

Abstract
The RNA-binding proteins involved in regulation of mRNA post-transcriptional processing and translation control the fates of thousands of mRNA transcripts and basic cellular processes. The best studied of these, HuR, is well characterized as a mediator of mRNA stability and translation, and more recently, as a factor in nuclear functions such as pre-mRNA splicing. Due to HuR’s role in regulating thousands of mRNA transcripts, including those for other RNA-binding proteins, HuR can act as a master regulator of cell survival and proliferation. HuR itself is subject to multiple post-translational modifications including regulation of its nucleocytoplasmic distribution. However, the mechanisms that govern HuR levels in the cell have only recently begun to be defined. These mechanisms are critical to cell health, as it has become clear in recent years that aberrant expression of HuR can lead alternately to decreased cell viability or to promotion of pathological proliferation and invasiveness. HuR is expressed as alternate mRNAs that vary in their untranslated regions, leading to differences in transcript stability and translatability. Multiple transcription factors and modulators of mRNA stability that regulate HuR mRNA expression have been identified. In addition, translation of HuR is regulated by numerous microRNAs, several of which have been demonstrated to have anti-tumor properties due to their suppression of HuR expression. This review summarizes the current state of knowledge of the factors that regulate HuR expression, along with the circumstances under which these factors contribute to cancer and inflammation.
Keywords: HuR, RNA binding proteins, Transcription, RNA stability, Translation, Protein stability, Cell stress, MicroRNAs, Cancer
Core tip: HuR is an RNA-binding protein that regulates post-transcriptional processing of thousands of mRNAs, including many that encode proteins that are critical to basic cellular functions. Thus, while loss of HuR can lead to cell death, pathological overexpression of HuR is associated with numerous types of cancer. However, the mechanisms that govern expression of HuR have only begun to be delineated. This review summarizes the current state of knowledge of these mechanisms and how they may contribute to cell survival and pathology.
INTRODUCTION
HuR is a ubiquitously expressed RNA-binding protein (RBP) of the embryonic lethal, altered vision (ELAV) family, and is one of the best-described regulators of mRNA fate. HuR produces broad cellular effects by binding to its target mRNAs, which number in the low thousands, and by aiding in mRNA splicing, stability, and most often translation, although a small subset of targets are translationally repressed by HuR. In recent years, transcriptome analysis has identified the mammalian mRNA targets of HuR[1,2], and has further identified HuR as a master regulator of other RNA binding proteins[3]. Because of its broad effects on so many aspects of post-transcriptional gene control, HuR may be considered as a “regulator of regulators”[3].
HuR binds its mRNA targets through sequences rich in uridine or adenosine/uridine (AREs), which are most typically present in non-coding regions of the transcript, particularly introns and the 3’ untranslated region (UTR). Under normal growth conditions, HuR is present primarily in the nucleus, but can shuttle to the cytoplasm to aid in mRNA processing. Translocation to and sequestration in the cytoplasm occurs under conditions of cellular stress (e.g., UV irradiation, nutrient or energy depletion, heat shock[4-7]) where it is believed to aid in coordinating mRNA turnover in a manner that protects cell survival[8]. HuR is also a potent promoter of cell proliferation and survival[9], but during lethal stresses, can aid in promoting caspase-mediated apoptosis[10]. Thus, HuR activity is critical for regulating pathways that mediate cell survival and death.
Aberrant overexpression of HuR can lead to cellular transformation, and indeed, heightened HuR levels have been observed in tumors from tissues throughout the body. Thus, tight regulation of HuR expression is key to promoting healthy cell survival while at the same time preventing pathological proliferation. Not surprisingly, regulation of HuR expression is intricately controlled at multiple levels of transcriptional, post-transcriptional, translational, and post-translational control. Here we will review the current state of knowledge of these mechanisms and discuss how HuR expression is controlled in physiologically adaptive ways such as response to cellular stress, and in situations that lead to pathological proliferation of cells and tumor formation.
PHYSIOLOGICAL EXPRESSION OF HUR AND RESPONSES TO STRESS
Regulation of HuR mRNA expression
The first studies of genetic regulation of HuR were performed in 2000 when the 5’ region of the mouse HuR gene was isolated and mapped. Primer extension experiments using mRNA from various tissues and cell lines revealed three products, suggesting the presence of multiple alternative transcriptional start sites. A SpeI-SmaI restriction fragment containing most of exon I and a few hundred bases of upstream sequence was demonstrated to contain transcriptional activity in reporter assays[11]. However, a definitive transcriptional activator was not identified until 2008, when it was revealed that HuR expression was mediated through the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt)/nuclear factor kappa B(NF-κB) pathway[12]. An NF-κB binding site in the HuR promoter was described as starting 133 bases upstream of the transcriptional start site, although a specific start sequence was not specified. Nonetheless, the activity of this binding site was clearly proven in various gastric carcinoma cell lines. Later studies from our own laboratory confirmed that PI3K/Akt/NF-κB regulation of HuR is also present in renal proximal tubule cells, and that this pathway is one arm of a positive feedback loop that results not only in transcriptional activation of HuR, but also in continued increases in Akt activity[13]. Therefore, without a “braking” mechanism for this signaling pathway, heightened levels of Akt and HuR can lead to tumorigenic conditions, as will be described below.
