

Abstract
Background
In recent years, JM-216/satraplatin (GPC Biotech, Inc.) has emerged as a novel oral platinum analogue with a better toxicity profile than cisplatin. Since satraplatin is more hydrophobic than cisplatin or oxaliplatin, it appears to demonstrate efficacy in cisplatin-resistant cell lines. The preclinical and clinical evaluation of satraplatin stimulated this review of the pharmacology and clinical trial data of this agent.
Methods
A literature review was conducted in the MEDLINE database from 1985 to present using the keywords ‘satraplatin’ or ‘JM-216’. The abstracts regarding satraplatin reported at the 2007 – 2009 American Society of Clinical Oncology meetings were also reviewed.
Results/conclusion
Satraplatin has a favorable toxicity profile, and appears to have clinical activity against a variety of malignancies such as breast, prostate and lung cancer. The oral route of administration and the intermittent schedule makes it very convenient for clinical use. Despite this, a FDA-approved indication has not yet been achieved. The only Phase III trial with satraplatin was conducted in pretreated metastatic castrate-resistant prostate cancer (CRPC), revealing an improvement in progression-free survival but no overall survival benefit. Future development would have to include designing trials in docetaxel-refractory metastatic CRPC, or in other malignancies where cisplatin is of benefit.
Keywords: JM-216, oral platinum, platin, prostate cancer, satraplatin
1. Introduction
The use of platinum-based chemotherapy started from accidental observations in the laboratory by Barnett Rosenberg, a professor of biophysics and chemistry at Michigan State University. During electrolysis of a platinum-based electrode, he noticed inhibition of multiplication of Escherichia coli cells [1]. This compound was later put to clinical use in the 1970s and was named cisplatin. It was approved by the FDA in 1978 for the treatment of testicular tumors. Over the years, cisplatin has become an invaluable component of therapy for some common solid tumors such as lung, head and neck and bladder cancers. The serious side effects of cisplatin such as kidney damage, neurotoxicity and ototoxicity, and the development of inherent insensitivity and/or resistance, stimulated the development of novel platinum analogues.
Carboplatin initially entered the clinic in London in 1982 [2], gaining popularity from numerous trials and analyses with results showing efficacy equivalent to cisplatin but a tolerable toxicity profile. Bone marrow suppression, principally thrombocytopenia, was the dose-limiting side effect of carboplatin. However, it demonstrated a significantly lower incidence of nephrotoxicity, neuropathy, ototoxicity and gastrointestinal side effects. In 1989, the FDA approved carboplatin for use in advanced ovarian cancer. Research into broadening the use of platinum-containing compounds in prostate, breast and colorectal cancers has continued over the last two decades. Oxaliplatin was found to have clinically significant activity in patients with advanced colon cancer. Phase III studies reported from 2000 – 2004 supported the utility of this drug in combination with conventional chemotherapy (infusional 5-fluorouracil/leucovorin, known as 5-FU/LV) for treatment of advanced colon cancer patients [3-6]. Finally, the FDA approved its use in combination with fluorouracil and leucovorin, initially in stage IV colorectal cancer, and later as adjuvant treatment of stage III colon cancer patients who have undergone complete resection of the primary tumor. Neuropathy and hematologic and gastrointestinal tract toxicities were among the most common adverse events reported with oxaliplatin therapy.
Platinum compounds have been used in the treatment of several human tumors including testicular, bladder, lung, head and neck, and cervical cancer. The disadvantages of currently marketed platinum analogues are that they must all be administered via intravenous infusion. They have severe, dose-limiting adverse effects such as neurotoxicity, ototoxicity, renal toxicity, nausea and vomiting. Intrinsic or acquired resistance to cisplatin/carboplatin was observed in ovarian cancer and other malignancies. The described mechanisms for this resistance were either failure of a sufficient amount of platinum to reach the target DNA and/or a failure to achieve cell death after platinum–DNA adduct formation [7,8]. These disadvantages in turn led to the development of the next generation of platinum analogues like satraplatin (Box 1), which have improved toxicity profiles, as well as reduced cross-resistance to cisplatin [8].
Box 1. Drug summary.
| Drug name | Satraplatin |
| Phase | Phase III |
| Indication | Cancer |
| Pharmacology description |
DNA antagonist |
| Route of administration |
Alimentary, p.o. |
| Chemical structure |
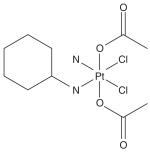 |
| Pivotal trial(s) | In a randomized Phase III trial in 50 patients with HRPC, satraplatin + prednisolone resulted in a 50% decline in prostate-specific antigen (PSA), and 22% on prednisone monotherapy Double-blind, randomized Phase III trial (SPARC) in Europe, S America and the US |
Pharmaprojects - copyright to Citeline Drug Intelligence(an Informa business).
Readers are referred to Informa-Pipeline (http://informa-pipeline.citeline.com) and Citeline (http://informa.citeline.com).
Satraplatin (formerly known as JM-216) was the first oral candidate from the JM/ICR collaboration after the initial successful clinical introduction of carboplatin. It is a fourth-generation platinum analogue with activity against cisplatin-resistant tumors and with a carboplatin-like toxicity profile (i.e., relatively milder in comparison to cisplatin). The lipophilicity and stability of JM-216 allows its oral administration, making it easier and convenient for the patients [9]. It has shown similar antitumor activity to that of cisplatin and carboplatin in in vitro and in vivo studies, as well as shown activity in some platinum resistant in vitro tumor models [10].
The clinical development of satraplatin, however, has encountered a number of hurdles resulting in the lack of FDA approval to date. The originally planned Phase III trial comparing satraplatin with placebo in chemotherapy-naive, metastatic, castrate-resistant prostate cancer (CRPC) revealed promising efficacy but was closed early due to a decision by the company. Later, when drug development of satraplatin resumed, docetaxel therapy had already established overall survival benefit and was FDA-approved in metastatic CRPC. Hence satraplatin had to be evaluated in pretreated metastatic CRPC, which remained a disease setting where no systemic agent had established role.
2. Pharmacology
2.1 Chemistry
Satraplatin or bis-(acetate)-ammine dichloro-(cyclohexylamine) platinum (IV) is a member from the mixed amine platinum IV dicarboxylate dichloride series [11,12]. The oxidations of platinum (II) complexes with hydrogen peroxide led to the formation of corresponding octahedral platinum dihydroxo complexes. These complexes, when stirred in the acid anhydrate solvent at ambient temperature for several hours, converted to dicarboxylate series complexes [12]. This reaction was subsequently used to prepare a series of products containing simple alkyl and aryl carboxylate ligands. Satraplatin has a single amine and cyclohexylamine group as its stable ligands, dichloro leaving groups and a pair of acetate trans ligands. Thus satraplatin has two chlorine atoms attached to the platinum as in cisplatin, but differs from it with two acetate groups attached, and a cyclohexyl moiety substituted on one of the amino groups [13]. This chemical structure makes it more lipophilic, reducing its water solubility. It is of a relatively low molecular weight, neutral and kinetically inert. It is relatively unstable in light and alkaline media but more stable in acids. The rate of reduction of satraplatin with 5 mM ascorbate is around 50 min, leaving adequate time for gastrointestinal absorption as parent platinum (IV) complex [10]. This complex then can be reduced elsewhere in the body to form reactive metabolites for target DNA.
