- and LITE-mediated epigenetic modifications.

(a) LITE epigenetic modifiers (epiLITE). (b) epiLITE AAV vectors. (c) epiLITE-mediated repression of endogenous Grm2 in neurons (n = 4 biological replicas). (d) epiLITE-mediated decrease in H3K9 histone acetylation at the Grm2 promoter (n = 4 biological replicas). (e, f) epiTALE-methyltransferases mediated decrease in Grm2 mRNA and corresponding enrichment of H3K9me1, H4K20me3, and H3K27me3 at the Grm2 promoter (n denoted within graph). (g, h) epiTALE histone deacetylases mediated repression of Grm2 and corresponding decreases in H4K8Ac and H3K9Ac marks at the Grm2 promoter (n denoted within graph). Mean ± s.e.m in all panels.
Spatiotemporally precise perturbation of transcription and epigenetic states in vivo using LITE enables researchers to test the causal role of gene regulation in diverse processes including development, learning, and disease. TALEs can be conveniently customized to target a wide range of genomic loci, and other DNA binding domains such as the RNA-guided Cas9 enzymes may be used in lieu of TALE to enable multiplexed transcriptional and epigenetic engineering of individual or groups of genomic loci in cells and whole organisms18-20. Novel modes of LITE modulation can also be achieved by replacing the effector module with functional domains such as chromatin modifying enzymes30. The LITE system enables a powerful set of novel capabilities for the optogenetic toolbox and establishes a highly generalizable and versatile platform for reverse-engineering the functional organization of mammalian genomes.
Methods
Design and construction of LITEs
All LITE constructs sequences can be found in Supplementary Sequences. We evaluated full-length CRY2 as well as a truncation consisting of the photolyase homology region alone (CRY2PHR, amino acids 1-498)6. For CIB1, we tested the full-length protein as well as an N-terminal domain-only fragment (CIBN, amino acids 1-170)6. The efficacy of each design is determined based on the level of light-dependent up-regulation of the endogenous target Neurog2 mRNA (Fig. 1b). In order to use AAV as a vector for the delivery of LITE components, we needed to ensure that the total viral genome size of each recombinant AAV, with the LITE transgenes included, did not exceed the packaging limit of 4.8 kb31. We shortened the TALE N- and C-termini (keeping 136 aa in the N-terminus and 63 aa in the C-terminus) and exchanged the CRY2PHR (1.5kb) and CIB1 (1kb) domains (TALE-CIB1 and CRY2PHR-VP64; Fig. 3a). TALE binding sequences were selected based on DNase I-sensitive regions in the promoter of each target gene. TALE targeting sequence are listed in Supplementary Table 6.
Neuro 2a culture and experiments
Neuro 2a cells (Sigma-Aldrich) were grown in media containing a 1:1 ratio of OptiMEM (Life Technologies) to high-glucose DMEM with GlutaMax and Sodium Pyruvate (Life Technologies) supplemented with 5% HyClone heat-inactivated FBS (Thermo Scientific), 1% penicillin/streptomycin (Life Technologies), and passaged at 1:5 every 2 days. 120,000 cells were plated in each well of a 24-well plate 18-20 h prior to transfection. 1 h before transfection, media was changed to DMEM supplemented with 5% HyClone heat-inactivated FBS and 1% penicillin/streptomycin. Cells were transfected with 1.0 μg total of construct DNA (at equimolar ratios) per well with 1.5 μL of GenJet (SignaGen Laboratories) transfection reagent according to the manufacturer’s instructions. Media was exchanged 24 h and 44 h post-transfection and light stimulation was started at 48 h. Stimulation parameters were: 5 mW/cm2, 466 nm, 7 % duty cycle (1 s light pulse 0.067 Hz) for 24 h unless indicated otherwise in figure legends. RNA was extracted using the RNeasy kit (Qiagen) according to manufacturer’s instructions and 1 μg of RNA per sample was reverse-transcribed using qScript (Quanta Biosystems). Relative mRNA levels were measured by quantitative real-time PCR (qRT-PCR) using TaqMan probes specific for the targeted gene as well as GAPDH as an endogenous control (Life Technologies, see Supplementary Table 7 for Taqman probe IDs). ΔΔCt analysis was used to obtain fold-changes relative to negative controls transduced with GFP only and subjected to light stimulation. Toxicity experiments were conducted using the LIVE/DEAD assay kit (Life Technologies) according to manufacturer’s protocol.
AAV vector production
The ssDNA-based genome of AAV is less susceptible to recombination, thus providing an advantage over RNA-based lentiviral vectors27 for the packaging and delivery of highly repetitive TALE sequences. 293FT cells (Life Technologies) were grown in antibiotic-free D10 media (DMEM high glucose with GlutaMax and Sodium Pyruvate, 10% heat-inactivated Hyclone FBS, and 1% 1M HEPES) and passaged daily at 1:2-2.5. The total number of passages was kept below 10 and cells were never grown beyond 85% confluence. The day before transfection, 1×106 cells in 21.5 mL of D10 media were plated onto 15 cm dishes and incubated for 18-22 hours or until ~80% confluence. For use as a transfection reagent, 1 mg/mL of PEI “Max” (Polysciences) was dissolved in water and the pH of the solution was adjusted to 7.1. For AAV production, 10.4 μg of pDF6 helper plasmid, 8.7 μg of pAAV1 serotype packaging vector, and 5.2 μg of pAAV vector carrying the gene of interest were added to 434 μL of serum-free DMEM and 130 μL of PEI “Max” solution was added to the DMEM-diluted DNA mixture. The DNA/DMEM/PEI cocktail was vortexed and incubated at room temperature for 15 min. After incubation, the transfection mixture was added to 22 mL of complete media, vortexed briefly, and used to replace the media for a 15 cm dish of 293FT cells. For supernatant production, transfection supernatant was harvested at 48 h, filtered through a 0.45 μm PVDF filter (Millipore), distributed into aliquots, and frozen for storage at −80°C.