A second level of regulation for HuR was identified as a consequence of the use of alternate transcriptional start sites. Our work on the role of HuR in renal proximal tubule cells during metabolic stress had identified two transcriptional start sites, at approximately 150 and 20 bases upstream of the coding region[14,15]. The 5’ UTR of these alternate transcripts are very different; the longer mRNA contains a G+C-rich 5’ UTR with a great deal of predicted secondary structure, while the shorter mRNA contains an A+T-rich sequence with very little secondary structure. In vitro translation assays demonstrated the shorter mRNA to be much more readily translated than the longer form[14]. During normal growth the alternate transcriptional start sites were used at roughly equal frequencies, but following metabolic stresses to kidney cells such as thapsigargin treatment or energy depletion, expression of the shorter transcript was increased[15]. Expression of this transcript was found to be regulated by multiple Smad 1/5/8 binding sites that were present in the 5’ UTR of the longer transcript. Expression from these sites was further shown to be responsive to bone morphogenetic protein 7 (BMP-7), which notably is a key regulator of renal development and recovery from ischemic stresses[16-20]. These findings suggest that metabolic stresses may prime cells to synthesize a more readily translatable form of HuR mRNA to aid in cell survival. Figure 1 depicts the transcriptional mediators and Akt activation pathway that lead to increased HuR mRNA expression.
Figure 1.
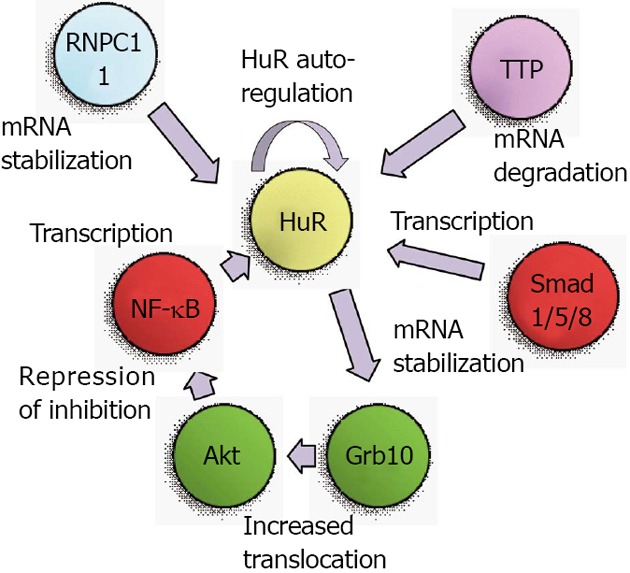
Translational and post-translational regulators of HuR protein levels. TTP: Tristetraprolin; Akt: Protein kinase B; NF-κB: Nuclear factor kappa B.
Production of transcripts with alternate 3’ UTR due to multiple polyadenylation sites is common in both rodents and humans, and the choice of polyadenylation sites may be used to achieve a specific biological outcome. In many cases, this choice can produce either a long 3’ UTR that contains AREs or a shorter 3’ UTR that lacks AREs[21]. In this way, it is expected that mRNAs from a single gene may be produced with lesser or greater stability. The HuR gene itself encodes two polyadenylation variants, a longer and more labile form containing functional AREs, and a shorter, more predominant form that lacks AREs[22]. It was subsequently demonstrated that HuR autoregulates its expression by virtue of control over the production of these variants. Briefly, HuR regulates its own expression through a negative feedback loop[23]. Nuclear HuR can bind its own pre-mRNA and increase production of the longer, more labile variant, thus keeping HuR levels at constant and relatively low physiological levels. These results also suggested that under conditions in which HuR is primarily cytoplasmic, the negative feedback loop may be interrupted, thus leading to increased HuR levels and potential oncogenic transformation of cells. Under some circumstances, HuR may also serve as a positive regulator of its own expression, as a role for HuR has been proposed in facilitating export of HuR mRNA from the nucleus in senescent cells[24]. More recent studies have implicated the RNA-binding protein RNPC1 as a mediator of HuR mRNA stability. RNPC1, like HuR, can bind to AREs and regulate transcript stability or translation. While it was demonstrated that RNPC1 stabilizes HuR mRNA by binding to its 3’ UTR, it is currently unclear whether RNPC1 and HuR bind the same sequences within the 3’ UTR[25]. As will be described below, the RBP tristetraprolin (TTP) can also bind to the HuR mRNA to promote its degradation, and the cellular levels of TTP can affect promotion of a tumor phenotype[26]. These studies indicate that regulation of HuR mRNA stability is likely to involve multiple protein binding sites and RBPs that can vary depending on the state of cellular health and growth. Figure 1 summarizes the transcriptional and post-transcriptional mediators that regulate expression of HuR mRNA.