2.2 Mechanism of action
Cisplatin and its derivatives, including satraplatin, mediate their action through the formation of DNA adducts and inter- and intra-strand crosslinks [14]. These adducts distort the DNA template with deceleration of cells in S phase followed with G2 phase arrest [15]. It also inhibits DNA replication and transcription and induces signal transduction pathway, leading to cell cycle arrest and apoptosis [16]. The asymmetrical stable ligands of satraplatin alter its DNA-adduct profile, leading to increased efficiency in inhibition of translational DNA synthesis [17] and less likelihood of being recognized by DNA-mismatch repair [18] or high-mobility group proteins [17,19]. The latter two mechanisms aid satraplatin to overcome cisplatin resistance.
2.3 Preclinical activity
Satraplatin demonstrated potent antitumor activity in vitro and in vivo against various tumor cell lines, including ovarian, cervical and lungs [20-22] that are sensitive or resistant to cisplatin. In human cervical cancer cell lines [21], the IC50 value for satraplatin was in the low to sub micromolar region 0.6 – 1.7μM, while for seven human ovarian cell lines [20], it was 1.7 μM (range 4.6 – 0.084) as compared to 3.5 μM (range 12.6 – 0.11) for cisplatin. The cytotoxic effect of satraplatin was demonstrated in selected cisplatin-resistant ovarian cancer cell lines [13,20], the androgen-sensitive LNCaP cell line, and the androgen-insensitive PC-3 and Du-145 cell lines. It also retained activity against tumor cell lines that were resistant to taxanes, doxorubicin, mitoxantrone, etoposide, and camptothecin, due to topoisomerase I, tubulin mutations or P-glycoprotein expression [16,22,23]. However, the agent was particularly inactive against the murine L1210 ascitic leukemia or its cisplatin-resistant subline [10,20].
The in vivo antitumor activity and toxicity data of satraplatin were obtained using the murine ADJ/PC6 plasmocytoma following either intraperitoneal or oral single-dose administration [24]. This model previously identified carboplatin as a viable alternative to cisplatin. With cisplatin, carboplatin and tetraplatin, both toxicity (50% lethal dose) and particularly antitumor efficacy (ED90) were reduced by the oral as well as intraperitoneal route, resulting in lower therapeutic indices. However, with satraplatin, toxicity was reduced 10-fold with oral administration but ED90 was similar for both routes of administration [20]. A human ovarian carcinoma xenograft model also demonstrated superior antitumor efficacy of orally administered satraplatin over cisplatin, carboplatin and tetraplatin. In both these models, the antitumor potency and therapeutic index were found to be better when satraplatin was administered in daily dosing for 5 days, as compared to single bolus dose [25].
Preclinical studies were conducted to demonstrate additive/synergistic tumor inhibition of satraplatin with other therapeutic modalities. The clinical success of combination chemotherapy involving parenterally administered cisplatin (or carboplatin) and VP-16 (etoposide) were the basis of these studies. The combination of orally administered satraplatin (JM-216) and etoposide was found to have therapeutic synergy when used in a murine tumor model [26]. The doses of each drug tolerated in the combination setting were much less than their individual maximal tolerated dose (MTD) levels. The MTD for five daily doses of JM-216 given orally, repeated at 3- or 4-week intervals, was 60 mg/kg/day. Another study, with concurrent ionizing radiation therapy, also suggested a synergistic effect of orally administered JM-216. Mice implanted with H460 human lung cancer xenograft were treated with 30 mg/kg satraplatin then, 1 h later, with 2 Gy radiation, for 5 consecutive days [27]. This combination regimen produced greater inhibition of tumor growth than either agent alone. These results were also supported by in vitro clonogenic assays using the same cell lines.
In summary, these preclinical studies provided the rationale for exploring the clinical activity of satraplatin as a single agent, and in combination regimens against a variety of tumor cell lines that were sensitive or resistant to other therapeutic agents.
2.4 Pharmacokinetics
From the preclinical studies, antitumor activity and therapeutic index were noted to be best for daily dosing for 5 days compared with single-dose, or once-daily, indefinite administration schedules. McKeage and colleagues, in their Phase I pharmacokinetic study, administered escalating doses of satraplatin upto 150 mg/m2/day for 5 days [28]. They observed linear correlations between plasma ultra-filtrate AUC and doses up to 120 mg/m2/day [28,29]. This relationship became nonlinear at higher doses or at a single dose > 200 mg/m2/day due to saturable gut absorption. There was no dose-dependent change in the proportion recovered in the urine over a wide dose range. Thus, the recommended dose for clinical trial was 80 – 120 mg/m2/day, given for 5 consecutive days, repeated every 3 – 4 weeks. The MTD was observed to be 140 mg/m2/day.
After oral administration of JM-216, < 2% of the intact drug was found in the plasma. Absorption was rapid, with peak plasma levels generally attained within 2 h. The plasma platinum ultra-filtrate levels peaked at around 2 h post-ingestion. AUCs on Day 5 were noted to be 1.7-fold higher (range, 1- to 2.6-fold) than on Day 1, suggestive of some accumulation of ultra-filterable platinum with repeat daily doses. The t½ was determined to be approximately 12 h when assayed 5 days after oral administration of a dose of 100 mg/m2 [28,29]. The majority of the platinum drug was bound to the plasma proteins and other blood components.
Metabolism and biotransformation have been studied in human tumor cell lines, rodents, and later, in human patients. Six metabolites were detected in mouse plasma following oral dosing of JM-216. Among these metabolites, JM-118, JM-383 and JM-518 showed potent cytotoxicity to the cancer cells in vitro. These also had antitumor activity when given intraperitoneally to the mice bearing ADJ/PC6 tumors [30]. The biotransformation profile was evaluated by HPLC followed by atomic absorption spectrophotometry (AA) in patient plasma ultrafiltrates [31]. In these ultra-filtrate samples, six platinum peaks of Pt IV and Pt II metabolites were observed, of which only four appeared during first 6 h. No parent JM-216 was detected. The major metabolite seen in all patients was the Pt II complex JM-118 (cis-amminedichloro-cyclohexylamine). JM-383 (bis (acetate) ammine (cyclohexylamine) dihydroxoplatinum IV) was a second metabolite. JM-559 and JM-518 were minor products (< 5% of the free platinum) of Pt IV species. The cytotoxicity profile of all three metabolites was very close to that of the parent drug in a panel of cisplatin-sensitive and -resistant human ovarian carcinoma cell lines. Two metabolites that were seen in the patients, but not in the in vitro incubation medium, suggested the possibility of enzymatic reaction [31].
More recent clinical studies have found that 20 – 30% of total platinum content in individual plasma ultra-filtrate samples is JM-118. A number of unidentified but additional platinum-containing species are also present, although these are not JM-383, JM-559 or JM-518 [32]. More recent studies using HPLC-inductively coupled plasma mass spectrometry (HPLC-ICPMS) have demonstrated that satraplatin-derived platinum became rapidly associated with red blood cells, at an accumulation t½ of 9.5 min (95% CI, 7.1 – 14.2 min) [33]. It has a disappearance t½ of 6.3 min and 5.3 h in fresh human whole blood and in fresh human plasma, respectively. At equilibrium, 62% of the total platinum in the blood was associated with the red blood cells, and 38% was associated with the plasma. Within plasma, 71% was bound to the plasma proteins while only 29% was found in ultra-filtrates or methanol extracts. The majority of platinum in red blood cells was associated with the red blood cell membrane. Thus these studies supported that it undergoes rapid biotransformation in whole blood in vitro, forming new forms of platinum, including JM-118 and platinum that becomes irreversibly bound to plasma proteins and the membranes of red blood cells [33,34]. Studies using human ovarian carcinoma cell lines of varying glutathione (GSH) content have showed that conjugation with GSH represents a major detoxification pathway for satraplatin [35].