Primary cortical neuron culture
Dissociated cortical neurons were prepared from C57BL/6N mouse embryos on E16 (Charles River Labs). Cortical tissue was dissected in ice-cold HBSS – (50 mL 10× HBSS, 435 mL dH2O, 0.3 M HEPES pH 7.3, and 1% penicillin/streptomycin). Cortical tissue was washed 3× with 20 mL of ice-cold HBSS and then digested at 37°C for 20 min in 8 mL of HBSS with 240 μL of 2.5% trypsin (Life Technologies). Cortices were then washed 3 times with 20 mL of warm HBSS containing 1 mL FBS. Cortices were gently triturated in 2 ml of HBSS and plated at 150,000 cells/well in poly-D-lysine coated 24-well plates (BD Biosciences). Neurons were maintained in Neurobasal media (Life Technologies), supplemented with 1× B27 (Life Technologies), GlutaMax (Life Technologies) and 1% penicillin/streptomycin.
Primary neuron transduction and light stimulation experiments
Primary cortical neurons were transduced with 250 μL of AAV1 supernatant on DIV 5. The media and supernatant were replaced with regular complete neurobasal the following day. Neurobasal was exchanged with Minimal Essential Medium (Life Technologies) containing 1× B27, GlutaMax (Life Technologies) and 1% penicillin/streptomycin 6 days after AAV transduction to prevent formation of phototoxic products from HEPES and riboflavin contained in Neurobasal during light stimulation. For co-transduction of primary neurons with two AAV vectors, the co-delivery efficiency is >80%, with individual components having transduction efficiencies between 83-92%.
Light stimulation was started 6 days after AAV transduction (DIV 11) with an intensity of 5 mW/cm2, duty cycle of 0.8% (250 ms pulses at 0.033Hz or 500 ms pulses at 0.016Hz), 466 nm blue light for 24 h unless indicated otherwise in figure legends. RNA extraction and reverse transcription were performed using the Cells-to-Ct kit according to the manufacturers instructions (Life Technologies). Relative mRNA levels were measured by quantitative real-time PCR (qRT-PCR) using TaqMan probes as described above for Neuro 2a cells.
Immunohistochemistry of primary neurons
For immunohistochemistry of primary neurons, cells were plated on poly-D-lysine/laminin coated coverslips (BD Biosciences) after harvesting. AAV1-transductions were performed as described above. Neurons were fixed 7 days post-transduction with 4% paraformaldehyde (Sigma Aldrich) for 15 min at RT. Blocking and permeabilization were performed with 10% normal goat serum (Life Technologies) and 0.5% Triton-X100 (Sigma-Aldrich) in DPBS (Life Technologies) for 1 h at room temperature. Neurons were incubated with primary antibodies overnight at 4°C, washed 3× with DPBS and incubated with secondary antibodies for 90 min at RT. For antibody providers and concentrations used, see Supplementary Table 8. Coverslips were finally mounted using Prolong Gold Antifade Reagent with DAPI (Life Technologies) and imaged on an Axio Scope A.1 (Zeiss) with an X-Cite 120Q light source (Lumen Dynamics). Image were acquired using an AxioCam MRm camera and AxioVision 4.8.2.
Western Blots
For preparation of total protein lysates, primary cortical neurons were harvested after light stimulation (see above) in ice-cold lysis buffer (RIPA, Cell Signaling; 0.1% SDS, Sigma-Aldrich; and cOmplete ULTRA protease inhibitor mix, Roche Applied Science). Cell lysates were sonicated for 5 min at ‘M’ setting with the Bioruptor water bath sonicator (Diagenode) and centrifuged at 21,000 × g for 10 min at 4°C. Protein concentration was determined using the RC DC protein assay (Bio-Rad). 30-40 μg of total protein per lane was separated under non-reducing conditions on 4-15% Tris-HCl gels (Bio-Rad) along with Precision Plus Protein Dual Color Standard (Bio-Rad) After wet electrotransfer to polyvinylidene difluoride membranes (Millipore) and membrane blocking for 45 min in 5% BLOT-QuickBlocker (Millipore) in Tris-buffered saline (TBS, Bio-Rad), western blots were probed with anti-mGluR2 (Abcam, 1:1.000) and anti-μ-tubulin (Sigma-Aldrich 1:20,000) overnight at 4°C, followed by washing and anti-mouse-IgG HRP antibody incubation (Sigma-Aldrich, 1:5,000 – 1:10,000). For further antibody details see Supplementary Table 8. Detection was performed via ECL Western blot substrate (SuperSignal West Femto Kit, Thermo Scientific). Blots were imaged with an AlphaImager system (Innotech), and quantified using ImageJ software 1.46r.
Production of concentrated and purified AAV1/2 vectors
Production of concentrated and purified AAV for stereotactic injection in vivo was performed using the same initial steps outlined above for production of AAV1 supernatant. However, for transfection, equal ratios of AAV1 and AAV2 serotype plasmids were used instead of AAV1 alone. Five 15cm plates were transfected per construct and cells were harvested with a cell-scraper 48 h post transfection. Purification of AAV1/2 particles was performed using HiTrap heparin affinity columns (GE Healthcare)32. We added a second concentration step down to a final volume of 100 μl per construct using an Amicon 500 μl concentration column (100 kDa cutoff, Millipore) to achieve higher viral titers. Titration of AAV was performed by qRT-PCR using a custom Taqman probe for WPRE (Life Technologies). Prior to qRT-PCR, concentrated AAV was treated with DNaseI (New England Biolabs) to achieve a measurement of DNaseI-resistant particles only. Following DNaseI heat-inactivation, the viral envelope was degraded by Proteinase K digestion (New England Biolabs). Viral titer was calculated based on a standard curve with known WPRE copy numbers.
Stereotactic injection of AAV1/2 and optical implant
All animal procedures were approved by the MIT Committee on Animal Care. Adult (10-14 weeks old) male C57BL/6N mice were anaesthetized by intraperitoneal (i.p.) injection of Ketamine/Xylazine (100 mg/kg Ketamine and 10 mg/kg Xylazine) and pre-emptive analgesia was applied (Buprenex, 1 mg/kg, i.p.). Craniotomy was performed according to approved procedures and 1μl of AAV1/2 was injected into ILC at 0.35/1.94/-2.94 (lateral, anterior and inferior coordinates in mm relative to bregma). During the same surgical procedure, an optical cannula with fiber (Doric Lenses) was implanted into ILC unilaterally with the end of the optical fiber located at 0.35/1.94/-2.64 relative to bregma. The cannula was affixed to the skull using Metabond dental cement (Parkell Inc) and Jet denture repair (Lang Dental) to build a stable, supporting cone. The incision was sutured and proper post-operative analgesics were administered for three days following surgery.