Expression patterns during development, aging, and cellular senescence
HuR was first described as the fourth member of the ELAV family of proteins that was originally identified in Drosophila. The three original members (currently called HuB, HuC, and HuD) were shown to have neuron-specific expression, but unexpectedly, a fourth member (now called HuR) was predicted by polymerase chain reaction and low-stringency cDNA library screening of vertebrates and was expressed in all tissues tested, including brain, kidney, lung, heart, liver, muscle, skin, testis, and ovary[27]. Shortly thereafter, a cDNA was isolated from HeLa cells and the corresponding mRNA was similarly found to be expressed in a wide variety of human tissues[28]. A murine version was described one year later[29]. Assays of mouse tissues from early embryogenesis to adulthood were performed to determine levels of HuR expression during vertebrate development. From the small number of embryonic and extra-embryonic tissues selected for assays, HuR was shown to have developmental age- and tissue-specific variability in expression. Interestingly, HuR levels strongly paralleled levels of AUF1, another RBP that binds AREs in target mRNAs, but promotes their degradation[30,31]. A more comprehensive examination of HuR levels in adult murine tissues showed that HuR protein was most strongly expressed in lymphoid tissues, intestine, and testes, with moderate expression in liver and uterus, and the lowest expression in brain, heart, lung, kidney, skeletal muscle, and ovary[32].
Early studies of HuR expression also examined its levels and effects on cell health during cellular aging. In multiple in vitro models of cellular senescence, HuR levels were shown to decrease, as did the half-lives of HuR’s corresponding mRNA targets. Further, HuR overexpression and knockdown of expression with antisense RNAs revealed a direct relationship between the levels of HuR and a “younger” cell phenotype[33]. However, a follow-up study examining the levels of mRNA regulatory proteins in human tissue arrays from individuals of various ages, revealed that HuR expression remained relatively unchanged with increasing age[34]. Therefore, the significance of the cellular senescence studies as they relate to human aging is unclear. The human tissue array study also confirmed previous murine studies in demonstrating tissue-specific levels of expression and the parallel expression of HuR and ARE-binding protein AUF1. Other studies, described below, indicate the importance of maintaining appropriate balances of RBPs that both degrade and stabilize ARE-containing mRNAs[26], so the parallel expression of HuR and AUF1 is likely a mechanism to ensure an appropriate balance of these mRNA transcripts.
Regulation of HuR translation and protein stability
In recent years, regulation of HuR biosynthesis by microRNAs (miRNAs) has been identified as a key process in controlling HuR levels. Multiple miRNAs, including miR-16 and miR-519, have been identified as inhibitors of HuR translation via direct binding of HuR mRNA. These miRNAs have been implicated in suppression of tumor cell growth through inhibition of HuR synthesis, and discussion of their function will be addressed below. In this section, we will discuss mechanisms that modulate HuR expression through regulators of protein stability and cleavage.
HuR is subject to multiple levels of post-translational regulation from diverse signals. As stated above, nucleocytoplasmic shuttling is an important mechanism by which HuR can be triggered to exert differential effects in cells. As previously reviewed, phosphorylation by various kinases, including checkpoint kinase 2 (Chk2), Cdk1, p38, and PKC, can regulate HuR levels in the cytoplasm and/or binding to target mRNAs[35]. HuR methylation by CARM1 (co-activator-associated arginine methyltransferase 1) can similarly affect HuR activity[36]. However, control of HuR protein levels and function through degradation or cleavage also has been shown to be key to HuR’s effects on cellular processes. Mild heat shock was demonstrated to rapidly decrease HuR protein without altering mRNA levels or translation rates. This loss of HuR was found to be due to ubiquitin-mediated proteolysis and is believed to enhance cell survival by altering the stability and/or translation of HuR target mRNAs[37]. Through a different pathway, ubiquitin-mediated proteolysis was also shown to cause HuR degradation when cancer cells are subjected to inhibition of glycolysis, which may represent an attempt to slow proliferation in the absence of cellular energy[38].
Post-translational regulation of HuR protein levels can be altered depending on the context of the stress. Mild stresses most often induce translocation of HuR from the nucleus to the cytoplasm, resulting in increased cell survival. It was reported that lethal stress such as treatment with the apoptosis inducer staurosporine also results in HuR translocation to the cytoplasm; however, once there, HuR may be cleaved by caspases, leading to an enhanced apoptotic response[39]. Similar caspase-mediated cleavage events were noted in cells subjected to chronic, but not acute, hypoxia. Further, one of the HuR cleavage products produced was demonstrated to bind the 3’ UTR of a HuR target mRNA (c-myc) and block its translation, leading to decreased cell viability[40]. A very recent study has demonstrated that under a lethal stress (staurosporine), HuR, which normally binds and stabilizes both pro- and anti-apoptotic mRNA targets, was cleaved and the resulting cleavage products bound and stabilized the pro-apoptotic mRNA caspase-9, but not the anti-apoptotic target prothymosin[10]. These results all suggest that the cleavage of HuR under lethal stress results in products that shift HuR’s function from a pro-survival factor to a pro-apoptosis activator.