JM-216 inhibition of CYP450 in human liver microsomes was investigated by measuring the inhibition potential (IC50 and Ki) on prototype reactions in a study by Ando and co-workers [36]. The IC50 values were 0.3 – 10 mM, indicating strong and nonspecific inhibitory effects of JM-216. The inhibition occurred in a noncompetitive manner. JM-216 inhibited the catalytic activities of several major CYP450 isozymes (CYP3A4, CYP2C8, CYP1A1, CYP1A2, CYP2E1 and CYP2D6) [35]. Thus dose reductions are necessary when substrates of these isozymes are used in combination regimens, such as with oral etoposide [26].
3. Clinical trials with satraplatin
In multiple clinical trials, satraplatin has been generally well tolerated. The most frequent adverse effects were noncumulative myelosuppression such as grade II – III neutropenia, thrombocytopenia and anemia. Among non-hematological events, nausea, vomiting and diarrhea were common. Unlike cisplatin, however, satraplatin was not associated with neurotoxicity, ototoxicity and nephrotoxicity. Satraplatin has been investigated in numerous clinical trials for a variety of human malignancies including trials in prostate, ovarian, cervical, lung, head and neck cancer.
3.1 Phase I studies
Multiple Phase I trials using a variety of dosing schedules have been conducted to evaluate the dosing and safety of satraplatin. These trials began in London during 1992 with single dose therapy given every 3 weeks [28]. This study was stopped due to nonlinear pharmacokinetics observed secondary to saturable absorption. The AUC and Cmax increased proportionately with increasing doses up to 120 mg/m2, but became nonlinear at doses > 200 mg/m2. The most important toxicity was myelosuppression (leukopenia and thrombocytopenia). There was no nephrotoxicity, ototoxicity, or neurotoxicity. Other Phase I trials used twice-daily dosing, every 3 weeks [37] and daily dosing for 14 days, given every 4 – 5 weeks [38]. Plasma pharmacokinetics was highly variable at all dose levels.
Linear pharmacokinetics were noticed with a daily 5-day dosing schedule, repeated every 3 – 4 weeks [29,39]. There was a significant correlation between the JM-216 dose and the platinum plasma ultra-filtrate AUC. The primary dose-limiting toxicity (DLT) was neutropenia and thrombocytopenia, although these were not cumulative and recovered by Day 28. Mild nausea, vomiting and diarrhea were commonly noted, as with the single-dose study. There was no neurotoxicity, ototoxicity and nephrotoxicity observed. Fokkema and colleagues studied satraplatin at doses of 120 mg/m2/day for 5 consecutive days repeated every 21 days with a maximum of six cycles among lung cancer patients [40]. They found no nephrotoxicity of JM-216. The dose recommended for further clinical development was 100 and 120 mg/m2/day for 5 days for previously treated and untreated patients, respectively, with cycles repeated every 21 or 28 days [10].
Phase I studies using combination regimens of satraplatin with either radiotherapy (in patients with NSCLC and squamous cell head and neck cancer) [41,42] or with paclitaxel were conducted [43]. The former studies evaluated the MTD among three escalating dose levels: 30, 45 and 60 mg/m2 given daily over 5 days, 3 weeks apart, with standard chest radiotherapy in lung cancer patients. The major DLT noted was myelosuppression. The recommended satraplatin dose for Phase II with concomitant radiotherapy was 30 mg/m2/day for 5 days. In the paclitaxel combination study, 43 patients received a total of 146 courses of the combination regimens. Generally, the combination was well tolerated, myelosuppression being the most common serious side effect. The doses recommended by the authors for Phase II testing were satraplatin 60 mg/m2/day for 5 days and paclitaxel 200 mg/m2 on Day 1, repeated every 21 days. The combination of satraplatin with oral uracil/ftorafur (UFT) with leucovorin, using a schedule of 14 consecutive days every 28 days, was studied in 20 patients [44]. Gastrointestinal side effects of nausea and vomiting were the DLTs, occurring at doses of 20 mg/day for satraplatin and 300 mg/day UFT. These doses were considerably lower than their individual MTDs, suggesting that shorter administration schedules needed to be explored.
3.2 Phase II studies
A Phase II study was conducted in previously untreated patients with locally advanced, unresectable or metastatic NSCLC [45]. The satraplatin dose used was 120 mg/m2/day for 5 days, repeated every 3 weeks. This study failed to demonstrate any sustained objective responses among the first 13 evaluable patients. There was some disease palliation noted, with stable disease among 6 (46%) patients compared with the single-agent response rates noted for cisplatin (20%) and carboplatin (12%) [46]. Hematological toxicities were few, with lower incidences of grade 2 neutropenia and thrombocytopenia.
A second multicenter Phase II trial evaluated satraplatin for its antitumor efficacy and tolerance in 27 patients with limited or extensive small-cell lung cancer (SCLC) [47]. It was initially dosed at 120 mg/m2/day for 5 days with repeated cycles every 21 days. This dose was escalated to 140 mg/m2/day in 13 patients, with no signs of severe toxicity noted at lower dose levels. Total 88 cycles were administered. A response rate of 38% with objective responses in 10 of 26 evaluable patients was noted, similar to that observed with cisplatin monotherapy in SCLC [48]. As with single-agent Phase I studies, the most common grade 3 and 4 toxicities were neutropenia (19.6%), lymphocytopenia (64.7%), and thrombocytopenia (29.8%). Nausea, vomiting and diarrhea were the most common non-hematological toxicities. There was no severe neurotoxicity or nephrotoxicity with satraplatin.
In a randomized Phase II trial in 40 patients with relapsed ovarian cancer, satraplatin demonstrated comparable antitumor efficacy to that of cisplatin or carboplatin [49]. Standard doses of cisplatin or carboplatin, along with 100 mg/m2/day for 5 days, were used. These doses were repeated every 28 days. An objective response was seen in 35% of the patients in each treatment group. A multicenter Phase II study of this agent as first- or second-line treatment of metastatic breast cancer was done in 40 patients [50]. 48% of patients had received prior adjuvant chemotherapy, while 60% had chemotherapy for metastatic breast cancer. A dose of 80 mg/m2/day satraplatin was given orally for 5 days every 21 days in two cycles. In 31 patients with measurable disease, four patients had a clinical benefit rate of 19% and two of them had partial responses. The most common toxicities were neutropenia (28%) and thrombocytopenia (25%). Median survival was 15 months. This study demonstrated that satraplatin had limited activity as a single agent in metastatic breast cancer, but recommended low-dose use in combination therapy in future trials in breast cancer.
Another Phase II study of JM-216 was conducted in 18 patients with advanced/recurrent squamous cancer of the cervix [51]. All these patients had received prior pelvic irradiation (RT); in addition, four had received cisplatin as concurrent therapy with radiation. The dose of satraplatin used was 30 mg/m2 daily for 14 days every 5 weeks. Nausea, vomiting and diarrhea were among the most common toxicities; 12 patients had a best response of stable disease, with no treatment-related deaths. Thus, this study also noted limited activity of satraplatin in patients with recurrent squamous cell carcinoma of the cervix. Further clinical evaluation was recommended, however, given that the study had small patient numbers and had used a low drug dose regimen.
3.3 Phase II and III clinical trials of satraplatin in metastatic CRPC
Hormonal therapy based on androgen deprivation is the most effective treatment modality for progressive prostate cancer patients. But subsequently, prostate cancer progresses despite castrate levels of testosterone achieved with hormone therapy or orchiectomy, hence being labeled as CRPC [52]. In metastatic CRPC, front-line treatment with a docetaxel-based regimen has demonstrated improved overall survival and progression-free survival (PFS). These observations have been demonstrated in two randomized Phase III trials (TAX 327 and Southwest Oncology Group [SWOG] 9916). In these trials, the combination of mitoxantrone plus prednisone was compared with docetaxel plus estramustine or docetaxel plus prednisone [53,54]. Despite the benefit observed with docetaxel therapy, eventually, disease progression occurs. Therefore, investigation of second-line therapy in metastatic CRPC is ongoing and represents a currently unmet need, since no systemic agent has proven efficacy in this setting. The success of orally available and less toxic satraplatin in various preclinical studies in hormone-sensitive and -resistant prostate cancer cells led to the clinical trials of satraplatin in CRPC.