Immunohistochemistry on ILC brain sections
Mice were injected with a lethal dose of Ketamine/Xylazine anaesthetic and transcardially perfused with PBS and 4% paraformaldehyde (PFA). Brains were additionally fixed in 4% PFA at 4°C overnight and then transferred to 30% sucrose for cryoprotection overnight at room temperature. Brains were then transferred into Tissue-Tek Optimal Cutting Temperature (OCT) Compound (Sakura Finetek) and frozen at −80°C. 18 μm sections were cut on a cryostat (Leica Biosystems) and mounted on Superfrost Plus glass slides (Thermo Fischer). Sections were post-fixed with 4% PFA for 15 min, and immunohistochemistry was performed as described for primary neurons above.
Light stimulation and mRNA level analysis in ILC
Neurons at the injection site were efficiently co-transduced by both viruses, with >80% of transduced cells expressing both TALE(Grm2)-CIB1 and CRY2PHR-VP64 (Fig. 3g and Extended Data Fig. 5b). 8 days post-surgery, awake and freely moving mice were stimulated using a 473 nm laser source (OEM Laser Systems) connected to the optical implant via fiber patch cables and a rotary joint. Stimulation parameters were the same as used on primary neurons: 5 mW (total output), 0.8% duty cycle (500 ms light pulses at 0.016 Hz) for a total of 12 h. Brain tissue from the fiber optic cannula implantation site was analyzed (Fig. 3h) for changes in Grm2 mRNA. Experimental conditions, including transduced constructs and light stimulation are listed in Supplementary Table 9.
After the end of light stimulations, mice were euthanized using CO2 and the prefrontal cortices (PFC) were quickly dissected on ice and incubated in RNA later (Qiagen) at 4°C overnight. 200 μm sections were cut in RNA later at 4°C on a vibratome (Leica Biosystems). Sections were then frozen on a glass coverslide on dry ice and virally transduced ILC was identified under a fluorescent stereomicroscope (Leica M165 FC). A 0.35 mm diameter punch of ILC, located directly ventrally to the termination of the optical fiber tract, was extracted (Harris uni-core, Ted Pella). The brain punch sample was then homogenized using an RNase-free pellet-pestle grinder (Kimble Chase) in 50 μl Cells-to-Ct RNA lysis buffer and RNA extraction, reverse transcription and qRT-PCR was performed as described for primary neuron samples.
Chromatin Immunoprecipitation
Neurons or Neuro 2a cells were cultured and transduced or transfected as described above. ChIP samples were prepared as previously described33 with minor adjustments for the cell number and cell type. Cells were harvested in 24-well format, washed in 96-well format, and transferred to microcentrifuge tubes for lysis. Sample cells were directly lysed by water bath sonication with the Biorupter sonication device for 21 minutes using 30s on/off cycles (Diagenode). qPCR was used to assess enrichment of histone marks at the targeted locus. qPCR primer sequences are listed in Supplementary Table 10.
Statistical analysis
All experiments were performed with a minimum of two independent biological replicates. Statistical analysis was performed with Prism (GraphPad) using Student’s two-tailed t-test when comparing two conditions, ANOVA with Tukey’s post-hoc analysis when comparing multiple samples with each other, and ANOVA with Dunnett’s post-hoc analysis when comparing multiple samples to the negative control.
Supplementary Material
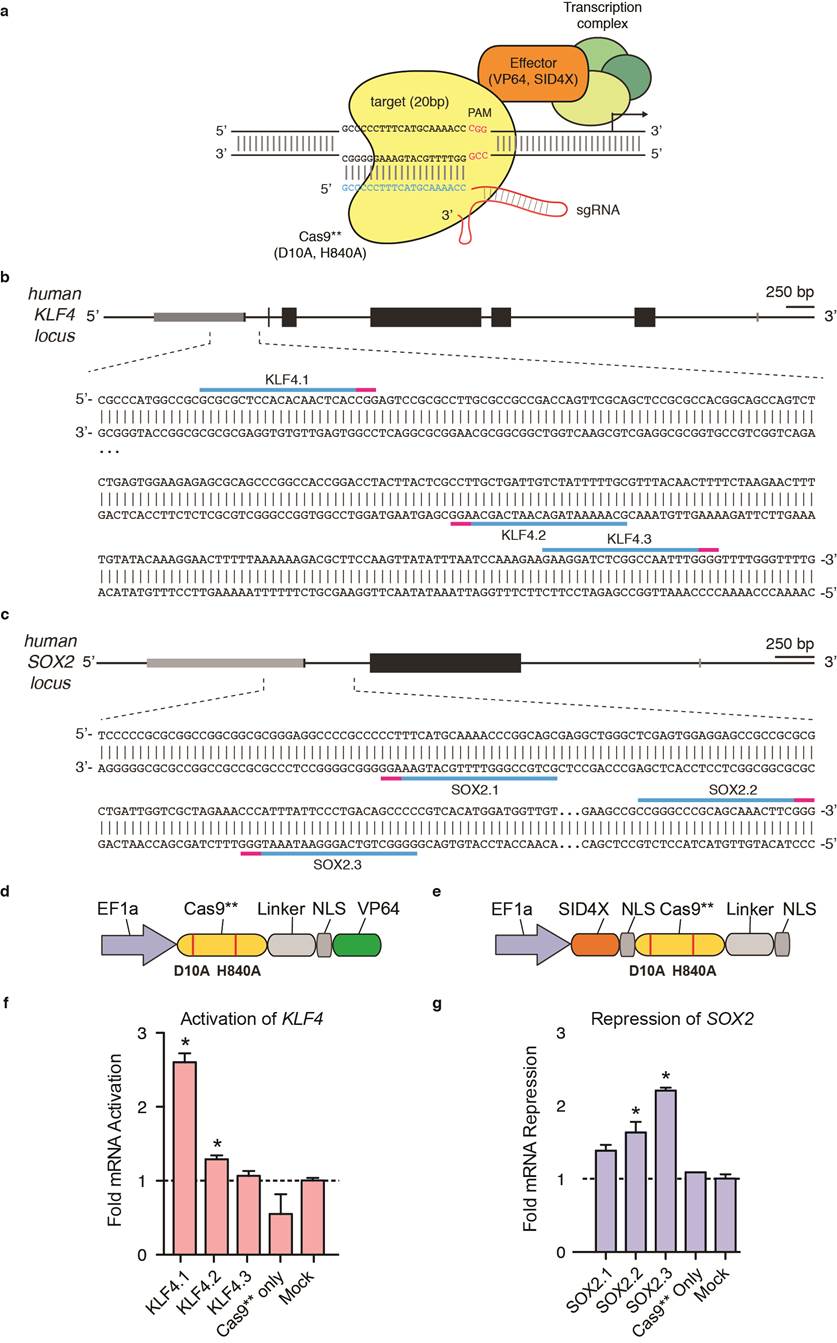
Extended Data Figure 1 | RNA-guided DNA binding protein Cas9 can be used to target transcription effector domains to specific genomic loci. (a) The RNA-guided nuclease Cas9 from the type II Streptococcus pyogenes CRISPR/Cas system can be converted into a nucleolytically-inactive RNA-guided DNA binding protein (Cas9**) by introducing two alanine substitutions (D10A and H840A). Schematic showing that a synthetic guide RNA (sgRNA) can direct Cas9**-effector fusion to a specific locus in the human genome. The sgRNA contains a 20bp guide sequence at the 5′ end which specifies the target sequence. On the target genomic DNA, the 20bp target site needs to be followed by a 5′-NGG PAM motif. (b, c) Schematics showing the sgRNA target sites in the human KLF4 and SOX2 loci respectively. Each target site is indicated by the blue bar and the corresponding PAM sequence is indicated by the magenta bar. (d, e) Schematics of the Cas9**-VP64 transcription activator and SID4X-Cas9** transcription repressor constructs. (f, g) Cas9**-VP64 and SID4X-Cas9** mediated activation of KLF4 and repression of SOX2 respectively. All mRNA levels were measured relative to GFP mock transfected 293FT cells (mean ± s.e.m.; n = 3 biological replicas).
{kind=link}
Extended Data Figure 8 | TALE SID4X repressor characterization and application in neurons. (a) A synthetic repressor was constructed by concatenating 4 SID domains (SID4X). To identify the optimal TALE-repressor architecture, SID or SID4X was fused to a TALE designed to target the mouse p11 gene. (b) Fold decrease in p11 mRNA was assayed using qRT-PCR. (mean ± s.e.m.; n = 3 biological replicas) (c) General schematic of constitutive TALE transcriptional repressor packaged into AAV. Effector domain SID4X is highlighted. hSyn: human synapsin promoter; 2A: Thosea asigna virus 2A self-cleaving peptide34; WPRE: woodchuck hepatitis post-transcriptional response element; bGH pA: bovine growth hormone poly-A signal. phiLOV2.135 (330bp) was chosen as a shorter fluorescent marker to ensure efficient AAV packaging. (d) A TALE targeting either the endogenous mouse loci Grm5 and Grm2 was fused to SID4X and virally transduced into primary neurons. SID4X-mediated target gene down-regulation is shown for each TALE relative to levels in control neurons expressing GFP only. (mean ± s.e.m.; n = 3-4 biological replicas)