PATHOLOGICAL OVEREXPRESSION OF HUR, REGULATION BY MICRO-RNAS, AND CANCER
HuR levels are elevated in numerous types of cancer
The importance of HuR to cell survival and proliferation is made evident by the many types of tissues in which elevated HuR levels are associated with cancer. These tumor types include breast, lung, ovarian and colon cancers[41] and numerous other tissues. Notably, while HuR is typically localized primarily to nuclei, high cytoplasmic levels of HuR are known to be associated with worse prognosis in numerous types of cancers including human lung adenocarcinoma[42], gall bladder carcinoma[43], urothelial carcinoma[44], ovarian cancer[45], breast cancer[46,47], cervical cancer[48], laryngeal squamous cell cancer[49], and colon cancer[50]. HuR has been shown to interact with and regulate a large number of mRNA transcripts with AREs that are involved in oncogenic cellular transformation. As previously reviewed, these include regulators of cell growth and division (e.g., c-myc, cyclins), gene products involved in invasion and metastasis (e.g., MMP-9), pro-survival mediators (e.g., prothymosin-α), and products that can trigger local angiogenesis [e.g., vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1 (HIF-1)][41].
Early work to examine the mechanisms behind regulation of HuR expression in cancer was performed in gastric tumor cells that expressed high levels of HuR. No genetic or epigenetic alterations were noted in these cells, but the elevated HuR expression was found to be dependent on excessive levels of PI3K-Akt signaling. Further, NF-κB, a downstream regulator of Akt, was shown to directly activate HuR transcription through a binding site in the HuR promoter. Akt was also implicated in promoting transport of HuR from the nucleus to the cytoplasm[12]. Although this study clearly implicated aberrant transcriptional control of HuR as contributing to cancer, most of our understanding of HuR’s regulation in cancer cells comes from analysis of its interactions with miRNAs. MiRNAs are small (about 22 nucleotides) noncoding RNA molecules that post-transcriptionally regulate gene expression by inducing mRNA degradation and/or suppressing translation. The interplay between HuR and microRNAs is complex, since defined miRNAs directly regulate HuR expression, and HuR is capable of inhibiting miRNA-mediated suppression or activation of target mRNAs. The latter effect occurs mostly in mRNAs that contain AREs downstream of miRNA binding sites in their 3’ UTR. It is postulated that miRNA target sites extensively overlap with HuR binding sites that are observed even in the intronic regions of various growth promoting gene transcripts[51], and the relationship between HuR and various miRNAs is usually functionally antagonistic. Competitive interaction usually results in an enhanced gene expression if HuR-mRNA binding dominates. However, when both HuR and miRNAs co-operatively bind transcripts, such mRNAs are usually expressed at lower levels[52]. MicroRNAs are differentially expressed in tumor cells and their interactions with RBPs such as HuR may eventually determine the outcome of tumor progression, chemotherapy and drug resistance. The variation of miRNA expression in primary versus metastatic tumors may partially explain the aggravated tumorigenic response in spite of interventional procedures in a number of cancers[53].
Regulation of HuR expression by microRNAs
The first miRNA demonstrated to regulate HuR was miR-519, as predicted by sequence analysis and confirmed by experimental procedures in 2008[54]. MiR-519 binding sites were identified in both the coding region and 3’ UTR of HuR. MiR-519 was shown to inhibit HuR expression in multiple tumor cell lines by suppressing HuR translation, but not HuR mRNA levels. Modulating the levels of miR-519 within cells affected HuR downstream targets. Not unexpectedly, decreasing the ability of miR-519 to bind HuR (through addition of antisense miR-519), increased HuR levels and the rate of cell division. In a subsequent study, HuR and miR-519 levels were examined in pairs of cancerous and adjacent healthy tissue[55]. HuR protein, but not mRNA, levels were increased in the cancer samples, and miR-519 levels were markedly reduced. miR-519 was also shown to inhibit tumor growth from HeLa cells injected into athymic mice, supporting the notion of miR-519 as a tumor suppressor that acts through HuR[55]. Notably, miR-519 levels were demonstrated to increase in a model of cellular senescence, suggesting that triggering of senescence through inhibition of HuR is a mechanism by which tumor suppression may occur[56].
In the last five years, several new miRNA regulators of HuR have been identified. MiR-16 was demonstrated to translationally repress HuR in breast cancer cells by interacting with the 3’ UTR of HuR mRNA[57]. This miRNA also suppresses translation of COX-2, tumor necrosis factor-α and Bcl-2[58,59], which, interestingly, are all tumor-promoting genes positively regulated by HuR. The complexity of the interaction between HuR and miR-16 was demonstrated to an even greater degree when it was shown that association of a HuR/miR-16 complex with AREs of several target transcripts could facilitate inhibition of miR-16 expression in colorectal cancer cells[60]. Thus, the tumor suppressor activity of miR-16 and the tumor-promoting activities of HuR appear to antagonize one another at multiple levels.