A Phase II study using satraplatin at the dose of 120 mg/m2/day for 5 days every 28 days was conducted in metastatic CRPC progressing on hormone therapy but untreated with chemotherapy [55]. Thirty-nine patients were enrolled and treated with a total of 155 courses of JM-216. Dose escalation and de-escalation were performed according to patient’s tolerance, with dose delays in 77% of courses and dose reductions in 31% of courses. Treatment was discontinued in five cases due to development of treatment-related intolerable toxicities. The most frequent grade 3 adverse events included thrombocytopenia (54%), neutropenia (52%), diarrhea (28%), anemia (24%), nausea (13%) and vomiting (16%). Altogether, 32 patients were assessed for PSA response; 36% of patients had stable disease and 26% had partial response, while 21% of patients had PSA progression. A dose reduction in future trials appeared to be indicated, since the majority of patients required a dose decrease to 100 mg/m2.
These encouraging results led to a randomized multicenter Phase III trial to determine the efficacy of front-line chemotherapy with satraplatin plus prednisone compared with that of prednisone alone [56]. The intended target sample size was 380 patients. But after 50 randomized patients, this study was terminated by the sponsoring company at the time (Bristol-Myers Squibb). In this study, a maximum of eight cycles were administered, with a satraplatin dose of 100 mg/m2/day for 1 – 5 days, repeated every 5 weeks along with prednisone 5 mg twice daily. The analysis of data collected on the 50 enrolled patients revealed a statistically significant increase in PSA response in the combination therapy arm compared with prednisone alone (p = 0.046). A > 50% decline in PSA was noted in 33.3% of patients treated with satraplatin and prednisone versus 8.7% of those treated with prednisone only; median PFS of 5.2 and 2.5 months, respectively, were observed (p = 0.023). Median survival observed was 14.9 months (95% CI, 13.7 – 28.4) with satraplatin plus prednisone and 11.9 months (95% CI, 8.4 – 23.1) with prednisone alone. This 3-month difference was not statistically significant (p = 0.579), probably owing to the small sample size. Overall, the satraplatin and prednisone regimen was well tolerated. Serious grade 3 hematologic toxicities included leukopenia (25.9%) and thrombocytopenia (29.6%). Nausea, vomiting, diarrhea, infections and hyperglycemia were the observed grade 3 non hematological toxicities (7.4%) [57]. PSA response rates and PFS were favorable in the combination therapy arm.
3.4 SPARC (Satraplatin and Prednisone Against Refractory Cancer) trial
When GPC Biotech acquired this compound, the impetus remained to explore satraplatin in pretreated metastatic CRPC, based on the promising results in the above trial and since this population had an unmet need. This led to the design of a Phase III registration trial for satraplatin in metastatic CRPC, called SPARC. This was a large, multicenter, multinational, double-blind, placebo-controlled Phase III trial [58]. It compared satraplatin plus prednisone versus placebo plus prednisone as second-line therapy in CRPC patients who had previously received one cytotoxic chemotherapy regimen. The need for a novel systemic therapy in this patient setting was reflected by the rapid accrual to this trial. The SPARC trial enrolled 950 patients over 12 months with a 2:1 randomization to satraplatin and prednisone versus placebo and prednisone. Over 50% of these subjects had received prior docetaxel therapy, and nearly 50% had received another form of therapy such as mitoxantrone, estramustine or cyclophosphamide. Treatment consisted of prednisone 5 mg twice daily on Days 1 – 35, plus either placebo or oral satraplatin 80 mg/m2 on Days 1 – 5 every 5 weeks. Recognizing the difficulty of using PSA progression or response as a surrogate for actual disease progression, SPARC utilized a composite end point of at least new lesions on bone scan, increased bone pain symptoms based on increasing pain medication use, and occurrence of skeletal events, to determine progression. The primary end points of the SPARC trial were PFS and time to tumor progression, and secondary end points were overall survival (OS) and time to pain progression.
A median PFS of 11.1 weeks in the combination arm versus 9.7 weeks in the prednisone arm, with improvement of 1.3 weeks or 9 days, was noted (95% CI, 0.57 – 0.77; p < 0.0001) [59]. In patients who had progressed with prior docetaxel therapy, median PFS on combination arm was 10.1 weeks versus 9.1 weeks in the prednisone-alone arm (95% CI, 0.54 – 0.83; p = 0.0006). This benefit, though statistically significant, is clinically irrelevant. At 6 months, the proportion of patients free of progression were 30% and 17%, favoring satraplatin; at 12 months, these rates were 17% versus 7% [60]. Pain response rates were 24.2% for the satraplatin-plus-prednisone arm compared with 13.8% for the prednisone arm (p < 0.005). Median time to pain progression was 66.1 weeks for the satraplatin arm compared with 22.3 weeks for the placebo arm (95% CI, 0.51 – 0.79, p < 0.001). This represented a 36% reduction in the relative risk of pain progression and was a clinically noteworthy outcome, especially given the durability of palliative response.
Dose reduction was required in 19.7% of patients, and 64.7% of the patients required dosing delays [57]. On satraplatin therapy, 0.6% cases developed neutropenic fever, 3.8% required platelet transfusion and 15.9% required red blood cell transfusions as compared to 0, 0.3 and 8%, respectively, in the prednisone-alone arm. Other grade 3 – 4 adverse events reported were diarrhea (2.1 vs 0%), deep venous thrombosis (1.6 vs 0%) and severe fatigue (4.9 vs 2.6%) [58]. The tolerability of satraplatin, and the durable pain response achieved, made the benefit-to-risk ratio favorable.
Despite the improvement in PFS and the palliation achieved, OS was not improved with satraplatin therapy. The median survival was 61.3 weeks in the satraplatin group and 61.4 weeks in the prednisone and placebo group, with a hazard ratio of 0.98 [61]. In patients who had received prior docetaxel (51% of the patients enrolled), a trend towards survival benefit favoring satraplatin was observed, with median survivals of 66.1 and 62.9 weeks with a hazard ratio of 0.78. In the docetaxel-refractory population or in the docetaxel-pretreated population, after adjusting for baseline prognostic factors of baseline Hb, LDH and alkaline phosphatase levels, the survival data achieved statistical significance favoring the satraplatin and prednisone regimen (p = 0.03). The patients who were refractory to docetaxel therapy, defined as progression during or within 50 days or 100 days of administration of docetaxel, also demonstrated a significant OS benefit with satraplatin therapy (p = 0.02, median survivals of 47 and 53 weeks) [61]. The data regarding prostate trials is summarized in Table 1.