{kind=link}
Extended Data Figure 9 | A diverse set of epiTALEs mediate transcriptional repression in neurons and Neuro 2a cells. (a) 24 different histone effector domains were each fused to a Grm2 targeting TALE. TALE-effector fusions were expressed in primary cortical mouse neurons using AAV transduction. Grm2 mRNA levels were measured using RT-qPCR relative to neurons transduced with GFP only. (*, p < 0.05; mean ± s.e.m.; n = 2-3 biological replicas) (b) A total of 32 epiTALEs were transfected into Neuro2A cells. 20 of them mediated significant repression of the targeted Neurog2 locus (* = p<0.05). (mean ± s.e.m.; n = 2-3 biological replicas)
{kind=link}
Extended Data Figure 10 | epiTALEs mediating transcriptional repression along with histone modifications in Neuro 2A cells. (a) TALEs fused to histone-deacetylating epigenetic effectors NcoR and SIRT3 targeting the murine Neurog2 locus in Neuro 2A cells were assayed for repressive activity on Neurog2 transcript levels. (mean ± s.e.m.; n = 2-3 biological replicas) (b) ChIP RT-qPCR showing a reduction in H3K9 acetylation at the Neurog2 promoter for NcoR and SIRT3 epiTALEs. (mean ± s.e.m.; n = 2-3 biological replicas) (c) The epigenetic effector PHF19 with known histone methyltransferase binding activity was fused to a TALE targeting Neurog2 mediated repression of Neurog2 mRNA levels. (mean ± s.e.m.; n = 2-3 biological replicas) (d) ChIP RT-qPCR showing an increase in H3K27me3 levels at the Neurog2 promoter for the PHF19 epiTALE. (mean ± s.e.m.; n = 2-3 biological replicas)
{kind=link}
Extended Data Figure 2 | Engineering of light stimulation parameters and activation domains of LITEs. (a) Illustration of the absorption spectrum of CRY2 in vitro. Cryptochrome 2 was optimally activated by 350-475 nm light23. A sharp drop in absorption and activation was seen for wavelengths greater than 480 nm. Spectrum was adapted from Ref. 23. (b) Impact of illumination duty cycle on LITE-mediated gene expression. Varying duty cycles (illumination as percentage of total time) were used to stimulate 293FT cells expressing LITEs targeting the KLF4 gene, in order to investigate the effect of duty cycle on LITE activity. KLF4 expression levels were compared to cells expressing GFP only. Stimulation parameters were: 466 nm, 5 mW/cm2 for 24 h. Pulses were performed at 0.067 Hz with the following durations: 1.7% = 0.25 s pulse, 7% = 1 s pulse, 27% = 4 s pulse, 100% = constant illumination. (mean ± s.e.m.; n = 3-4 biological replicas) (c) The transcriptional activity of CRY2PHR:: CIB1 LITE was found to vary according to the intensity of 466 nm blue light. Neuro 2a cells were stimulated for 24 h hours at a 7% duty cycle (1s pulses at 0.066Hz) (d) Light-induced toxicity measured as the percentage of cells positive for red-fluorescent ethidium homodimer-1 versus calcein-positive cells. All Neurog2 mRNA levels were measured relative to cells expressing GFP only (mean ± s.e.m.; n = 3-4 biological replicas) (e) We compared the activation domains VP16 and p65 in addition to VP64 to test the modularity of the LITE CIB1-effector component. Neurog2 up-regulation with and without light by LITEs using different transcriptional activation domains (VP16, VP64, and p65). Neuro-2a cells transfected with LITE were stimulated for 24 h with 466 nm light at an intensity of 5mW/cm2 and a duty cycle of 7% (1 s pulses at 0.066 Hz). All three domains produced a significant light-dependent Neurog2 mRNA up-regulation (p < 0.001,). We selected VP64 for subsequent experiments due to its lower basal activity in the absence of light-stimulation. (mean ± s.e.m.; n = 3-4 biological replicas)
{kind=link}
Extended Data Figure 3 | Chemical induction of endogenous gene transcription. (a) Schematic showing the design of a chemical inducible two hybrid TALE system based on the abscisic acid (ABA) receptor system. ABI and PYL dimerize upon the addition of ABA and dissociates when ABA is withdrawn. (b) Time course of ABA-dependent Neurog2 up-regulation. 250 μM of ABA was added to HEK 293FT cells expressing TALE(Neurog2)-ABI and PYL-VP64. Fold mRNA increase was measured at the indicated time points after the addition of ABA. (c) Decrease of Neurog2 mRNA levels after 24 h of ABA stimulation. All Neurog2 mRNA levels were measured relative to expressing GFP control cells (mean ± s.e.m.; n = 3-4 biological replicas).
{kind=link}
Extended Data Figure 4 | Efficient AAV production using cell supernatant. (a) Lentiviral and AAV vectors carrying GFP were used to test transduction efficiency. (b) Primary embryonic cortical neurons were transduced with 300 and 250 μL supernatant derived from the same number of AAV or lentivirus-transfected 293FT cells. Representative images of GFP expression were collected at 7 d.p.i. Scale bars = 50 μm. (c) The depicted process was developed for the production of AAV supernatant and subsequent transduction of primary neurons. 293FT cells were transfected with an AAV vector carrying the gene of interest, the AAV1 serotype packaging vector (pAAV1), and helper plasmid (pDF6) using PEI. 48 h later, the supernatant was harvested and filtered through a 0.45 μm PVDF membrane. Primary neurons were then transduced with supernatant and remaining aliquots were stored at −80°C. Stable levels of AAV construct expression were reached after 5-6 days. AAV supernatant production following this process can be used for production of up to 96 different viral constructs in 96-well format (employed for TALE screen in neurons shown in Fig. 2c)