miR-125a was first reported to inhibit cell growth and promote apoptosis by translationally repressing HuR in breast cancer cells[61]. In another study, miR-125 was shown to inhibit phosphorylation of Akt in breast cancer cells[62]. This suppression of Akt activation could interfere with the growth-promoting environment through various downstream pathways, one of which is the transcriptional activation of HuR expression through Akt/NF-κB signaling[12,13]. Thus, miR-125 may inhibit HuR expression at multiple levels, through direct translational suppression and through indirect inhibition of transcription. Overexpression of another miRNA, miR-34a, was shown to suppress HuR protein levels in prostate cancer cells, thus modulating cell proliferation and drug resistance in those cells[63]. However, no potential binding sites for miR-34a were found by in silico analysis of the HuR 3’ UTR, suggesting that miR-34a may regulate HuR through binding in other regions of the transcript or through other mechanisms. MiR-9 similarly acts as a tumor suppressor by directly binding the 3’ UTR of HuR, thus suppressing HuR expression and expression of its downstream targets[64]. HuR has also recently been reported to be a target of miR-146, a potent anti-inflammatory molecule[65]. HuR was shown to be a direct target of miR-146, which suppresses both HuR mRNA and protein levels. HuR is established as a regulator of mRNAs involved in inflammation[35,66], as well as a positive regulator of NF-κB activity[13,67]. Thus, one pathway through which miR-146 exerts its anti-inflammatory effects is through suppression of HuR.
While the miRNAs described above all directly bind to HuR mRNA and inhibit synthesis of the protein, other miRNAs can positively regulate HuR synthesis through indirect mechanisms. MiR-29a, a miRNA abundant in breast cancer cells, binds to and degrades the mRNA transcript of tristetraprolin (TTP), another RBP that works to promote decay of target mRNAs. Because HuR mRNA is a target of TTP-mediated degradation, miR-29a’s overall effects are to reduce TTP levels while increasing HuR expression. This imbalance in the HuR/TTP ratio correlated with increased expression of ARE-containing, tumor-promoting mRNAs. Importantly, inhibition of miR-29a reversed the imbalance, suggesting this microRNA as a potential target for inhibition in breast cancer[26]. This study demonstrates that miRNA-mediated regulation of other RBPs is critical to the overall activity of HuR and provides insight into ways in which a network of RBPs can control cell fate. Figure 2 summarizes the effects of miRNAs along with post-translational mechanisms in regulating cellular levels of HuR.
Figure 2.
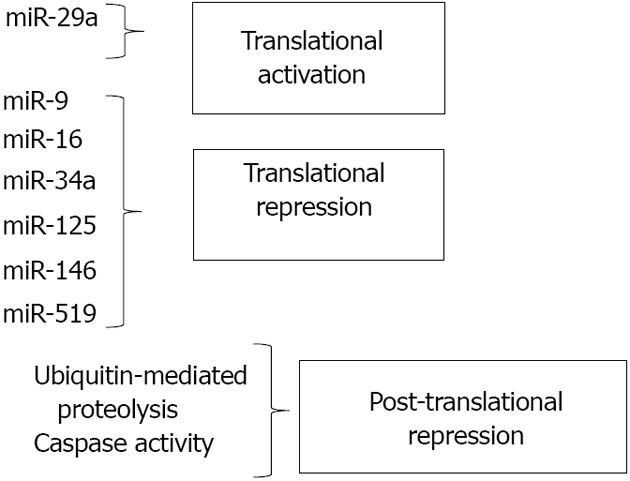
Regulators of HuR mRNA Expression.
SUMMARY AND FUTURE DIRECTIONS
The diverse molecular functions of HuR in both normal and malignant tissues have prompted researchers to probe the role of this master regulator in various types of human cancer, inflammation, and other diseases. Elucidating the mechanisms of transcriptional, post-transcriptional, translational, and post-translational regulation of HuR in both cellular stress and disease can provide critical insights into HuR’s overarching control of cellular processes. Although our knowledge of these mechanisms has expanded rapidly over the last decade, there are many aspects of HuR expression that still require study. The full range of transcription factors that regulate HuR expression is not yet elucidated, and how aberrant transcription of HuR might lead to cancer and other diseases is largely unexplored. Further, the functional relevance of HuR mRNAs with alternate 5’ and 3’ UTRs is still not well developed. Multiple studies, including our own, have noted uncoupling of HuR mRNA and protein levels in both normal and malignant tissues[29,66,68], and the mechanisms behind this uncoupling must still be investigated. However, this phenomenon is likely to be related, at least in part, to the presence of alternate mRNA HuR transcripts with different translatabilities and mRNA half-lives. How these alternate mRNAs are generated and translated during normal cell growth, cellular stress, and oncogenesis must still be determined. It is clear that HuR itself is likely to be subjected to post-transcriptional regulation by a variety of RBPs that are still undefined.
Increasing evidence demonstrates that miRNAs are involved in a complex, intricate network with HuR to post-transcriptionally regulate genes involved in development, stress, cell cycle, and cell survival, and this interplay might very well regulate a multitude of disease pathways. Fine tuning of such regulatory networks between a large repertoire of miRNAs and RBPs very well might be a trigger or a switch dictating the fate of every cell in the human body. Although it is clear from murine knockout studies that HuR expression is critical for organismal development and survival[69], little is known about how differences in HuR expression among various tissues and at different stages of development affect these processes.