Table 1. Summary of Phase II and Phase III trials of satraplatin in metastatic CRPC.
| Study | Phase | Tumor/ histology |
Combination drugs |
Doses of combination drugs |
No. of pts |
Results |
|---|---|---|---|---|---|---|
| Sternberg et al., 2005 [56] |
III | CRPC | S + Pr vs Pr | S 100 mg/m2/day Days 1 – 5, every 5 weeks for 8 cycles; Pr 10 mg twice daily |
50 | S + Pr vs P PSA response: 33.3 vs 8.7% PFS: 5.2 vs 2.5 months MS:14.9 vs 11.9 months |
| Sartor and Petrylak et al., 2008 [60] |
III | CRPC | S + Pr vs P + Pr |
S 80 mg/m2/day × 5 q 5 weeks + Pr 5 mg b.i.d. (Days 1 – 35) Pr 5mg b.i.d. (Days 1 – 35) + placebo |
950 | S + Pr vs P + Pr PFS: 11.1 vs 9.7 weeks MS: 61.3 vs 61.4 weeks MPP: 6.1 vs 22.3 PRR: 24.2 vs 13.8 |
| Petrylak et al., 2009 (Subgroup of SPARC) [61] |
III | D-refractory CRPC |
S + Pr vs P + Pr |
S 80 mg/m2/day × 5 q 5 weeks + Pr 5 mg b.i.d. (Days 1 – 35) Pr 5 mg b.i.d. (Days 1 – 35) + placebo |
488 | Survival benefits*: S + Pr vs P + Pr Prior D : 66.1 vs 62.9 R1: 53.1 vs 47.0 R2: 53.9 vs 47.0 |
| Vaishampayan et al., 2009 [62] |
II | CRPC | B + S + Pr in D-pretreated patients |
S 80 mg/m2 for Days 1 – 5, B 10 mg/kg on Day 1 and 15 mg/kg on Day 15 of each 35-day cycle and Pr 5 mg b.i.d. |
19 | PSA decline: 7 patients MDR: 3/6 patients PD: 11/16 patients |
Improved survival on S + Pr after stratification and prespecified baseline prognostic factors (LDH, hemoglobin and alkaline phosphatase); R1 group, progression while on D or within 100 days of the last dose; R2 group, progression while on D or within 50 days of the last dose.
B: Bevacizumab; CPRC: Metastatic castrate-resistant prostate cancer; D: Docetaxel; LDH:; MDR: Measurable disease response; MPP: Median time to pain progression;
MS: Median survival; PD: Progression of disease; PFS: Progression-free survival; Pr: Prednisone; PRR: Pain response rate; PSA: Prostate-specific antigen; S: Satraplatin;
SPARC: Satraplatin and prednisone against refractory cancer [Trial].
Satraplatin-based combination regimens have been evaluated; however, this has only been in small Phase II trials. The Phase II trials of satraplatin-based combinations are summarized in Tables 1 and 2. We are currently conducting a Phase II trial of satraplatin, prednisone and bevacizumab in metastatic CRPC progressing after docetaxel therapy. Preliminary results of the study were reported at the American Society of Clinical Oncology (ASCO) genito-urinary cancers symposium in February 2009 [62]. Of the patients enrolled, 19 were evaluable for response, with a promising preliminary response rate. Of the six patients with measurable disease, three demonstrated a response, and a ≥ 30% PSA decline rate of 25% was noted in the 19 evaluable patients. The combination was tolerable, with the main severe toxicity being pulmonary embolism in two patients; this was probably related to bevacizumab. Other combinations of satraplatin, with paclitaxel, gemcitabine and capecitabine, have been investigated, with Phase II doses being established and preliminary evidence of efficacy seen. Further investigation of combination therapies would be justified if regulatory approval of satraplatin is achieved.
Table 2. Phase I/II clinical trials with satraplatin in numerous solid tumor malignancies [67-71].
| Study | Type | Tumor type | Combination drugs |
Doses of combination drugs | No. of patients |
Results |
|---|---|---|---|---|---|---|
| Sessa et al., 1998 [67] |
I | AST | S and C | S 60 mg/m2/day Days 1 – 5 C 1650 mg/m2/day Days 8 – 21 of 35-day cycle |
38 | MTD: S – 70 mg/m2 C – 2000 mg/m2 |
| Wisinski et al., 2008 [68] |
I | AST | S and C | S 60 mg/m2/day Days 1 – 5 with escalation in dose by 20 mg/m2 as tolerated C 1650 mg/m2/day from Days 1 – 14 of 21-day cycle |
22 | MTD – 100 mg/m2 Stable disease – 54% |
| Paola et al., 2009 [69] |
I | AST | S and Gem | Group A* – Gem 800 mg + S 40 mg Group B – Gem 800 mg + S 60 mg Gem given on Days 1, 8 and 15 q28 d; S from day 1 – 5 |
17 | Good antitumor activity with acceptable toxicity profile |
| Leal et al., 2008 [70] |
I | AST | S and Tax | Tax 60 mg/m2 S 50 mg/m2/day, Days 1 – 5 of a 21-day cycle |
23 | Encouraging results; recommended for Phase II trials‡ with or without G-CSF |
| Thompson et al., 2008 [71] |
II | NSCLC | S and P | S 80 mg/m2/day Days 1 – 5, P 200 mg/m2 i.v. Day 1 every 28-day cycle for 6 cycles |
22 | ORR – 21% (1 complete and 6 partial responses) SD – 24% (8 patients) PD – 42% (14 patients) |
Group A (6 patients), with prior chemotherapy – Gem 800 mg + S 40 mg; Group B (6 patients), with no prior chemotherapy – Gem 800 mg + S 60 mg.
Recommended Phase II dose in repeated three cycles: without G-CSF – Tax, 60 mg/m2 i.v. Day 1 with SA 40 mg/m2 p.o. Days 1 – 5; with G-CSF, Tax 70 mg/m2 i.v. Day 1 with SA 50 mg/m2 p.o. Days 1 – 5.
AST: Advanced solid tumors; C: Capecitabine; Gem: Gemcitabine; MTD: Maximum tolerated dose; NSCLC: Non-small cell lung cancer; ORR: Overall response rate;
P: Paclitaxel; PD: Progression of disease; S: Satraplatin; SD: Stable disease; Tax: Docetaxel.
4. Conclusion
Satraplatin has the advantages over cisplatin of a better toxicity profile and oral route of administration; however, its efficacy in a specific malignancy remains to be proven. Future investigation in the cisplatin-resistant setting, or in docetaxel-refractory metastatic CRPC, would be warranted to develop a niche for this agent in human cancer therapeutics.
5. Expert opinion
Platinum agents have demonstrated a broad spectrum of activity against a wide range of solid tumor malignancies such as testis, lung, cervix, head and neck and bladder cancers. Indeed, most of the cures achieved in solid tumors are largely due to cisplatin therapy. However, cisplatin is difficult to tolerate, with side effects such as nausea, emesis, neuropathy, ototoxicity and nephrotoxicity. Carboplatin was developed as a platinum analogue with a more favorable toxicity profile. This was established as a replacement for cisplatin in lung cancer. The side-effect profile was milder, but myelosuppression was more common with carboplatin than cisplatin. Oxaliplatin was later introduced and became an established therapy in colorectal cancer, a malignancy where cisplatin and carboplatin have no proven efficacy. Oxaliplatin does, however, have significant neuropathy as one of its toxicities. Satraplatin has the advantage of being the only platinum analogue with bioavailability via the oral route of administration, and a convenient schedule of 5 days every 3 – 5 weeks. It is also very well tolerated, and has a favorable toxicity profile, with < 2% chance of clinically relevant myelosuppression and a small incidence of gastrointestinal toxicity. The agent would therefore be considered attractive for clinical application.
The clinical development of satraplatin has encountered a number of hurdles, resulting in the lack of FDA approval to date. Clinical trials comparing with cisplatin or carboplatin were not warranted, since Phase II trials revealed that the agent had only modest clinical efficacy in lung and cervical cancers. The initial strategy of exploring this agent in metastatic CRPC appeared justified. At the time of the initial evaluation of satraplatin, no systemic therapy had demonstrated survival benefit in metastatic CRPC. Unfortunately, despite demonstrating superior efficacy favoring the satraplatin arm in frontline therapy of metastatic CRPC, this study was closed early due to a decision by the company. During the course of the 950-patient registration trial of satraplatin, docetaxel therapy demonstrated a survival benefit in metastatic chemotherapynaive CRPC. [53,54]. Since then, active evaluation of numerous biologic and chemotherapy agents commenced in the docetaxel-pretreated metastatic CRPC population [63,64]. Satraplatin then also had to be evaluated in chemotherapy-pretreated metastatic CRPC, which remained a disease setting where no systemic agent had proven benefit to date.