{kind=link}
Extended Data Figure 5 Characterizing LITEs in neurons and in vivo. (a) Impact of light duty cycle on primary neuron health. The effect of light stimulation on primary cortical neuron health was compared for duty cycles of 7%, 0.8%, and no light conditions. Calcein was used to evaluate neuron viability. Bright-field images show cell morphology and integrity. Primary cortical neurons were stimulated with the indicated duty cycle for 24 h with 5 mW/cm2 of 466 nm light. Representative images, scale bar = 50 μm. Pulses were performed in the following manner: 7% duty cycle = 1 s pulse at 0.067 Hz, 0.8% duty cycle = 0.5 s pulse at 0.0167 Hz. (b) Co-transduction efficiency of LITE components by AAV1/2 in vivo in mouse infralimbic cortex. Cells transduced by TALE(Grm2)-CIB1 alone, CRY2PHR-VP64 alone, or co-transduced were calculated as a percentage of all transduced cells (mean ± s.e.m.; n = 9 fields from 3 animals). (c) Grm2 mRNA levels were determined in primary neurons transfected with individual LITE components. Primary neurons expressing TALE(Grm2)-CIB1 alone led to a similar increase in Grm2 mRNA levels as unstimulated cells expressing the complete LITE system. (mean ± s.e.m.; n = 3-4 biological replicas)
{kind=link}
Extended Data Figure 6 | Effects of LITE Component Engineering on Activation, Background Signal, and Fold Induction. Protein modifications were employed to find LITE components resulting in reduced background transcriptional activation while improving induction ratio by light. Protein alterations are discussed in detail below. In brief, nuclear localization signals and mutations in an endogenous nuclear export signal were used to improve nuclear import of the CRY2PHR-VP64 component. Several variations of CIB1 intended to either reduce nuclear localization or CIB1 transcriptional activation were pursued in order to reduce the contribution of the TALE-CIB1 component to background activity. The results of all tested combinations of CRY2PHR-VP64 and TALE-CIB1 are shown above. The table to the left of the bar graphs indicates the particular combination of domains/mutations used for each condition. Each row of the table and bar graphs contains the component details, Light/No light activity, and induction ratio by light for the particular CRY2PHR/CIB1 combination. Combinations that resulted in both decreased background and increased fold induction compared to LITE 1.0 are highlighted in green in the table column marked “+” (t-test p<0.05). See Supplementary Discussions for detailed explanation of each modification. (mean ± s.e.m.; n = 2-3 biological replicas)
{kind=link}
Extended Data Figure 7 | Strategies for optimizing the LITE system. (a) In the absence of light, the TALE-CIB1 LITE component resides in the cytoplasm due to the absence of a nuclear localization signal, NLS (or the addition of a weak nuclear export signal, NES). The CRY2PHR-VP64 component containing a NLS on the other hand is actively imported into the nucleus on its own. (b) In the presence of blue light, TALE-CIB1 binds to CRY2PHR. The strong NLS present in CRY2PHR-VP64 now mediates nuclear import of the complex of both LITE components, enabling them to activate transcription at the targeted locus. In addition to the LITE 2.0 constructs, several CRY2PHR-VP64::TALE-CIB1 combinations from the engineered LITE component screen were of particular note. LITE 1.9.0, which combined the α-importin NLS effector construct with a mutated endogenous NLS and Δ276-307 TALE-CIB1 construct, exhibited an induction ratio greater than 9 and an absolute light activation of more than 180. LITE 1.9.1, which combined the unmodified CRY2PHR-VP64 with a mutated NLS, Δ318-334, AD5 NES TALE-CIB1 construct, achieved an induction ratio of 4 with a background activation of 1.06. A selection of other LITE 1.9 combinations with background activations lower than 2 and induction ratios ranging from 7 to 12 were also highlighted. (mean ± s.e.m.; n = 2-3 biological replicas)
{kind=link}
Acknowledgements
We thank C. Jennings for comments, F.A. Ran for help with illustrations, C. Lin for editing, M. Cunniff for technical assistance and W. Yan for computational analysis, and members of the Zhang Lab for discussion, support, and advice. S.K. is supported by a Hubert Schoemaker Fellowship from the McGovern Institute for Brain Research at MIT. M.H. is supported by a postdoctoral fellowship from the Human Frontiers Science Program. G.M.C is support by a NIH NHGRI CEGS grant (P50-HG005550). F.Z. is supported by a NIH Transformative R01 award (R01-NS073124), a NIH Director’s Pioneer Award (DP1-MH100706), the Keck, McKnight, Vallee, Damon Runyon, Searle Scholars, Klingenstein, and Simons Foundations, Bob Metcalfe and Jane Pauley. Sequence, protocol, and reagent information are available through the Zhang Lab Website at http://www.genome-engineering.org.