In this review, we have used HuR’s role in cancer to illustrate how alteration of its expression can contribute to human disease. MiRNA and HuR interactions are in the limelight for their outcomes in the field of cancer, but it is also clear that alterations in HuR expression are associated with other physiological and pathological processes. For example, expression of HuR mRNA, as well as two of its targets, the VEGF and HIF-1α mRNAs, was shown to increase in animal models of hypoxia[70]. Subsequently, we demonstrated increased levels of HuR protein expression in rat kidneys subjected to ischemia-reperfusion injury, and only in the regions of the kidney susceptible to damage[14]. Heightened HuR levels have also been associated with a number of vascular pathologies[71]. However, the mechanisms behind these changes in HuR levels have not been determined. Through in vitro studies of normal, stressed, and cancer cells, there now exists a catalogue of mechanisms by which HuR expression is regulated. These findings should provide a strong basis for understanding the molecular changes that result in altered HuR expression in other diseases. Work to extend these findings to physiological and disease processes at the whole animal level should now be undertaken.
Footnotes
P- Reviewers: Demonacos C, Teng RJ S- Editor: Zhai HH L- Editor: A E- Editor: Yan JL
References
- 1.Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dassi E, Zuccotti P, Leo S, Provenzani A, Assfalg M, D’Onofrio M, Riva P, Quattrone A. Hyper conserved elements in vertebrate mRNA 3’-UTRs reveal a translational network of RNA-binding proteins controlled by HuR. Nucleic Acids Res. 2013;41:3201–3216. doi: 10.1093/nar/gkt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol. 2000;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallouzi IE, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci USA. 2000;97:3073–3078. doi: 10.1073/pnas.97.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaman I, Fernandez J, Sarkar B, Schneider RJ, Snider MD, Nagy LE, Hatzoglou M. Nutritional control of mRNA stability is mediated by a conserved AU-rich element that binds the cytoplasmic shuttling protein HuR. J Biol Chem. 2002;277:41539–41546. doi: 10.1074/jbc.M204850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyaraj S, Dakhlallah D, Hill SR, Lee BS. HuR stabilizes vacuolar H+-translocating ATPase mRNA during cellular energy depletion. J Biol Chem. 2005;280:37957–37964. doi: 10.1074/jbc.M502883200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Roretz C, Di Marco S, Mazroui R, Gallouzi IE. Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wiley Interdiscip Rev RNA. 2011;2:336–347. doi: 10.1002/wrna.55. [DOI] [PubMed] [Google Scholar]
- 9.Abdelmohsen K, Lal A, Kim HH, Gorospe M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle. 2007;6:1288–1292. doi: 10.4161/cc.6.11.4299. [DOI] [PubMed] [Google Scholar]
- 10.von Roretz C, Lian XJ, Macri AM, Punjani N, Clair E, Drouin O, Dormoy-Raclet V, Ma JF, Gallouzi IE. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ. 2013;20:154–168. doi: 10.1038/cdd.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King PH, Fuller JJ, Nabors LB, Detloff PJ. Analysis of the 5’ end of the mouse Elavl1 (mHuA) gene reveals a transcriptional regulatory element and evidence for conserved genomic organization. Gene. 2000;242:125–131. doi: 10.1016/s0378-1119(99)00537-5. [DOI] [PubMed] [Google Scholar]
- 12.Kang MJ, Ryu BK, Lee MG, Han J, Lee JH, Ha TK, Byun DS, Chae KS, Lee BH, Chun HS, et al. NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology. 2008;135:2030–2042, 2042.e1-3. doi: 10.1053/j.gastro.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Singh M, Martinez AR, Govindaraju S, Lee BS. HuR inhibits apoptosis by amplifying Akt signaling through a positive feedback loop. J Cell Physiol. 2013;228:182–189. doi: 10.1002/jcp.24120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayupova DA, Singh M, Leonard EC, Basile DP, Lee BS. Expression of the RNA-stabilizing protein HuR in ischemia-reperfusion injury of rat kidney. Am J Physiol Renal Physiol. 2009;297:F95–F105. doi: 10.1152/ajprenal.90632.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeyaraj SC, Singh M, Ayupova DA, Govindaraju S, Lee BS. Transcriptional control of human antigen R by bone morphogenetic protein. J Biol Chem. 2010;285:4432–4440. doi: 10.1074/jbc.M109.062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, Loveday K, Klahr S, Sampath TK, Morrissey J. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol. 2000;279:F130–F143. doi: 10.1152/ajprenal.2000.279.1.F130. [DOI] [PubMed] [Google Scholar]