Unfortunately, the SPARC Phase III trial conducted in metastatic CRPC pretreated with one prior chemotherapy regimen did not reveal overall survival benefit, despite showing statistically significant benefit in pain control and PFS. The reasons for this are likely to be as follows:
The composite end point used for progression had not been validated as a surrogate for overall survival in any prior study.
Prior docetaxel therapy was not a prerequisite of eligibility, and only half (51%) of the patients enrolled had prior docetaxel therapy. Hence the control arm had a better opportunity of salvage with docetaxel, and would have demonstrated a survival better than expected. In fact, there was a trend towards survival benefit in a prespecified analysis of the docetaxel-pretreated patient subgroup, with median survival of 66.1 weeks in the satraplatin arm and 62.9 weeks in the placebo and prednisone arm (HR = 0.78; log-rank p = 0.06; Cox proportional hazards, p = 0.039) [60]. Further, within the docetaxel-pretreated group, survival analysis was conducted in two subgroups defined as R1 (which included the patients progressing while on docetaxel, or within 100 days of the last dose) and R2 (which included patients progressing while on, or within 50 days of, the last dose). The OS was statistically significant, favoring the satraplatin-based arm in the R1 group (log-rank p = 0.03; Cox proportional hazards, p = 0.009) and revealed a trend towards benefit in the R2 group (log-rank p = 0.09; Cox proportional hazards, p = 0.029) [61]. Additionally, when adjustments for the known validated prognostic factors such as hemoglobin, LDH and alkaline phosphatase were performed, OS benefit was demonstrated with satraplatin and prednisone therapy [60].
Based on the above facts and subset data analysis, further development of satraplatin in metastatic CRPC would still be recommended employing the following strategies:
Better patient selection, based on validated criteria such as lack of PSA decline of > 30% with four cycles/12 weeks of docetaxel therapy, or progression within 100 days after discontinuing docetaxel.
Using criteria predictive of docetaxel refractoriness, such as tubulin III expression in metastatic CRPC trial designs. Both beta tubulin III and ERCC overexpression are predictive of worse outcome with carboplatin and paclitaxel therapy in NSCLC [65].
Clinical eligibility criteria for the evaluation of satraplatin in other malignancies such as urothelial, head and neck or lung cancers should involve progression of disease following prior cisplatin or carboplatin therapy, hence qualified as platinum-resistant disease.
Cisplatin resistance such as ERCC1 to be used as entry criteria. ERCC1 positivity is predictive of cisplatin resistance in the therapy of lung cancer [66].
Hence the consideration of a personalized strategy with selecting the ERCC1-positive and/or beta tubulin III-overexpressing tumors for satraplatin therapy seems justified. Conducting trials in enriched populations as described above is likely to improve the chances of detecting a clinically relevant difference and may reduce sample size and subsequently the cost. For combination therapies, concurrent radiation, or satraplatin with either of bevacizumab, capecitabine or taxanes, appear to be worthy of exploration.
In conclusion, this well-tolerated and convenient-to-use agent continues to make attempts to carve a niche for itself in the therapy of human malignancies. Based on the single-agent clinical data and possible efficacy in cisplatin-resistant tumors, further investigation is warranted.
Footnotes
Declaration of interest
UN Vaishampayan has received research support and honoraria from GPC Biotech, Inc. (the licensee of satraplatin). The paper was funded by Wayne State University and the Karmanos Cancer Institute, Detroit, MI.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Trazaska S. Cisplatin. Chem Eng News. 2005;83:65. [Google Scholar]
- 2.Harrap KR. Preclinical studies identifying Carboplatin as a viable cisplatin alternative. Cancer Treat Rev. 1985;12:21–33. doi: 10.1016/0305-7372(85)90015-5. [DOI] [PubMed] [Google Scholar]
- 3.De Gramont A, Figer A, Metal S. Leucovorin and fluorouracil with or without Oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–47. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 4.Giacchetti S, Perpoint B, Zidani N, et al. Phase III multicenter randomized trial of Oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18:136–47. doi: 10.1200/JCO.2000.18.1.136. [DOI] [PubMed] [Google Scholar]
- 5.Rothenberg ML, Oza AM, Bigelow RH, et al. Superiority of Oxaliplatin and fluorouracil-leucovorin compared with either therapy alone in patients with progressive colorectal cancer after irinotecan and fluorouracil-leucovorin interim results of a Phase III trial. J Clin Oncol. 2003;21:2059–69. doi: 10.1200/JCO.2003.11.126. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and Oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004;22:23–9. doi: 10.1200/JCO.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 7.Kelland LR. Overcoming resistance to platinum therapy in patients with advanced cancer. Am J Cancer. 2002;1:247–55. [Google Scholar]
- 8.Ho YP, Au-Yeung SC, To KK. Platinum based anticancer agents: innovative design strategies and biological perspectives. Med Res Rev. 2003;23:633–55. doi: 10.1002/med.10038. [DOI] [PubMed] [Google Scholar]
- 9.Harrap KR, Murrer BA, Giandomenico C, et al. Ammine/amine platinum IV dicarboxylates: a novel class of complexes which circumvent intrinsic cisplatin resistance. In: Howell SB, editor. Platinum and other metal coordination complexes in cancer chemotherapy. Plenum; New York: 1991. pp. 391–9. [Google Scholar]
- 10.Kelland LR. An update on Satraplatin: the first orally available platinum anticancer drug. Expert Opin Investig Drugs. 2000;9:1373–82. doi: 10.1517/13543784.9.6.1373. [DOI] [PubMed] [Google Scholar]
- 11.Giandomenico CM, Abrams MJ, Murrer BA, et al. Synthesis and reactions of a new class of orally active PT (IV) antitumor complexes. In: Howell SB, editor. Platinum and other metal co-ordination compounds in cancer chemotherapy. Plenum Press; New York, USA: [Google Scholar]
- 12.Ju Lee Eun, Jun Moo-Jin, Soo Sohn Youn. Synthesis and properties of Novel Platinum Complexes involving asymmetric chiral diamines as carrier ligands. Bull Korean Chem Soc. 1999;20:1295–8. [Google Scholar]
- 13.Choy H. Satraplatin: an orally available platinum analog for the treatment of cancer. Expert Rev Anticancer Ther. 2006;6:973–82. doi: 10.1586/14737140.6.7.973. [DOI] [PubMed] [Google Scholar]
- 14.Mellish KJ, Barnard CF, Murrer BA, et al. DNA-binding properties of novel cis- and trans platinum-based anticancer agents in 2 human ovarian carcinoma cell lines. Int J Cancer. 1995;62:717–23. doi: 10.1002/ijc.2910620612. [DOI] [PubMed] [Google Scholar]