Footnotes
Competing Financial Interests
A patent application has been filed relating to this work, and the authors plan on making the reagents widely available to the academic community through Addgene and to provide software tools via the Zhang lab website (www.genome-engineering.org).
Author Contributions
S.K., M.D.B., and F.Z. developed the concept and designed experiments. S.K., M.D.B., A.T., P.D.H., M.H., and D.A.S. carried out LITE-related experiments and analyzed data. L.C. and P.D.H. developed the SID4× effector domain, the P11-targeting TALEs, and the abscisic acid induction system. R.P. developed the Cas9 transcription activator and repressor systems. S.K., A.T., M.D.B., P.D.H., and F.Z. wrote the manuscript with input from M.H., L.C., and G.C.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Deisseroth K. Optogenetics. Nature methods. 2011;8:26–29. doi: 10.1038/nmeth.f.324. doi:10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang F, et al. The microbial opsin family of optogenetic tools. Cell. 2011;147:1446–1457. doi: 10.1016/j.cell.2011.12.004. doi:10.1016/j.cell.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. doi:10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nature biotechnology. 2009;27:941–945. doi: 10.1038/nbt.1569. doi:10.1038/nbt.1569. [DOI] [PubMed] [Google Scholar]
- 5.Strickland D, et al. TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nature methods. 2012;9:379–384. doi: 10.1038/nmeth.1904. doi:10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy MJ, et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nature methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. doi:10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nature biotechnology. 2002;20:1041–1044. doi: 10.1038/nbt734. doi:10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- 8.Ye H, Daoud-El Baba M, Peng RW, Fussenegger M. A synthetic optogenetic transcription device enhances blood-glucose homeostasis in mice. Science. 2011;332:1565–1568. doi: 10.1126/science.1203535. doi:10.1126/science.1203535. [DOI] [PubMed] [Google Scholar]
- 9.Polstein LR, Gersbach CA. Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. Journal of the American Chemical Society. 2012;134:16480–16483. doi: 10.1021/ja3065667. doi:10.1021/ja3065667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugaj LJ, Choksi AT, Mesuda CK, Kane RS, Schaffer DV. Optogenetic protein clustering and signaling activation in mammalian cells. Nature methods. 2013 doi: 10.1038/nmeth.2360. [DOI] [PubMed] [Google Scholar]
- 11.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. doi:10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 12.Boch J, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. doi:10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 13.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. doi:10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F, et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nature biotechnology. 2011;29:149–153. doi: 10.1038/nbt.1775. doi:10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, et al. Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis. Science. 2008;322:1535–1539. doi: 10.1126/science.1163927. doi:10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- 16.Beerli RR, Segal DJ, Dreier B, Barbas CF., 3rd Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong L, Zhou R, Kuo Y.-c., Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3:968. doi: 10.1038/ncomms1962. doi: http://www.nature.com/ncomms/journal/v3/n7/suppinfo/ncomms1962_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bikard D, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic acids research. 2013 doi: 10.1093/nar/gkt520. doi:10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. doi:10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. doi:10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. doi:10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee R, et al. The signaling state of Arabidopsis cryptochrome 2 contains flavin semiquinone. The Journal of biological chemistry. 2007;282:14916–14922. doi: 10.1074/jbc.M700616200. doi:10.1074/jbc.M700616200. [DOI] [PubMed] [Google Scholar]
- 24.Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. doi:10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 26.Liang F-S, Ho WQ, Crabtree GR. Engineering the ABA Plant Stress Pathway for Regulation of Induced Proximity. Sci. Signal. 2011;4:rs2. doi: 10.1126/scisignal.2001449. doi:10.1126/scisignal.2001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holkers M, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic acids research. 2013;41:e63. doi: 10.1093/nar/gks1446. doi:10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang F, et al. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. doi:10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H, et al. Photoexcited CRY2 Interacts with CIB1 to Regulate Transcription and Floral Initiation in Arabidopsis. Science. 2008;322:1535–1539. doi: 10.1126/science.1163927. doi:10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- 30.de Groote ML, Verschure PJ, Rots MG. Epigenetic Editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic acids research. 2012;40:10596–10613. doi: 10.1093/nar/gks863. doi:10.1093/nar/gks863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Yang H, Colosi P. Effect of Genome Size on AAV Vector Packaging. Mol Ther. 2009;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McClure C, Cole KL, Wulff P, Klugmann M, Murray AJ. Production and titering of recombinant adeno-associated viral vectors. J Vis Exp. 2011:e3348. doi: 10.3791/3348. doi:10.3791/3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blecher-Gonen R, et al. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nature protocols. 2013;8:539–554. doi: 10.1038/nprot.2013.023. [DOI] [PubMed] [Google Scholar]
- 34.Szymczak AL, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nature biotechnology. 2004;22:589–594. doi: 10.1038/nbt957. doi:10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 35.Christie JM, et al. Structural tuning of the fluorescent protein iLOV for improved photostability. The Journal of biological chemistry. 2012;287:22295–22304. doi: 10.1074/jbc.M111.318881. doi:10.1074/jbc.M111.318881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Figure 1 | RNA-guided DNA binding protein Cas9 can be used to target transcription effector domains to specific genomic loci. (a) The RNA-guided nuclease Cas9 from the type II Streptococcus pyogenes CRISPR/Cas system can be converted into a nucleolytically-inactive RNA-guided DNA binding protein (Cas9**) by introducing two alanine substitutions (D10A and H840A). Schematic showing that a synthetic guide RNA (sgRNA) can direct Cas9**-effector fusion to a specific locus in the human genome. The sgRNA contains a 20bp guide sequence at the 5′ end which specifies the target sequence. On the target genomic DNA, the 20bp target site needs to be followed by a 5′-NGG PAM motif. (b, c) Schematics showing the sgRNA target sites in the human KLF4 and SOX2 loci respectively. Each target site is indicated by the blue bar and the corresponding PAM sequence is indicated by the magenta bar. (d, e) Schematics of the Cas9**-VP64 transcription activator and SID4X-Cas9** transcription repressor constructs. (f, g) Cas9**-VP64 and SID4X-Cas9** mediated activation of KLF4 and repression of SOX2 respectively. All mRNA levels were measured relative to GFP mock transfected 293FT cells (mean ± s.e.m.; n = 3 biological replicas).