- 17.Vukicevic S, Basic V, Rogic D, Basic N, Shih MS, Shepard A, Jin D, Dattatreyamurty B, Jones W, Dorai H, et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest. 1998;102:202–214. doi: 10.1172/JCI2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Chen Q, Simon TC, Strebeck F, Chaudhary L, Morrissey J, Liapis H, Klahr S, Hruska KA. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–2049. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 19.Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Müller GA, Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 20.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 21.Khabar KS, Bakheet T, Williams BR. AU-rich transient response transcripts in the human genome: expressed sequence tag clustering and gene discovery approach. Genomics. 2005;85:165–175. doi: 10.1016/j.ygeno.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Al-Ahmadi W, Al-Ghamdi M, Al-Haj L, Al-Saif M, Khabar KS. Alternative polyadenylation variants of the RNA binding protein, HuR: abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Res. 2009;37:3612–3624. doi: 10.1093/nar/gkp223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai W, Zhang G, Makeyev EV. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 2012;40:787–800. doi: 10.1093/nar/gkr783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi J, Chang N, Liu X, Guo G, Xue L, Tong T, Gorospe M, Wang W. Reduced nuclear export of HuR mRNA by HuR is linked to the loss of HuR in replicative senescence. Nucleic Acids Res. 2010;38:1547–1558. doi: 10.1093/nar/gkp1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho SJ, Jung YS, Zhang J, Chen X. The RNA-binding protein RNPC1 stabilizes the mRNA encoding the RNA-binding protein HuR and cooperates with HuR to suppress cell proliferation. J Biol Chem. 2012;287:14535–14544. doi: 10.1074/jbc.M111.326827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Ahmadi W, Al-Ghamdi M, Al-Souhibani N, Khabar KS. miR-29a inhibition normalizes HuR over-expression and aberrant AU-rich mRNA stability in invasive cancer. J Pathol. 2013;230:28–38. doi: 10.1002/path.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Good PJ. A conserved family of elav-like genes in vertebrates. Proc Natl Acad Sci USA. 1995;92:4557–4561. doi: 10.1073/pnas.92.10.4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996;271:8144–8151. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- 29.Okano HJ, Darnell RB. A hierarchy of Hu RNA binding proteins in developing and adult neurons. J Neurosci. 1997;17:3024–3037. doi: 10.1523/JNEUROSCI.17-09-03024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gouble A, Morello D. Synchronous and regulated expression of two AU-binding proteins, AUF1 and HuR, throughout murine development. Oncogene. 2000;19:5377–5384. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- 31.Lafon I, Carballès F, Brewer G, Poiret M, Morello D. Developmental expression of AUF1 and HuR, two c-myc mRNA binding proteins. Oncogene. 1998;16:3413–3421. doi: 10.1038/sj.onc.1201895. [DOI] [PubMed] [Google Scholar]
- 32.Lu JY, Schneider RJ. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J Biol Chem. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M. Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol. 2001;21:5889–5898. doi: 10.1128/MCB.21.17.5889-5898.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masuda K, Marasa B, Martindale JL, Halushka MK, Gorospe M. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging (Albany NY) 2009;1:681–698. doi: 10.18632/aging.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed) 2012;17:189–205. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Park S, Kilburn B, Jelinek MA, Henschen-Edman A, Aswad DW, Stallcup MR, Laird-Offringa IA. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J Biol Chem. 2002;277:44623–44630. doi: 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- 37.Abdelmohsen K, Srikantan S, Yang X, Lal A, Kim HH, Kuwano Y, Galban S, Becker KG, Kamara D, de Cabo R, et al. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J. 2009;28:1271–1282. doi: 10.1038/emboj.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu PC, Chuang HC, Kulp SK, Chen CS. The mRNA-stabilizing factor HuR protein is targeted by β-TrCP protein for degradation in response to glycolysis inhibition. J Biol Chem. 2012;287:43639–43650. doi: 10.1074/jbc.M112.393678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazroui R, Di Marco S, Clair E, von Roretz C, Tenenbaum SA, Keene JD, Saleh M, Gallouzi IE. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J Cell Biol. 2008;180:113–127. doi: 10.1083/jcb.200709030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talwar S, Jin J, Carroll B, Liu A, Gillespie MB, Palanisamy V. Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J Biol Chem. 2011;286:32333–32343. doi: 10.1074/jbc.M111.255927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2:11–13. doi: 10.4161/rna.2.1.1552. [DOI] [PubMed] [Google Scholar]
- 42.Lauriola L, Granone P, Ramella S, Lanza P, Ranelletti FO. Expression of the RNA-binding protein HuR and its clinical significance in human stage I and II lung adenocarcinoma. Histol Histopathol. 2012;27:617–626. doi: 10.14670/HH-27.617. [DOI] [PubMed] [Google Scholar]
- 43.Sun DP, Lin CY, Tian YF, Chen LT, Lin LC, Lee SW, Hsing CH, Lee HH, Shiue YL, Huang HY, et al. Clinicopathological significance of HuR expression in gallbladder carcinoma: with special emphasis on the implications of its nuclear and cytoplasmic expression. Tumour Biol. 2013;34:3059–3069. doi: 10.1007/s13277-013-0872-2. [DOI] [PubMed] [Google Scholar]
- 44.Liang PI, Li WM, Wang YH, Wu TF, Wu WR, Liao AC, Shen KH, Wei YC, Hsing CH, Shiue YL, et al. HuR cytoplasmic expression is associated with increased cyclin A expression and poor outcome with upper urinary tract urothelial carcinoma. BMC Cancer. 2012;12:611. doi: 10.1186/1471-2407-12-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denkert C, Weichert W, Pest S, Koch I, Licht D, Köbel M, Reles A, Sehouli J, Dietel M, Hauptmann S. Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res. 2004;64:189–195. doi: 10.1158/0008-5472.can-03-1987. [DOI] [PubMed] [Google Scholar]