- 15.O’Neill CF, Koberle B, Masters JR, et al. Gene-specific repair of Pt/DNA lesions and induction of apoptosis by the oral platinum drug JM216 in three human ovarian carcinoma cell lines sensitive and resistant to cisplatin. Br J Cancer. 1999;81:1294–303. doi: 10.1038/sj.bjc.6694381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wosikowski K, Caligiuri M, Rattel B, et al. Efficacy of satraplatin (JM216) and its metabolites is maintained in taxanes-resistant tumors. Proceedings of the First International Conference on Cancer Therapeutics: Molecular Targets, Pharmacology and Clinical Applications. 2004 [Google Scholar]
- 17.Vaisman A, Lim S, Patrick SM, et al. Effect of DNA polymerases and high mobility group protein I on the carrier ligand specificity of translesion synthesis past platinum-DNC adducts. Biochemistry. 1999;38:11026–39. doi: 10.1021/bi9909187. [DOI] [PubMed] [Google Scholar]
- 18.Fink D, Nebel S, Aebi S, et al. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–6. [PubMed] [Google Scholar]
- 19.Wei M, Cohen SM, Silverman AP, et al. Effets of spectator ligands on the specific recognition of intrastrand platinum- DNA cross-links by high mobility group box and TATA-binding proteins. J Biol Chem. 2001;276:38774–80. doi: 10.1074/jbc.M106374200. [DOI] [PubMed] [Google Scholar]
- 20.Kelland LR, Abel G, McKeage MJ, et al. Preclinical antitumor evaluation of bis-acetato-ammine-dichloro-cyclohexylamine platinum (IV): an orally active platinum drug. Cancer Res. 1993;53:2581–86. [PubMed] [Google Scholar]
- 21.Mellish KJ, Kelland LR, Harrap KR. In vitro platinum drug chemo-sensitivity of human cervical squamous cell carcinoma cell lines with intrinsic and acquired resistance to cisplatin. Br J Cancer. 1993;68:240–50. doi: 10.1038/bjc.1993.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Twentyman PR, Wright KA, Mistry P, et al. Sensitivity to novel platinum compounds of panels of human lung cancer cell lines with acquired and inherent resistance to cisplatin. Cancer Res. 1992;52:5674–80. [PubMed] [Google Scholar]
- 23.Wosikowski K, Lamphere L, Unteregger G, et al. Preclinical antitumor activiy of the oral platinum analog satraplatin. Cancer Chemother Pharmacol. 2007;60:589–600. doi: 10.1007/s00280-007-0502-z. [DOI] [PubMed] [Google Scholar]
- 24.Goddard PM, Valenti MR, Harrap KR. The role of murine tumor models and their acquired platinum-resistant counterparts in the evaluation of novel platinum antitumor agents: a cautionary note. Ann Oncol. 1991;2:535–40. doi: 10.1093/oxfordjournals.annonc.a058017. [DOI] [PubMed] [Google Scholar]
- 25.McKeage MJ, Kelland LR, Boxall FE, et al. Schedule dependency of orally administered bis-acetato-ammine-dichloro-cyclohexylamine-platinum (IV) (JM216) in vivo. Cancer Res. 1994;54:4118–22. [PubMed] [Google Scholar]
- 26.Rose WC. Combination chemotherapy involving orally administered etoposide and JM-216 in murine tumor models. Cancer Chemother Pharmacol. 1997;40:51–6. doi: 10.1007/s002800050624. [DOI] [PubMed] [Google Scholar]
- 27.Amorino GP, Mohr PJ, Hercules SK, et al. Combined effects of the orally active cisplatin analogue, JM216 and radiation therapy in antitumor therapy. Cancer Chemother Pharmacol. 2000;46:423–6. doi: 10.1007/s002800000169. [DOI] [PubMed] [Google Scholar]
- 28.McKeage MJ, Mistry P, Ward J, et al. A phase I and pharmacology study of an oral platinum complex, JM216: dose-dependent pharmacokinetics with single dose administration. Cancer Chemother Pharmacol. 1995;36:451–8. doi: 10.1007/BF00685793. [DOI] [PubMed] [Google Scholar]
- 29.McKeage MJ, Raynaud F, Ward J, et al. Phase I and pharmacokinetic study of an oral platinum complex given daily for 5 days in patients with cancer. J Clin Oncol. 1997;15:2691–700. doi: 10.1200/JCO.1997.15.7.2691. [DOI] [PubMed] [Google Scholar]
- 30.Raynaud FI, Boxall FE, Goddard P, et al. Metabolism, protein binding and in vivo activity of the oral platinum drug JM 216 and its biotransformation products. Anticancer Res. 1996;16:1857–62. [PubMed] [Google Scholar]
- 31.Raynaud FI, Mistry P, Donaghue A, et al. Biotransformation of the platinum drug JM216 following oral administration to cancer patients. Cancer Chemother Pharmacol. 1996;38:155–62. doi: 10.1007/s002800050464. [DOI] [PubMed] [Google Scholar]
- 32.Kelland L. Broadening the clinical use of platinum drug-based chemotherapy with new analogues: satraplatin and picoplatin. Expert Opin Investig Drugs. 2007;16:1009–21. doi: 10.1517/13543784.16.7.1009. [DOI] [PubMed] [Google Scholar]
- 33.Carr JL, Tingle MD, Mekeage MJ. Rapid biotransformation of satraplatin by human red blood cells in vivo. Cancer Chemother Pharmacol. 2002;50:9–15. doi: 10.1007/s00280-002-0462-2. [DOI] [PubMed] [Google Scholar]
- 34.Carr JL, Tingle MD, McKeage MJ. Satraplatin activation by hemoglobin, cytochrome C and liver microsomes in vitro. Cancer Chemother Pharmacol. 2006;57:483–90. doi: 10.1007/s00280-005-0069-5. [DOI] [PubMed] [Google Scholar]
- 35.Raynaud FI, Odell DE, Kelland LR. Intracellular metabolism of the orally active platinum drug JM216: influence of glutathione levels. Br J Cancer. 1996;74:380–6. doi: 10.1038/bjc.1996.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ando Y, Shimizu T, Nakamura K, et al. Potent and non specific inhibition of cytochrome P450 by JM216, a new oral platinum agent. Br J Cancer. 1998;78:1170–4. doi: 10.1038/bjc.1998.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beale P, Raynaud F, Hanwell J, et al. Phase I study of oral JM216 given twice daily. Cancer Chemother Pharmacol. 1998;42:142–8. doi: 10.1007/s002800050797. [DOI] [PubMed] [Google Scholar]
- 38.Sessa C, Minoia C, Ronchi A, et al. Phase I clinical and pharmacokinetic study of the oral platinum analog JM216 given daily for 14 days. Annals Oncol. 1998;9:1315–22. doi: 10.1023/a:1008441416790. [DOI] [PubMed] [Google Scholar]
- 39.Kurata T, Tamura T, Sasaki Y, et al. Pharmacokinetic and pharmacodynamic analysis of bis-acetato-ammine-dichloro-cyclohexylamine-platinum (IV) (JM216) administered once a day for five consecutive days: a phase I study. Jpn J Clin Oncol. 2000;30:377–84. doi: 10.1093/jjco/hyd102. [DOI] [PubMed] [Google Scholar]
- 40.Fokkema E, de Vries EG, Meijer S, et al. Lack of nephrotoxicity of new oral platinum drug JM216 in lung cancer patients. Cancer Chemother Pharmacol. 2000;45:89–92. doi: 10.1007/PL00006749. [DOI] [PubMed] [Google Scholar]
- 41.Hoffman P, Mauer A, Haraf D, et al. Oral JM216 plus concomitant radiotherapy (RT) for patients (pts) with advanced malignancies of the chest. Proc ASCO. 1998;17:488a. [Google Scholar]
- 42.Cmelak AJ, Choy H, Murphy BA, et al. Phase I study of JM216 with concurrent radiation in non-small cell lung cancer and squamous cell head and neck cancer. Proc ASCO. 1999;18:393a. [Google Scholar]