Extended Data Figure 8 | TALE SID4X repressor characterization and application in neurons. (a) A synthetic repressor was constructed by concatenating 4 SID domains (SID4X). To identify the optimal TALE-repressor architecture, SID or SID4X was fused to a TALE designed to target the mouse p11 gene. (b) Fold decrease in p11 mRNA was assayed using qRT-PCR. (mean ± s.e.m.; n = 3 biological replicas) (c) General schematic of constitutive TALE transcriptional repressor packaged into AAV. Effector domain SID4X is highlighted. hSyn: human synapsin promoter; 2A: Thosea asigna virus 2A self-cleaving peptide34; WPRE: woodchuck hepatitis post-transcriptional response element; bGH pA: bovine growth hormone poly-A signal. phiLOV2.135 (330bp) was chosen as a shorter fluorescent marker to ensure efficient AAV packaging. (d) A TALE targeting either the endogenous mouse loci Grm5 and Grm2 was fused to SID4X and virally transduced into primary neurons. SID4X-mediated target gene down-regulation is shown for each TALE relative to levels in control neurons expressing GFP only. (mean ± s.e.m.; n = 3-4 biological replicas)
Extended Data Figure 9 | A diverse set of epiTALEs mediate transcriptional repression in neurons and Neuro 2a cells. (a) 24 different histone effector domains were each fused to a Grm2 targeting TALE. TALE-effector fusions were expressed in primary cortical mouse neurons using AAV transduction. Grm2 mRNA levels were measured using RT-qPCR relative to neurons transduced with GFP only. (*, p < 0.05; mean ± s.e.m.; n = 2-3 biological replicas) (b) A total of 32 epiTALEs were transfected into Neuro2A cells. 20 of them mediated significant repression of the targeted Neurog2 locus (* = p<0.05). (mean ± s.e.m.; n = 2-3 biological replicas)
Extended Data Figure 10 | epiTALEs mediating transcriptional repression along with histone modifications in Neuro 2A cells. (a) TALEs fused to histone-deacetylating epigenetic effectors NcoR and SIRT3 targeting the murine Neurog2 locus in Neuro 2A cells were assayed for repressive activity on Neurog2 transcript levels. (mean ± s.e.m.; n = 2-3 biological replicas) (b) ChIP RT-qPCR showing a reduction in H3K9 acetylation at the Neurog2 promoter for NcoR and SIRT3 epiTALEs. (mean ± s.e.m.; n = 2-3 biological replicas) (c) The epigenetic effector PHF19 with known histone methyltransferase binding activity was fused to a TALE targeting Neurog2 mediated repression of Neurog2 mRNA levels. (mean ± s.e.m.; n = 2-3 biological replicas) (d) ChIP RT-qPCR showing an increase in H3K27me3 levels at the Neurog2 promoter for the PHF19 epiTALE. (mean ± s.e.m.; n = 2-3 biological replicas)
Extended Data Figure 2 | Engineering of light stimulation parameters and activation domains of LITEs. (a) Illustration of the absorption spectrum of CRY2 in vitro. Cryptochrome 2 was optimally activated by 350-475 nm light23. A sharp drop in absorption and activation was seen for wavelengths greater than 480 nm. Spectrum was adapted from Ref. 23. (b) Impact of illumination duty cycle on LITE-mediated gene expression. Varying duty cycles (illumination as percentage of total time) were used to stimulate 293FT cells expressing LITEs targeting the KLF4 gene, in order to investigate the effect of duty cycle on LITE activity. KLF4 expression levels were compared to cells expressing GFP only. Stimulation parameters were: 466 nm, 5 mW/cm2 for 24 h. Pulses were performed at 0.067 Hz with the following durations: 1.7% = 0.25 s pulse, 7% = 1 s pulse, 27% = 4 s pulse, 100% = constant illumination. (mean ± s.e.m.; n = 3-4 biological replicas) (c) The transcriptional activity of CRY2PHR:: CIB1 LITE was found to vary according to the intensity of 466 nm blue light. Neuro 2a cells were stimulated for 24 h hours at a 7% duty cycle (1s pulses at 0.066Hz) (d) Light-induced toxicity measured as the percentage of cells positive for red-fluorescent ethidium homodimer-1 versus calcein-positive cells. All Neurog2 mRNA levels were measured relative to cells expressing GFP only (mean ± s.e.m.; n = 3-4 biological replicas) (e) We compared the activation domains VP16 and p65 in addition to VP64 to test the modularity of the LITE CIB1-effector component. Neurog2 up-regulation with and without light by LITEs using different transcriptional activation domains (VP16, VP64, and p65). Neuro-2a cells transfected with LITE were stimulated for 24 h with 466 nm light at an intensity of 5mW/cm2 and a duty cycle of 7% (1 s pulses at 0.066 Hz). All three domains produced a significant light-dependent Neurog2 mRNA up-regulation (p < 0.001,). We selected VP64 for subsequent experiments due to its lower basal activity in the absence of light-stimulation. (mean ± s.e.m.; n = 3-4 biological replicas)
Extended Data Figure 3 | Chemical induction of endogenous gene transcription. (a) Schematic showing the design of a chemical inducible two hybrid TALE system based on the abscisic acid (ABA) receptor system. ABI and PYL dimerize upon the addition of ABA and dissociates when ABA is withdrawn. (b) Time course of ABA-dependent Neurog2 up-regulation. 250 μM of ABA was added to HEK 293FT cells expressing TALE(Neurog2)-ABI and PYL-VP64. Fold mRNA increase was measured at the indicated time points after the addition of ABA. (c) Decrease of Neurog2 mRNA levels after 24 h of ABA stimulation. All Neurog2 mRNA levels were measured relative to expressing GFP control cells (mean ± s.e.m.; n = 3-4 biological replicas).