- 46.Denkert C, Weichert W, Winzer KJ, Müller BM, Noske A, Niesporek S, Kristiansen G, Guski H, Dietel M, Hauptmann S. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin Cancer Res. 2004;10:5580–5586. doi: 10.1158/1078-0432.CCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 47.Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, Furneaux H, Hla T, Haglund C, Ristimäki A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005;65:2157–2161. doi: 10.1158/0008-5472.CAN-04-3765. [DOI] [PubMed] [Google Scholar]
- 48.Lim SJ, Kim HJ, Kim JY, Park K, Lee CM. Expression of HuR is associated with increased cyclooxygenase-2 expression in uterine cervical carcinoma. Int J Gynecol Pathol. 2007;26:229–234. doi: 10.1097/01.pgp.0000236946.82334.07. [DOI] [PubMed] [Google Scholar]
- 49.Cho NP, Han HS, Soh Y, Lee KY, Son HJ. Cytoplasmic HuR over-expression is associated with increased cyclooxygenase-2 expression in laryngeal squamous cell carcinomas. Pathology. 2007;39:545–550. doi: 10.1080/00313020701684391. [DOI] [PubMed] [Google Scholar]
- 50.Denkert C, Koch I, von Keyserlingk N, Noske A, Niesporek S, Dietel M, Weichert W. Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclooxygenase-2. Mod Pathol. 2006;19:1261–1269. doi: 10.1038/modpathol.3800645. [DOI] [PubMed] [Google Scholar]
- 51.Uren PJ, Burns SC, Ruan J, Singh KK, Smith AD, Penalva LO. Genomic analyses of the RNA-binding protein Hu antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. J Biol Chem. 2011;286:37063–37066. doi: 10.1074/jbc.C111.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srikantan S, Tominaga K, Gorospe M. Functional interplay between RNA-binding protein HuR and microRNAs. Curr Protein Pept Sci. 2012;13:372–379. doi: 10.2174/138920312801619394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vang S, Wu HT, Fischer A, Miller DH, MacLaughlan S, Douglass E, Steinhoff M, Collins C, Smith PJ, Brard L, et al. Identification of ovarian cancer metastatic miRNAs. PLoS One. 2013;8:e58226. doi: 10.1371/journal.pone.0058226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci USA. 2008;105:20297–20302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdelmohsen K, Kim MM, Srikantan S, Mercken EM, Brennan SE, Wilson GM, Cabo Rd, Gorospe M. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle. 2010;9:1354–1359. doi: 10.4161/cc.9.7.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY) 2010;2:333–343. doi: 10.18632/aging.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu F, Zhang X, Lei Y, Liu X, Liu Z, Tong T, Wang W. Loss of repression of HuR translation by miR-16 may be responsible for the elevation of HuR in human breast carcinoma. J Cell Biochem. 2010;111:727–734. doi: 10.1002/jcb.22762. [DOI] [PubMed] [Google Scholar]
- 58.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 60.Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167–180. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo X, Wu Y, Hartley RS. MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol. 2009;6:575–583. doi: 10.4161/rna.6.5.10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007;282:1479–1486. doi: 10.1074/jbc.M609383200. [DOI] [PubMed] [Google Scholar]
- 63.Kojima K, Fujita Y, Nozawa Y, Deguchi T, Ito M. MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate. 2010;70:1501–1512. doi: 10.1002/pros.21185. [DOI] [PubMed] [Google Scholar]
- 64.Leucci E, Zriwil A, Gregersen LH, Jensen KT, Obad S, Bellan C, Leoncini L, Kauppinen S, Lund AH. Inhibition of miR-9 de-represses HuR and DICER1 and impairs Hodgkin lymphoma tumour outgrowth in vivo. Oncogene. 2012;31:5081–5089. doi: 10.1038/onc.2012.15. [DOI] [PubMed] [Google Scholar]
- 65.Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med. 2013;5:949–966. doi: 10.1002/emmm.201202318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3’ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- 67.Rhee WJ, Ni CW, Zheng Z, Chang K, Jo H, Bao G. HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells. Proc Natl Acad Sci USA. 2010;107:6858–6863. doi: 10.1073/pnas.1000444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeyaraj SC, Dakhlallah D, Hill SR, Lee BS. Expression and distribution of HuR during ATP depletion and recovery in proximal tubule cells. Am J Physiol Renal Physiol. 2006;291:F1255–F1263. doi: 10.1152/ajprenal.00440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, Harokopos V, Aidinis V, Hemberger M, Kontoyiannis DL. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol. 2009;29:2762–2776. doi: 10.1128/MCB.01393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Avivi A, Shams I, Joel A, Lache O, Levy AP, Nevo E. Increased blood vessel density provides the mole rat physiological tolerance to its hypoxic subterranean habitat. FASEB J. 2005;19:1314–1316. doi: 10.1096/fj.04-3414fje. [DOI] [PubMed] [Google Scholar]
- 71.Pullmann R, Juhaszova M, López de Silanes I, Kawai T, Mazan-Mamczarz K, Halushka MK, Gorospe M. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J Biol Chem. 2005;280:22819–22826. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]