- 43.Jones SF, Greco FA, Hainsworth JD, et al. Phase I study of JM216 in combination with paclitaxel in patients with advanced malignancies. Proc ASCO. 1999;18:216. [Google Scholar]
- 44.Demario MD, Ratain MJ, Vogelzang NJ, et al. A phase I study of oral uracil/ftorafur (UFT) plus leucovorin and bis-acetato-ammine-dichloro-cyclohexylamine-platinum IV (216) each given over 14 days every 28 days. Cancer Chemother Pharmacol. 1999;43:385–8. doi: 10.1007/s002800050911. [DOI] [PubMed] [Google Scholar]
- 45.Judson I, Cerny T, Epelbaum R, et al. Phase II trial of the oral platinum complex JM216 in non-small cell lung cancer: an EORTC early clinical studies group investigation. Ann Oncol. 1997;8:604–6. doi: 10.1023/a:1008245709924. [DOI] [PubMed] [Google Scholar]
- 46.Schrump DS, Altorki NK, Henschke CL, et al. Non-small cell lung cancer. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. 7th edition Lippincot Williams & Wilkins; PA, USA: 2005. pp. 753–810. [Google Scholar]
- 47.Fokkema E, Groen HJ, Bauer J, et al. Phase II study of oral platinum drug JM216 as first line treatment in patients with small cell lung cancer. J Clin Oncol. 1999;17:3822–7. doi: 10.1200/JCO.1999.17.12.3822. [DOI] [PubMed] [Google Scholar]
- 48.Grant SC, Gralla RJ, Kris MG, et al. Single-agent chemotherapy trials in small –cell lung cancer, 1970 to 1990: the case for studies in previously treated patients. J Clin Oncol. 1992;10:484–98. doi: 10.1200/JCO.1992.10.3.484. [DOI] [PubMed] [Google Scholar]
- 49.Bristol-Myers Squibb Report. Jun 9, 1998. Accession No. 910068667. A randomized phase II study of satraplatin (JM216) or standard platinum therapy in patients with late relapses of epithelial ovarian cancer (CA 142-006) [Google Scholar]
- 50.Smith JW, McIntyre KJ, Acevedo PJ, et al. Results of a phase II open-label, nonrandomized trial of oral satraplatin in patients with metastatic breast cancer. Breast Cancer Res Treat. 2009;118:361–7. doi: 10.1007/s10549-009-0410-5. [DOI] [PubMed] [Google Scholar]
- 51.Trudeau M, Stuart G, Hirte H, et al. A phase II trial of JM-216 in cervical cancer: an NCIC CTG study. Gynecol Oncol. 2002;84:327–31. doi: 10.1006/gyno.2001.6409. [DOI] [PubMed] [Google Scholar]
- 52.Muthuramalingam SR, Patel K, Protheroe A. Management of patients with hormone refractory prostate cancer. Clin Oncol (R Coll Radiol) 2004;16:505–16. doi: 10.1016/j.clon.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 53•.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. This and reference 54 were two Phase III trials that established the efficacy of chemotherapy in metastatic CRPC.
- 54•.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. Together with reference 53, established the efficacy of chemotherapy in metastatic CRPC.
- 55.Latif T, Wood L, Connell C, et al. Phase II study of oral bis (aceto) ammine dichloro (cyclohexamine) platinum (IV) (JM-216, BMS-182751) given daily for 5 days in hormone refractory prostate cancer (HRPC) Invest New Drugs. 2005;23:79–84. doi: 10.1023/B:DRUG.0000047109.76766.84. [DOI] [PubMed] [Google Scholar]
- 56.Sternberg CN, Whelan P, Hetherington J, et al. Phase III trial of satraplatin, an oral platinum plus prednisone vs prednisone alone in patients with hormone refractory prostate cancer. Oncology. 2005;68:2–9. doi: 10.1159/000084201. [DOI] [PubMed] [Google Scholar]
- 57.Sternberg CN, Hetherington J, Paluchowska B, et al. Randomized phase III trial of a new oral platinum, satraplatin (JM-216) plus prednisone or prednisone alone in patients with hormone refractory prostate cancer [abstract 1586] Proc Am Soc Clin Oncol. 2003;22:395. doi: 10.1159/000084201. [DOI] [PubMed] [Google Scholar]
- 58.Armstrong A, George DJ. Satraplatin in the treatment of hormone refractory metastatic prostate cancer. Ther Clin Risk Manag. 2007;3:877–83. [PMC free article] [PubMed] [Google Scholar]
- 59•.Petrylak DP, Sartor OA, Witjes JA, et al. A Phase III randomized double blind trial of satraplatin and prednisone vs placebo and prednisone for patients with hormone refractory prostate cancer (HRPC) [abstract #145]; Proc Am Soc Clin Oncol Prostate Cancer Symp 2007 Initial report of the Phase III registration trial of satraplatin in metastatic CRPC. [Google Scholar]
- 60.Sartor OA, Petrylak DP, Witjes JA, et al. Satraplatin (S) demonstrates significant clinical benefits for the treatment of patients with HRPC: results of a randomized phase III trial; Proc Am Soc Clin Oncol; Chicago, IL, USA. May 2008; Abstract 5003. [Google Scholar]
- 61.Petrylak DP, Sartor AO, Witjes F, et al. Satraplatin in patients with advanced hormone-refractory prostate cancer (HRPC): Overall survival (OS) results from the phase III satraplatin and prednisone against refractory cancer (SPARC) trial; GU ASCO symposium; Orlando, FL, USA. Feb 2009; Abstract 163. [Google Scholar]
- 62.Vaishampayan U, Heilbrun L, Smith D, et al. Phase II trial of satraplatin, prednisone and bevacizumab in metastatic castrate resistant prostate cancer pretreated with docetaxel; GU ASCO symposium; Orlando, FL, USA. Feb 2009; Abstract 190. [Google Scholar]
- 63.Papatsoris AG, Karamouzis MV, Papavassiliou AG. Novel biological agents for the treatment of Hormone-Refractory Prostate Cancer (HRPC) Curr Med Chem. 2005;12:277–96. doi: 10.2174/0929867053363306. [DOI] [PubMed] [Google Scholar]
- 64.Garmey EG, Sartor O, Halabi S, et al. Second-line chemotherapy for advanced hormone-refractory prostate cancer. Clin Adv Hematol Oncol. 2008;6:118–22. [PubMed] [Google Scholar]
- 65.Azuma K, Komohara Y, Sasada T, et al. Excision repair cross-complementation group 1 predicts progression-free and overall survival in non-small cell lung cancer patients treated with platinum-based chemotherapy. Cancer Sci. 2007;98:1336–43. doi: 10.1111/j.1349-7006.2007.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azuma K, Sasada T, Kawahara A, et al. Expression of ERCC1 and class III beta-tubulin in non-small cell lung cancer patients treated with carboplatin and paclitaxel. Lung Cancer. 2009;6:326–33. doi: 10.1016/j.lungcan.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 67.Sessa C, Hess D, Bauer J, et al. J Clin Oncol 26: 2008 ASCO Annual Meeting. May 20 supplement; abstr 2560. [Google Scholar]
- 68.Wisinski K, Mulcahy M, Kuzel TM, et al. J Clin Oncol 26: 2008 ASCO Annual Meeting. May 20 supplement; abstr 13554. [Google Scholar]
- 69.Paola D, Alonso S, Alessio D, et al. J Clin Oncol 27, 2009 ASCO Annual Meeting (abstr e13534) [Google Scholar]
- 70.Leal TB, Wilding G, Eickhoff J, et al. J Clin Oncol 26: 2008 ASCO Annual Meeting. May 20 supplement (abstract 2570. [Google Scholar]
- 71.Thompson DS, Spigel DR, Hainsworth JD, et al. J Clin Oncol 26: 2008 ASCO Annual Meeting. May 20 supplement; abstr 19023. [Google Scholar]