Extended Data Figure 4 | Efficient AAV production using cell supernatant. (a) Lentiviral and AAV vectors carrying GFP were used to test transduction efficiency. (b) Primary embryonic cortical neurons were transduced with 300 and 250 μL supernatant derived from the same number of AAV or lentivirus-transfected 293FT cells. Representative images of GFP expression were collected at 7 d.p.i. Scale bars = 50 μm. (c) The depicted process was developed for the production of AAV supernatant and subsequent transduction of primary neurons. 293FT cells were transfected with an AAV vector carrying the gene of interest, the AAV1 serotype packaging vector (pAAV1), and helper plasmid (pDF6) using PEI. 48 h later, the supernatant was harvested and filtered through a 0.45 μm PVDF membrane. Primary neurons were then transduced with supernatant and remaining aliquots were stored at −80°C. Stable levels of AAV construct expression were reached after 5-6 days. AAV supernatant production following this process can be used for production of up to 96 different viral constructs in 96-well format (employed for TALE screen in neurons shown in Fig. 2c)
Extended Data Figure 5 Characterizing LITEs in neurons and in vivo. (a) Impact of light duty cycle on primary neuron health. The effect of light stimulation on primary cortical neuron health was compared for duty cycles of 7%, 0.8%, and no light conditions. Calcein was used to evaluate neuron viability. Bright-field images show cell morphology and integrity. Primary cortical neurons were stimulated with the indicated duty cycle for 24 h with 5 mW/cm2 of 466 nm light. Representative images, scale bar = 50 μm. Pulses were performed in the following manner: 7% duty cycle = 1 s pulse at 0.067 Hz, 0.8% duty cycle = 0.5 s pulse at 0.0167 Hz. (b) Co-transduction efficiency of LITE components by AAV1/2 in vivo in mouse infralimbic cortex. Cells transduced by TALE(Grm2)-CIB1 alone, CRY2PHR-VP64 alone, or co-transduced were calculated as a percentage of all transduced cells (mean ± s.e.m.; n = 9 fields from 3 animals). (c) Grm2 mRNA levels were determined in primary neurons transfected with individual LITE components. Primary neurons expressing TALE(Grm2)-CIB1 alone led to a similar increase in Grm2 mRNA levels as unstimulated cells expressing the complete LITE system. (mean ± s.e.m.; n = 3-4 biological replicas)
Extended Data Figure 6 | Effects of LITE Component Engineering on Activation, Background Signal, and Fold Induction. Protein modifications were employed to find LITE components resulting in reduced background transcriptional activation while improving induction ratio by light. Protein alterations are discussed in detail below. In brief, nuclear localization signals and mutations in an endogenous nuclear export signal were used to improve nuclear import of the CRY2PHR-VP64 component. Several variations of CIB1 intended to either reduce nuclear localization or CIB1 transcriptional activation were pursued in order to reduce the contribution of the TALE-CIB1 component to background activity. The results of all tested combinations of CRY2PHR-VP64 and TALE-CIB1 are shown above. The table to the left of the bar graphs indicates the particular combination of domains/mutations used for each condition. Each row of the table and bar graphs contains the component details, Light/No light activity, and induction ratio by light for the particular CRY2PHR/CIB1 combination. Combinations that resulted in both decreased background and increased fold induction compared to LITE 1.0 are highlighted in green in the table column marked “+” (t-test p<0.05). See Supplementary Discussions for detailed explanation of each modification. (mean ± s.e.m.; n = 2-3 biological replicas)
Extended Data Figure 7 | Strategies for optimizing the LITE system. (a) In the absence of light, the TALE-CIB1 LITE component resides in the cytoplasm due to the absence of a nuclear localization signal, NLS (or the addition of a weak nuclear export signal, NES). The CRY2PHR-VP64 component containing a NLS on the other hand is actively imported into the nucleus on its own. (b) In the presence of blue light, TALE-CIB1 binds to CRY2PHR. The strong NLS present in CRY2PHR-VP64 now mediates nuclear import of the complex of both LITE components, enabling them to activate transcription at the targeted locus. In addition to the LITE 2.0 constructs, several CRY2PHR-VP64::TALE-CIB1 combinations from the engineered LITE component screen were of particular note. LITE 1.9.0, which combined the α-importin NLS effector construct with a mutated endogenous NLS and Δ276-307 TALE-CIB1 construct, exhibited an induction ratio greater than 9 and an absolute light activation of more than 180. LITE 1.9.1, which combined the unmodified CRY2PHR-VP64 with a mutated NLS, Δ318-334, AD5 NES TALE-CIB1 construct, achieved an induction ratio of 4 with a background activation of 1.06. A selection of other LITE 1.9 combinations with background activations lower than 2 and induction ratios ranging from 7 to 12 were also highlighted. (mean ± s.e.m.; n = 2-3 biological replicas)